


 , Vittorio Sciruicchio 1, Pasquale Parisi 3
, Vittorio Sciruicchio 1, Pasquale Parisi 31 Children Epilepsy and EEG Center, Presidio Ospedaliero (PO), San Paolo Hospital, 70132 Bari, Italy
2 Child Neuropsychiatry Unit Department, Pro.M.I.S.E. “G D'Alessandro", University of Palermo, 90127 Palermo, Italy
3 Department of Neuroscience, Mental Health & Sense Organs (NESMOS), Faculty of Medicine & Psychology, Sant’Andrea Hospital, 00198 Rome, Italy
4 Child Neuropsychiatry Department, Istituto Mediterraneo Eccellenza Pediatrica (ISMEP) - Azienda di Rilievo Nazionale di Alta Specializzazione (ARNAS) Civico Palermo, 90100 Palermo, Italy
5 Department of Maternal and Child Health, Policlinico Umberto I Hospital, Sapienza University of Rome, 00161 Rome, Italy
6 Pediatric Department, Azienda Ospedaliero Universitaria Pisana (AOUP) Santa Chiara Hospital, 56126 Pisa, Italy
†These authors contributed equally.
Abstract
Autonomic symptoms may be local and general clinical manifestations of both epilepsy and migraine caused by the dysfunction of brain areas best known as the central autonomic network. Despite their prevalence, autonomic signs are often misdiagnosed and their treatment is undervalued. This review aims to describe the autonomic manifestations reported during seizures and migraineur attacks according to their presentation, focusing on the role of the central autonomic network (CAN) and on the parasympathetic outflow that often-induced cranial autonomic symptoms (CAS) during migraineur attacks. Further, our purpose is to analyze the pathophysiological meanings and whether their presence influences the prognosis and therapy of these disorders.
Keywords
- migraine
- epilepsy
- autonomic nervous system
- headache
- seizures
- cranial autonomic symptoms
The autonomic nervous system (ANS) constitutes a highly intricate neuronal network that permeates the entire human body [1, 2, 3]. It assimilates information from both the internal body and the external environment, subsequently orchestrating bodily functions to ensure the maintenance of homeostasis. The ANS is essentially divided into two main components: the central autonomic network (CAN) and the peripheral nervous system (PNS) [2, 3]. The CAN comprises cortical structures such as orbitofrontal cortex, anterior cingulate cortex, the insular cortex, along with hypothalamus, the amygdala, and various brainstem nuclei (including the periaqueductal gray matter, parabrachial complex nucleus, nucleus of the solitary tract, medullary raphe and ventrolateral reticular formation of the medulla) some of which modulate sympathetic output while others oversee parasympathetic output [2, 3]. The PNS is tasked with receiving autonomic inputs from visceral receptors and relaying this information to the CAN. Autonomic outputs are bifurcated into the sympathetic and parasympathetic autonomic systems, which operate in both complementary and opposing manners [4]. Given the ANS’s role in regulating a multitude of organ-level functions, disturbances within the ANS can elicit a wide array of symptoms [1]. This paper aims to encapsulate the prevailing scientific insights on the involvement of the ANS in migraine and epilepsy and to examine the characteristic autonomic symptoms associated with these conditions.
Headache is one of the most prevalent neurological disorders, even in very young children, including infants [5, 6]. It is a significant cause of disability and a common reason for admissions to the emergency department in both adults and children [7, 8]. It ranks as the fifth most common cause overall and the third most common among women aged 15–64 [7]. Headaches are categorized into primary types, such as migraine, tension-type headache, and trigeminal autonomic cephalalgias (TACs), and secondary types, which are defined by their etiology [9, 10]. Genetic, biochemical and environmental factors are involved in pathophysiology [11, 12, 13, 14, 15]. The ANS is crucial in determining the onset and progression of primary headaches, particularly migraine and TACs, and in the development of cranial and systemic autonomic symptoms in secondary headaches. The ANS operates through central and peripheral pathways. In primary headaches, the trigeminovascular system and the trigeminal autonomic reflex are responsible for cranial autonomic symptoms (CAS), including forehead sweating, facial flushing, miosis, lacrimation, conjunctival injection, eyelid edema, ptosis, irritability, rhinorrhea, nasal congestion, ear redness, ear swelling, throat swelling, and voice changes [16].
Autonomic dysfunctions in secondary headaches are likely triggered by the autonomic baroreflex system [17]. This system adjusts heart rate, blood pressure, and vascular tone to maintain cerebral blood flow during changes in body posture through a combination of peripheral (via the glossopharyngeal and vagus nerves) and central mechanisms, dependent on body orientation. Dysfunction in this reflex can lead to fluctuating blood pressure levels and headaches [17].
During an attack of primary headache, the pain-sensitive trigeminal afferents from the ophthalmic branch of the trigeminal nerve are activated. After synapsing in the trigeminocervical complex (TCC) within the brainstem, they send pain signals to subcortical and cortical areas, facilitating pain perception through trigeminovascular activation. The TCC is connected with the CAN, including the locus coeruleus, raphe nucleus, periaqueductal gray, thalamus, hypothalamus, and cortex, regulating pain processing and perception, which allows for the descending modulation of the TCC [16, 18]. The trigeminal autonomic reflex arises from efferent fibers in the TCC that project to the superior salivatory nucleus, where preganglionic parasympathetic neurons are activated before synapsing in the sphenopalatine ganglion. Postganglionic parasympathetic fibers act on the lacrimal glands and the nasal and palatal mucosa, triggering lacrimation, nasal congestion, or rhinorrhea [16, 17, 18].
