
 , Zhenyu Fan 1,*
, Zhenyu Fan 1,*1 Department of Pharmacy, Affiliated Hospital of Nantong University, 226001 Nantong, Jiangsu, China
2 Department of Pathology, Affiliated Hospital of Nantong University, 226001 Nantong, Jiangsu, China
3 Medical Research Center, Affiliated Hospital 2 of Nantong University, 226001 Nantong, Jiangsu, China
†These authors contributed equally.
Abstract
Objectives: Endothelial-to-mesenchymal transition (EndoMT) is a
significant biological phenomenon wherein endothelial cells undergo a loss of
their endothelial traits and progressively acquire mesenchymal characteristics.
Consequently, this transformation leads to both a compromised ability to maintain
lumen permeability and alterations in vascular structure, which hampers the
preservation of blood-brain barrier integrity. This study aimed to investigate
inflammation-induced EndoMT and its etiology, with the goal of impeding the
infiltration of peripheral inflammation into the central nervous system.
Materials and Methods: Lipolysaccharide (LPS) was administered
intraperitoneally to mice several times to establish a chronic inflammatory
model. A cellular inflammatory model was established by LPS in human brain
microvascular endothelial cells (HBMECs). The mRNA expressions of inflammatory
cytokines interleukin-1
Keywords
- EndoMT
- Sestrin2
- autophagy
- inflammation
The brain blood vessels serve as a vital interface for the exchange of nutrients, metabolites and gases between the peripheral and central nervous system (CNS), as well as a protective barrier against exogenous substances and neurotoxic components in blood [1]. Vascular endothelial cells, which form a flattened epithelial layer along the internal surfaces of the heart, blood vessels, and lymphatics, exhibit phagocytic capabilities towards foreign bodies, bacteria, necrotic and senescent tissues, and actively participate in the immune response of an organism [2]. When blood is contaminated with harmful substances, such as endotoxins and inflammatory factors, endothelial cells are initially subjected to abnormal blood environments, leading to impaired structure and function of these cells. Previous research has demonstrated that under the influence of various stimuli, endothelial cells undergo a transformation, losing their original characteristics and becoming deficient in tight junction proteins, thereby undergoing an endothelial-to-mesenchymal transition (EndoMT). This transition is detrimental to maintaining blood-brain barrier homeostasis and has the potential to facilitate the infiltration of peripheral inflammation into the CNS. Inhibition of EndoMT has significant alleviating effect on CNS disease [3, 4, 5]. However, the majority of reports about EndoMT are focused on peripheral vascular and organ injuries, and EndoMT in the CNS requires further exploration.
Sestrins, a group of stress-induced proteins, exhibit a high degree of
conservation across various organisms and are integral to cellular adaptation in
response to stress [6]. The diverse functions of Sestrins are observed in
different organs and tissues. Sestrin2, a homologous protein to Sestrin1, is a
remarkably conserved antioxidant protein initially recognized as hypoxia-inducing
gene 95. Research has demonstrated that Sestrin2 accumulates in cells subjected
to stress and assumes a crucial function in suppressing the generation of
reactive oxygen species (ROS), consequently safeguarding cells against oxidative
harm [7]. As a stress-induced metabolic regulator, Sestrin2 restrains oxidative
stress and pro-inflammatory signaling pathways through mechanisms reliant on
adenosine 5
Male C57BL/6J mice (6–8-week-old) were obtained from the Experimental Animal Center of Nantong University. For the duration of the experiment, the mice were housed in a controlled environment that ensured pathogen-free conditions, consistent temperature and relative humidity. Treated mice were injected with 1 mg/kg/day LPS (L8880, Solarbio, Beijing, China) intraperitoneally for 5 days, while control mice were injected intraperitoneally with an equivalent volume of physiological saline for the same duration. The mice were subjected to anesthesia by isoflurane (R510-22-10, Rayward, Shenzhen, Guangdong, China) and underwent heart perfusion using physiological saline. Brain samples were gathered for further analysis. It is important to note that the animal experiments conducted in this study received approval and strictly adhered to the regulations established by the Animal Ethics Committee of Nantong University (S20230315-005).
Human brain microvascular endothelial cells (HBMECs) were obtained from
YingBioTech (Shanghai, China) with certification by short tandem repeat (STR)
analysis and tested to be free of mycoplasma. HBMECs were cultured in Roswell
Park Memorial Institute (RPMI) 1640 medium (A1049101, Gibco, New York, NY, USA) supplemented with 10% fetal bovine serum (FBS, Z7186FBS, Zeta Life, CA,
USA) and 1% penicillin-streptomycin (C125C5, NCM Biotech, Suzhou, Jiangsu, China) at 37 °C in a 5% CO
GenPharma (Shanghai, China) synthesized the siRNAs for Sestrin2, which were
transfected into HBMECs for 48 h with GP-transfect-Mate (G04008, GenePharma,
Shanghai, China). Sestrin2 siRNA sequences were presented as below.