Furthermore, activation of the parasympathetic nervous system results in the release of several neuropeptides into the bloodstream (including calcitonin gene-related peptide - Calcitonin Gene-Related Peptide (CGRP), vasoactive intestinal peptide, substance P and pituitary adenylate cyclase-activating peptide-38), involved in vasodilation and mast cell degranulation [18, 19, 20, 21]. The release of proinflammatory substances by trigeminal sensory fibers increases neuroinflammation, intensifying the level and persistence of pain. Additionally, hypofunction of the sympathetic system is crucial in miosis and/or ptosis during a headache attack, potentially triggered by the compression of sympathetic fibers downstream of the post-ganglionic sympathetic fibers by perivascular edema caused by neurogenic inflammation [17, 22, 23, 24]. This paper briefly reviews the complex peripheral and central pathogenetic mechanisms that precipitate migraine attacks and associated symptoms, as detailed in more comprehensive texts.
Migraine is characterized by the occurrence of recurrent painful episodes lasting 4 to 72 hours (or 2 to 48 hours in children under 14 years), typically of moderate to severe intensity, with a predominantly unilateral presentation, though bilateral in children [9]. The pain is pulsating in nature and exacerbated by routine physical activity. It is commonly accompanied by nausea and vomiting, as well as photophobia and phonophobia [9]. The migraine attack consists of several phases: prodromal symptoms, aura, the headache phase, and postdrome symptoms. The International Classification of Headache Disorders, 3rd edition (ICHD-3), categorizes migraines into those without aura and those with aura, and identifies chronic migraine as headaches occurring on 15 or more days per month for over three months [9].
At the core of migraine pathogenesis is the trigeminal-vascular system, with the activation of CGRP playing a crucial role. This peptide significantly influences the development of migraine, highlighting the importance of the trigeminal-vascular system (TVS) and CGRP [25]. The efficacy of preventive therapies with anti-CGRP monoclonal antibodies and CGRP receptor antagonists, known as gepants, further emphasizes their critical role in managing migraine attacks [25, 26, 27].
In summary, while numerous neuropeptides, chemical mediators, and systems are involved in migraine, substantial evidence underscores the significant involvement of the ANS in this complex condition. The ANS plays a key role in coordinating the various aspects of migraine, from the initial prodromal symptoms to the concluding postdrome.
Migraine is a multifaceted syndrome, not only manifesting as pain but also encompassing a wide array of signs and symptoms that affect mood, cognition, sleep, arousal, and nutritional patterns. Certain symptoms, known as prodromes, can emerge hours before the migraine attack, serving as early indicators for the patient [28].
Prodromic symptoms encompass sensory hypersensitivity to light, noise, and movement; mood disturbances; sleep irregularities; and neuroendocrine changes such as increased thirst, cravings, yawning, and frequent urination [28]. A significant body of neuroimaging research suggests that the hypothalamus, with its extensive connections, plays a pivotal role in the perception and transmission of pain [18]. The nociceptive pathways of the trigeminovascular neurons relay information to the hypothalamic nuclei and, through connections to the cortex, brainstem, and autonomic preganglionic neurons, contribute to many of migraine’s common manifestations, including affective, autonomic, endocrine, and general physiological responses. Nociceptive activation may be initiated by both exogenous and endogenous factors, affecting the meninges (trigeminovascular activation) and the hypothalamus, thus triggering prodromal symptoms and migraine attack [18, 29].
In the premonitory phase, the activation of meningeal nociceptors results from the release of pro-inflammatory neuropeptides, tied to an increase in parasympathetic tone in the ganglionic parasympathetic neurons within the superior salivatory nucleus, following hypothalamic activation. Moreover, the significance of autonomic activation is underscored by the occurrence of cranial CAS prior to, and independently of, the headache phase, illustrating that the 24 headache is not a prerequisite for the emergence of CAS [30].
In this brief summary, the role of the dopaminergic system in the onset of prodromal symptoms (see yawning, feeling of fatigue, drowsiness, nausea, etc.) should not be forgotten, but this in-depth study is beyond the scope of this review [18].
Migraine auras represent a constellation of symptoms indicative of cortical dysfunction, encompassing neurologic, gastrointestinal, and autonomic manifestations. These phenomena may precede or accompany a migraine attack [9, 18]. The visual aura is the most prevalent form of aura and is typically localized to the occipital lobe of the brain. Cortical spreading depression (CSD) initiates as a localized disturbance that progressively expands, potentially extending to subcortical levels [18]. Recent evidence suggests that CSD may also originate from deeper brain structures, both from a clinical perspective [31] and a functional standpoint, culminating in parasympathetic activation [32].
This novel insight posits that CSD could trigger mechanisms leading to parasympathetic activation. The observed parasympathetic hypertonia during migraine aura episodes provides a plausible explanation for the increased prevalence of autonomic and localized symptoms in individuals experiencing migraines with aura [17, 32]. These symptoms can include but are not limited to, excessive sweating, syncope, abdominal pain, and tearing.
Although the activation of trigeminal sensory fibers, mediated by the TVS and the release of neuropeptides such as CGRP and pituitary adenylate cyclase-activating polypeptide (PACAP), is the primary pathophysiological mechanism underlying migraine pain, the onset and persistence of pain are also influenced by the release of proinflammatory mediators from intracranial vessels. This process is triggered by the activation of both parasympathetic and sympathetic fibers [16, 17, 33].
Additionally, autonomic activation via the autonomic trigeminal reflex may not only manifest clinical signs and symptoms but also contribute to the development of pain. During migraine attacks, the occurrence of CAS, is due to the connections of the TVS with the superior salivatory nucleus and the sphenopalatine ganglion, while the third-order neurons of the TVS are involved in specific autonomic behaviors. In primary headaches, enhanced cranial parasympathetic outflow often leads to trigeminal autonomic symptoms. CAS, typically unilateral in TACs, have been observed more mildly and bilaterally in migraines recently [34].
CAS result from the trigeminal autonomic reflex (TAR), a protective reflex activated by nociceptive stimulation of the trigeminal nerve, which contributes to the intensity of headache pain. Consequently, TAR may be a target for various preventive and acute headache treatments and predict the response, including triptans, botulinum toxins, oxygen therapy and high-frequency stimulation of the sphenopalatine ganglion, all of which alleviate autonomic symptoms by acting on the TAR [35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45].
Barbanti et al. [46], twenty years ago, noted the presence of CAS during migraine attacks, even unilaterally, and their correlation with the response to triptans. Similar findings have emerged from studies on CAS in children with migraines; CAS may be considered a risk factor for the chronicization of migraine, and the early presence of CAS in youth has been suggested to contribute to the persistence of headache into adulthood [47].
CAS in migraines may occur in all phases of the attack, are more likely to be bilateral, especially in children [48, 49, 50], and are described as less severe than in TACs [34]. Recently, besides the commonly recognized CAS, additional symptoms like red ear, throat swelling, voice change, grittiness, and scratchiness have been associated with CAS [51]. Symptoms such as visual blurring, one of the most common, may stem from cranial autonomic dysfunction caused by an imbalance in sympathetic and parasympathetic signaling. Moreover, potential sympathetic hypofunction might lead to miosis and hyperexcitability in the accommodative response. Observations of mydriasis at the photophobia threshold, impaired pupillary constriction, and re-dilation latency suggest a mixed cranial autonomic dysfunction [17].
Studies indicate that migraines with CAS may feature more severe, frequent, and prolonged attacks, but also a better response to triptan therapy [36, 37]. In our Fig. 1 (Ref. [16]), we list various CAS along with their prevalence range in migraines as reported in the literature [16]. In Fig. 2 (Ref. [16]), we show the anatomical pathways involved in inducing cranial autonomic symptoms in migraine attacks. It’s important to note that CAS are hallmark signs and symptoms of TACs, which include disorders like cluster headache, hemicrania continua, paroxysmal hemicrania, and short-lasting unilateral neuralgiform headache attack syndromes, all defined by recurrent episodes of unilateral facial pain lasting 15 to 180 minutes, accompanied by CAS [9]. TACs are linked to hypothalamic dysfunction involving the paraventricular and suprachiasmatic nuclei, crucial for circadian periodicity, leading to TVS and TAR activation and specific autonomic-behavioral patterns. In cluster headaches, CAS are usually unilateral and occur before and during attacks, especially nasal congestion, lacrimation, and conjunctival injection [26]. These shared manifestations between migraines and the presence of aura and general vegetative signs in cluster headaches may suggest alternative pathophysiological hypotheses (see the modular theory) [52]. Recently, an interesting study reported a high prevalence of CAS even in a population with episodic tension headache, although lower than in the migraine population. This finding, if confirmed by other studies, would point to difficulties in the differential diagnosis between the two forms. However, it should be noted that these subjects reported more disability than subjects with CAS-negative episodic tension-type headache. This observation suggests reflections on the relationship between tension-type headache and migraine as a continuum and not separate disorders. Certainly further studies, both clinical and instrumental, are needed to confirm or exclude this hypothesis [53].
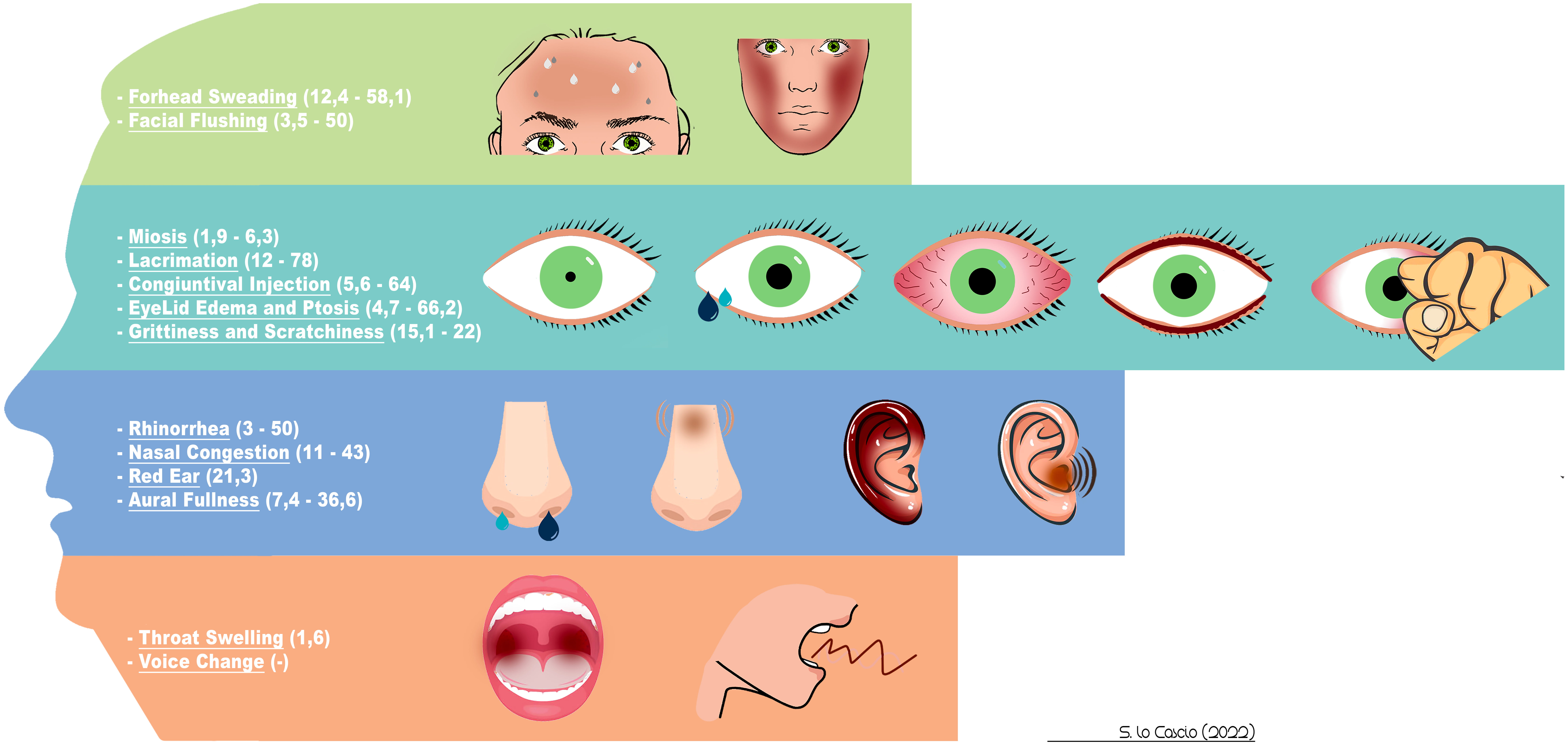 Fig. 1.
Fig. 1.Autonomic cranial symptoms and prevalence. Schematic and figurative illustration of all autonomic cranial symptoms in migraine and their prevalence (%) based on literature review. Illustration by Salvatore Lo Cascio. From Lo Cascio et al. Cranial Autonomic Symptoms and Migraine: What Relationship and What Meaning? A Review J. Integr. Neurosci. 2022; 21(6): 166 https://doi.org/10.31083/j.jin2106166[16].
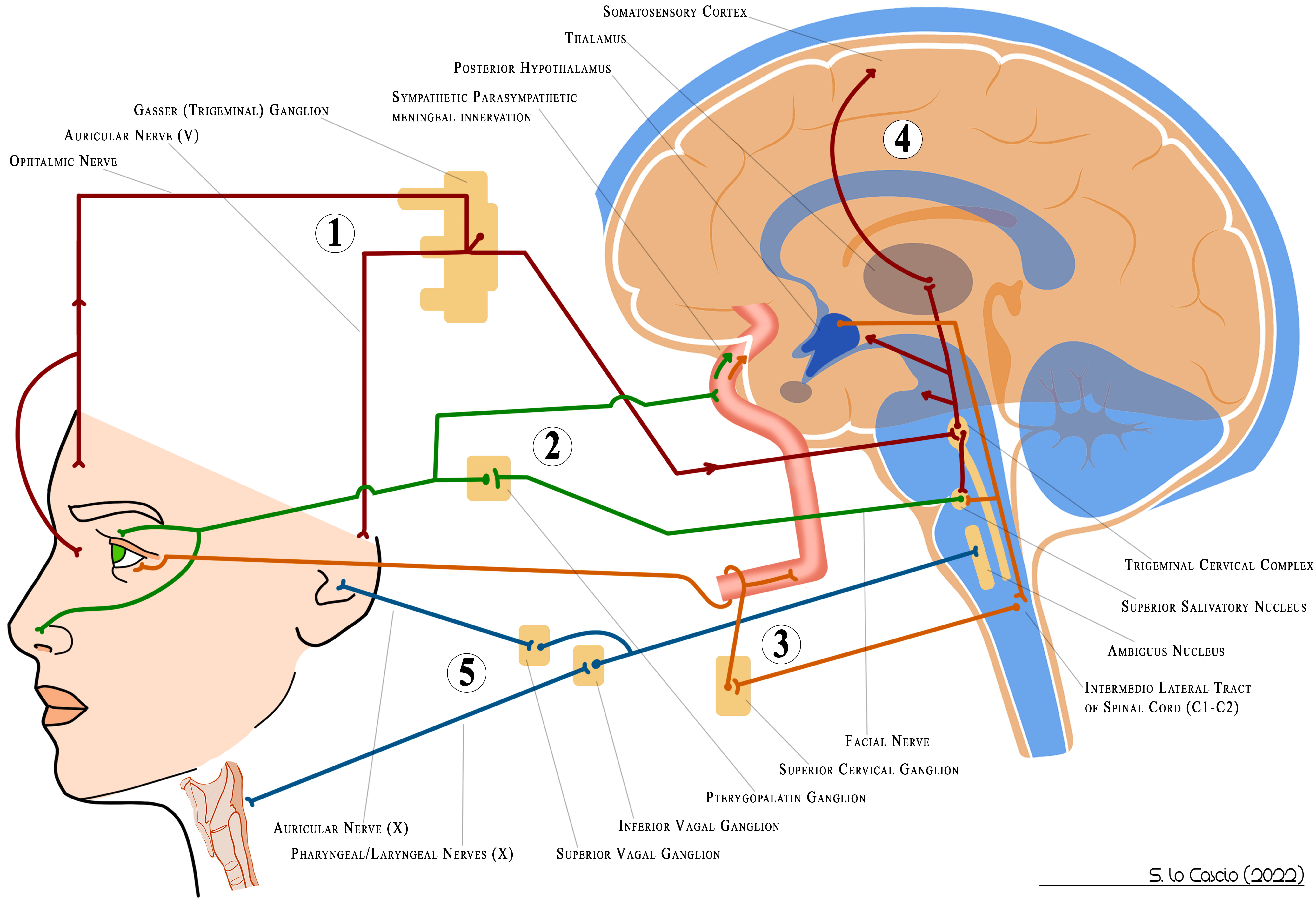 Fig. 2.
Fig. 2.Anatomical pathways involved in inducing cranial autonomic symptoms during migraine attacks. ① Trigeminal Reflex. ② Cranial parasympathetic activation mediated by Facial Nerve. ③ Sympathetic modulation. ④ Central nervous system (CNS) activation, thalamus and somatosensory cortex to pain, posterior hypothalamus to autonomic modulation. ⑤ Possible anatomic pathway for aural fullness, throat swelling and voice change. Illustration by Salvatore Lo Cascio. From Lo Cascio et al. Cranial Autonomic Symptoms and Migraine: What Relationship and What Meaning? A Review J. Integr. Neurosci. 2022; 21(6): 166 https://doi.org/10.31083/j.jin2106166[16].
Research indicates that visceral symptoms, such as nausea, vomiting, constipation, diarrhea, stomach fullness, bloating, belching, frequent defecation, and urination, are prevalent across all stages of a migraine attack. The presence of these visceral symptoms is linked to more severe and debilitating migraines [54]. Thus, identifying the phenotypic presentation of associated migraine symptoms, particularly during the pre-attack phase, could facilitate early and effective management strategies.
Migraines accompanied by aura are often associated with more extensive autonomic dysfunction than those without aura, with nausea being the most frequently reported autonomic symptom. The underlying mechanisms of nausea are multifaceted, encompassing disturbances in gastrointestinal motor and sensory functions, autonomic dysfunction, and central nervous system regulation. Vomiting, a notable symptom during migraine episodes, results from the activation of the nucleus tractus solitarius, which communicates with the dorsal motor nucleus of the vagus [18]. This last, in turn, orchestrates parasympathetic and sympathetic efferent pathways to the gastrointestinal tract and extends within the spinal nerves to the diaphragm and abdominal muscles, playing a crucial role in the manifestation of emesis during migraine attacks [18, 29].
Currently, only a limited number of studies concentrate on the postdrome phase of migraines. Although the ICHD-3 [9], describes the postdrome as “a symptomatic phase lasting up to 48 hours following the resolution of pain in migraine attacks, with or without aura”, few research efforts adhere strictly to this definition. These studies vary in their definitions and methodologies for assessment. Interestingly, many symptoms reported during the postdrome phase mirror those experienced during the prodrome, often beginning even during the painful phase of the attack [55, 56, 57]. The main ones are shown in the Table 1.
| Prodromes | Aura | Pain Phase | Postdromes |
| Food Craving | Visual | Headache | Astenia |
| Yawning | Sensitive | Nausea | Sadness |
| Odor Sensibility | Language | Vomit | Abdominal Pain |
| Thirsty | Motor | Phonophobia | Ocular Pain |
| Irritability | Cognitive | Photophobia | Nausea |
| Sleepness | Others | Osmophobia | Sleepness |
| Polyuria | Affettive Symptoms | Cognitive Difficulties | |
| Intolerance Light And Sound | Cognitive Symptoms | Neck Pain | |
| Cas | Cas | Fatigue | |
| Mood Change | Allodynia | Mood Change | |
| Neck Pain | Yawning | ||
| Vertigo | Gastrointestinal Symptoms |
It has been proposed that the frontal lobes and the hypothalamus play a crucial role in the pathophysiology of the postdrome phase of migraines [56]. Functional neuroimaging studies have revealed a reduction in blood flow across various cortical and subcortical regions, notably within the hypothalamus, frontal lobes, and limbic areas [56]. The array of symptoms observed during the postdrome phase suggests the engagement of the central autonomic system, mediated by the hypothalamus through its connections with parasympathetic, sympathetic, and brainstem projections [56].
Significant research has been undertaken to pinpoint markers of autonomic activation during the interictal phase of migraine, employing a range of methods including the assessment of cardiovascular reflexes, evaluation of pupillary and vascular reactivity, and the administration of drugs that mimic sympathetic and parasympathetic activity [58]. However, the outcomes of these studies have been inconsistent, revealing evidence of both hyperfunction and hypofunction within the sympathetic and parasympathetic systems. A recent review concludes that, as of now, “there is currently no apparent autonomic deficit considered intrinsic to migraine” [59].
Epileptic seizures are characterized as “a temporary manifestation of signs and/or symptoms due to abnormal, excessive, or synchronous neuronal activity in the brain [60, 61]”, often accompanied by autonomic symptoms. These symptoms may either supplement other seizure manifestations or serve as the primary feature of the seizure. Clinically significant autonomic disorders may accompany all seizure types (generalized, focal, and/or unknown onset) across all phases (early ictal, ictal, and post-ictal). Moreover, since ictal autonomic signs are either forgotten or unrecognized, they are often overlooked [62]. The most prevalent forms of epilepsy that involve autonomic phenomena include temporal lobe epilepsy (TLE) and self-limited epilepsy with autonomic seizures (SeLEAS). However, complex autonomic dysfunction is also a defining characteristic of some developmental and epileptic encephalopathies (DEE), and both primary and secondary involvement of the ANS is crucial in the etiology of sudden unexpected death in epilepsy (SUDEP) [63]. Autonomic symptoms can range from mild seizure manifestations to severe, life-threatening events [64]. This wide spectrum of symptoms is believed to be mediated by cortical discharges that utilize the CAN pathways [65], predominantly located in the non-dominant hemisphere [66]. Autonomic signs typically manifest early during the ictal phase when the cortex engages the CAN at the outset. Changes in autonomic function may precede ictal electroencephalographic activity by several seconds [67]. There is often preictal tachycardia, observed in approximately one-third of cases, along with an increase in sympathetic activity during the preictal and early ictal phases [68]. A recent retrospective study introduced a modified index reflecting sympathetic tone, showing promising results in seizure detection. An algorithm based on these changes achieved a sensitivity of 88% in identifying seizures in a small cohort of patients with temporal lobe epilepsy [69]. Alterations in heart rate have been documented to occur early in or even before a seizure, suggesting a potential predictive role [70]. However, understanding of the mechanisms behind these changes remains limited. Pre-ictal heart rate patterns were more commonly observed in mesial TLE, which involves areas closely linked to CAN centers, compared to lateral TLE or other lobe origins. Typically, pre-ictal heart rate increases occur within a 5–60 second window before seizure onset [71]. On the contrary, autonomic symptoms may appear post ictal or later during the seizure [72]. Autonomic seizures, characterized by symptoms stemming from dysfunctions in systems controlled by the ANS [73], are more common in children, likely because of a subcortical lower seizure threshold, attributed to the presumed immaturity of the CAN [74, 75]. Certain autonomic symptoms can hint at the seizure’s origin, although some signs may arise due to the spread of discharge [76]. They can result from sympathetic nervous system overactivity, though the parasympathetic nervous system, especially in cardiovascular autonomic dysfunction, also contributes to some symptoms [77].
Autonomic dysfunctions are often identified by recurring, stereotypical symptoms that impact various systems including cardiovascular, neuroendocrine, respiratory, genitourinary, sexual, gastrointestinal, as well as skin and pupillary reactions [78, 79]. Cardiovascular changes, such as ictal sinus tachycardia, ictal bradycardia-asystole, postictal ventricular fibrillation and atrial flutter/atrial fibrillation, observed in both generalized and focal seizures, are implicated as potential mechanisms contributing to SUDEP [80, 81]. Cardiac responses are modulated by the insula: the right side for sympathetic activation and the left side for parasympathetic regulation [82, 83]. Notably, ictal sinus tachycardia is documented in 82% of epilepsy patients [84], and focal to bilateral tonic-clonic seizures are associated with a more significant increase in ictal heart rate compared to focal seizures with impaired consciousness [28]. Ictal bradycardia, less frequent than tachycardia, is reported in 5 to 8% of seizures [85, 86]. Ictal bradycardia is often linked to seizures in the left hemisphere, likely due to the left insular cortex’s influence in triggering this symptom [82, 83]. Although rare, ictal asystole, followed by syncope, is noted in individuals with temporal lobe epilepsy [87, 88].
Respiratory symptoms frequently occur during focal seizures originating in the lower brain stem, where central respiratory centers are under the forebrain cortical areas’ control, including the hippocampal formation, basal forebrain, insula, anterior cingulate gyrus, and motor area. Temporal lobe epilepsy often involves apnea and oxygen desaturation, with the severity of desaturation correlating with the onset in the temporal lobe, seizure lateralization to the right hemisphere, duration, and speed of contralateral seizure spread [89, 90, 91]. the highest risk of seizure-related respiratory impairments is observed in TLE patients experiencing contralateral diffusion [92]. Exaggerated autonomic stimulation, leading to excessive respiratory secretions, may trigger concurrent hypersalivation and retching, indicating direct CAN activation. These symptoms usually originate from the temporal lobe’s mesial portion, with uncertain lateralization [93, 94]. The same cortical area, particularly the insular cortex, is implicated in gastrointestinal auras in 83% of cases, representing the most common symptoms of focal epilepsy in adults [95, 96].
During focal seizures, pallor, sweating, flushing, and piloerection, frequently paired with sensations of cold and warmth, may occur. Mostly in younger individuals, seizures origin from the left temporal lobe are characterized by ictal pallor [97]. Ictal hypersalivation is frequently seen in self-limited focal epilepsies of childhood, such as self-limited epilepsy with centrotemporal spikes and self-limited epilepsy with autonomic seizures [98, 99]. In a small study involving ten adults, the researchers demonstrated that this rare indicator serves as a distinguishing marker for mesial temporal seizures, primarily originating from the non-dominant hemisphere [100].
TLE [101] accounts for approximately 40% of all adult epilepsy cases [102] and 60–75% of those with drug-resistant epilepsy [103]. TLE encompasses a diverse set of conditions unified by the location of the epileptogenic zone within the temporal lobe, whether in the lateral or mesial regions. the most prevalent form is the mesial TLE and it is arguably the most recognized electro-clinical pattern among all epilepsies [104, 105]. Typically, the symptomatology of focal seizures begins with an epileptic aura, which are subjective symptoms indicating the initial seizure discharge in the brain and may sometimes be the sole clinical manifestation.
It is established that regions such as the anterior cingulum, insular cortex, posterior orbitofrontal cortex, supplementary sensorimotor area, and amygdala play a role in altering respiratory and heart rates, inducing mydriasis, piloerection, and genitourinary symptoms [105, 106, 107, 108, 109]. The recurrence of uncontrolled seizures, as observed in drug-resistant TLE, may lead to epilepsy-associated autonomic dysfunction, because of the persistent activation of the CAN. As the disease progresses and seizure frequency increases, this autonomic dysfunction becomes more pronounced in patients with TLE [110]. This is evident in the triadic symptoms of retching, nausea, and vomiting, which are characteristic of SeLEAS, although infrequent in temporal epilepsy [111].
The symptom-generating zone for abdominal auras, the most common type of autonomic aura, includes the anterior insular cortex, mesial temporal structures, frontal operculum, and supplementary motor area [112]. Focal aware seizures may also feature autonomic symptoms, along with emotional (e.g., fear), cognitive (e.g., deja vu, jamais vu), or sensory (e.g., olfactory, gustatory, visual, auditory) manifestations. Sensory auras, especially those that are olfactory and often reported as disagreeable odors coupled with gustatory sensations, known as “uncinate fits”, are historically associated with TLE, yet only occur in about 5% of patients [112] and involve the amygdala, olfactory bulb, orbitofrontal cortex and insular cortex [109, 111]. Visual auras can be simple or complex, with the former resulting from activation of visual association areas and adjacent contralateral primary visual cortex, and the latter involving the temporo-occipital junction or basal temporal cortex [113].
In mesial TLE, fear is a common emotional symptom, primarily associated with the amygdala, although other areas such as the mesial frontal regions, parietal and occipital lobes have also been implicated [114, 115].
Symptoms such as epigastric sensations, cold shivering, piloerection, and urinary incontinence, especially during focal to bilateral seizures, may indicate frontal lobe involvement [116]. Additionally, differentiating between frontal and temporal lobe epilepsy can be challenging, particularly when ictal tachycardia is observed. A recent investigation into the differences between TLE and frontal lobe epilepsy (FLE) using ultra-short-term heart rate variability (HRV) analysis revealed distinct HRV profiles during pre-ictal, ictal, and post-ictal phases between the groups: patients with TLE showed increased sympathetic or vagal activity in the pre-ictal and post-ictal phases, whereas, during the ictal period, FLE patients experienced significant changes in sympathetic tone [117].
Among the first epilepsy syndromes to manifest with autonomic signs and a
possible autonomic status epilepticus in children is SeLEAS, previously referred
to as Panayiotopoulos syndrome or early-onset benign occipital epilepsy
[118, 119, 120, 121, 122]. The typical onset age ranges from 3 to 6 years, with the majority of
cases (70%) manifesting at around 5 years of age [121, 122]. SeLEAS represents
5% of all epilepsies in children aged 1 to 14 years and accounts for 13% of
epilepsies in the 3 to 6-year age group [122, 123, 124]. The diagnosis requires the
presence of focal autonomic seizures, which may or may not include impaired
awareness [109, 119]. Initial autonomic symptoms most commonly involve pallor,
retching, nausea, general discomfort, abdominal pain or flushing. Occurring in
about 75% of affected children [118] vomiting is the most frequent autonomic
symptom; in some cases symptoms may be limited to nausea or retching. Seizures
often progress to include eye and/or head deviation, generalized hypotonia, and
either focal clonic (hemiclonic) or focal to bilateral tonic–clonic movements.
These seizures are typically prolonged (lasting over 30 minutes) and in 70% of
cases, they begin during sleep [121, 125, 126]. Electroencephalography reveals
multifocal spikes characterized by high amplitude sharp-slow wave complexes
(
It’s rare, but SeLEAS can progress to epileptic encephalopathy with spike-and-wave activation during sleep (EE-SWAS). While likely genetically influenced, no specific causative gene variants have been identified to date. There is an observed higher prevalence of febrile seizures among first-degree relatives and case reports of siblings with other forms of SeLFEs [128]. Typically, prophylactic antiepileptic drug treatment are not needed for most patients as the disease is a remarkably benign condition with a favorable prognosis [129].
Autonomic symptoms can be observed in various DEEs, often serving as indicators of epileptic events or, more commonly, as markers closely associated with the extent of functional disabilities [130]. Rett syndrome and Cyclin-dependent kinase-like 5 (CDKL5) Deficiency Disorder, are two neurodevelopmental disorders predominantly affecting females, characterized by frequent occurrences of seizures and paroxysmal autonomic symptoms [131, 132, 133]. Autonomic disturbances in these conditions include peripheral vasomotor dysfunctions, breathing irregularities during wakefulness, apnoea, and cardiac dysautonomia, increasing the risk of arrhythmias [133, 134].
Dravet syndrome, an epileptic encephalopathy manifesting in the first year of life, is characterized by various seizure types [135]. While the diagnosis is based on clinical observations, most affected individuals have mutations in the Sodium voltage-gated channel alpha subunit 1 (SCN1A) gene, which encodes neuronal sodium channels [85]. Issues with temperature regulation are reported in half of the patients; compared to healthy controls, in Dravet syndrome patients’ additional autonomic dysfunctions like pupillary dilation, abnormal sweating, flushing, gastroparesis, and alterations in heart rate are more frequently observed [135, 136]. In addition, autonomic symptoms are commonly observed as the initial sign of focal or focal to bilateral tonic/tonic-clonic seizures, in patients affected by SCN8A mutations, to the extent that they are considered hallmarks of this DEE [136]. A sequence of autonomic symptoms is frequently noted: some symptoms appearing within the first seconds (such as facial flushing, sometimes accompanied by bradycardia, sialorrhea, and hypopnea), and some others like tachycardia, perioral cyanosis, polypnea, and pallor emerging later during the seizure [136]. Prolonged apnea requiring ventilatory support, occurring during a tonic seizure is considered distinctive of individuals with this mutation [137]. Additionally, Trivisano et al. [138] recently reported a patient affected by DEE due to SCN8A mutation featuring ictal asystole that necessitated the implantation of a cardiac pacemaker. Ictal bradycardia has also been reported in patients suffering from DEE secondary to mutations in the Fibroblast Growth-Factor Homologous Factor 1 (FHF1) gene, which encodes for small cytosolic proteins. These proteins interact with the cytoplasmic tails of voltage-gated sodium channels (Nav1.6), encoded by SCN8A, and enhance excitability by increasing the voltage dependence of fast inactivation of neuronal sodium channels. In this condition, sporadic cases of SUDEP are also reported [139].
Finally, autonomic signs are frequently reported in other DEEs [140, 141] and in other genetically determined neurodegenerative diseases with epilepsy, often increasing the morbidity of affected patients.
In this context, dysautonomic features may serve as biomarkers for the severity of DEE [130] and should be evaluated during follow-up.
In summarizing the critical insights garnered from our review, it’s evident that the ANS is crucial in the pathophysiology of both migraine and epilepsy. This complex and multifaceted neural network, essential for regulating unconscious and involuntary functions, maintains total body homeostasis. Dysfunctions within the ANS, attributable to various diseases, manifest a broad spectrum of symptoms across different conditions, including migraine and epilepsy. These conditions exhibit diverse autonomic signs and symptoms, underscoring the intricate involvement of the ANS. A deeper understanding of the specific autonomic symptoms and their correlation with different migraine phases or seizure types is instrumental in enhancing diagnosis, prevention, and treatment strategies for these disorders. Recognizing the connection between autonomic symptoms and these neurological conditions facilitates a more nuanced approach to patient care, allowing for targeted interventions that address the underlying autonomic dysfunctions. However, our current understanding of the mechanisms driving ANS activation and symptom generation in migraine and epilepsy remains incomplete. There is a pressing need for more research and evidence to elucidate the physiological pathways leading to ANS dysfunctions. Such efforts will not only improve our comprehension of these complex interactions but also pave the way for developing innovative therapeutic strategies aimed at mitigating the impact of ANS-related symptoms in affected individuals.
DDA, FC, AF, VR, VS and PP designed the research study. DDA, FC and AF drafted the manuscript. SLC designed the figures. DDA, FC, AF, SLC, EC, GT, AS and GB performed the literature searches. VR, VS and PP critically revised the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.