Sestrin2 siRNA 1, 5
Sestrin2 overexpressive lentivirus was constructed by GenePharma (Shanghai, China). HBMECs were plated in a 6-well plate at the confluence of 50% and infected by Sestrin2 overexpressive lentivirus and control lentivirus with polybrene for 48 h. Then, HBMECs were exposed to LPS (100 ng/mL) for 24 h.
Mouse brain sections of 15 µm containing cortical regions were obtained by
freezing section. Then, permeability of cell membrane was increased by 0.3%
Triton (P0096, Beyotime, Shanghai, China) in phosphate buffer saline (PBS). Brain
sections were blocked in 10% sheep serum (ZLI-9056, ZSGB-BIO, Beijing, China) at room temperature for 1 h followed by
incubation in platelet endothelial cell adhesion molecule-1 (CD31) (1:100, ab24590, Abcam, Cambridge,
UK) and alpha smooth muscle actin (
A radio-immunoprecipitation assay buffer (P0013C, Beyotime, Shanghai, China) was
used to lyse mouse cortex tissues and HBMECs followed by the measurement of
protein concentration using a bicinchoninic acid (BCA) analysis kit (P0012S,
Beyotime). After separation on 10% polyacrylamide gels, proteins were
transferred to polyvinylidene fluoride (PVDF) (IPVH00010, Millipore, Boston, MA, USA) membranes, which were then
incubated with primary antibodies overnight at 4 °C after blocking in
5% nonfat milk. The next day, PVDF membranes were incubated in goat anti-rabbit
secondary antibody (1:2000, SA00001-2, Proteintech) and goat anti-mouse secondary
antibody (1:2000, SA00001-1, Proteintech) labeled by horseradish peroxidase at
room temperature for 1 h. Blot images were detected using a chemiluminescence
system and the band signal was analyzed using ImageJ software 1.51j8 (National
Institutes of Health, Bethesda, MD, USA). Primary antibodies were
purchased from Proteintech and details were presented as inducible nitric oxide
synthase (iNOS) (1:1000, 18985-1-AP), Sestrin2 (1:1000, 21346-1-AP),
glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:5000, 60004-1-Ig),
Total RNA was extracted using Trizol reagent (T9108, Takara, Tokyo, Japan).
First, RNA was purified with gDNA wiper solution and reverse transcribed into
cDNA with a HiScript III 1st Strand cDNA Synthesis Kit (R312-01, Vazyme, Nanjing,
China). Then, target mRNA was detected by real-time polymerase chain reaction
(PCR) with Taq Pro Universal SYBR qPCR Master Mix (Q712-02, Vazyme). The PCR
conditions were 95 °C for 30 s, followed by 40 cycles at 95 °C
for 10 s and 60 °C for 30 s. Target RNA levels were normalized to GAPDH.
The quantitative expression level was analyzed using the
2
HBMECs were digested with 0.25% trypsin (C100C1, NCM Biotech, Suzhou, Jiangsu, China) containing ethylene diamine tetraacetic acid (EDTA) followed by centrifugation at 1200 rpm for 5 min. Cell pellet was resuspended with fixation fluid for electron microscope (G1102, Servicebio, Wuhan, Hubei, China) after discarding supernatant. Astrocytes were washed using sucrose-sodium cacodylate buffer and soaked in osmium tetroxide-sodium cacodylate for 2 h. Then, astrocytes were stained using 2% aqueous uranyl acetate after washing in water. Subsequently, samples were dehydrated in an ethanol series and embedded in Epon 812 (90529-77-4, SPI Science, West Chester, PA, USA). Ultrathin sections of 80 nm were obtained using a ultramicrotome with a diamond knife. Grids were poststained with 2% saturated uranyl acetate in 1% lead citrate and 50% ethanol. Photographs were captured using an electron microscope camera (TECNAI G2 20 TWIN, Thermo fisher, Waltham, MA, USA).
The statistical analyses were conducted using GraphPad Prism 6.0 (GraphPad
Software, San Diego, CA, USA). A Student’s t-test was used to compare the
difference between two groups and one-way analyses of variance (ANOVA) was used
to compare the differences among multiple groups. The statistical difference was
considered significant if p
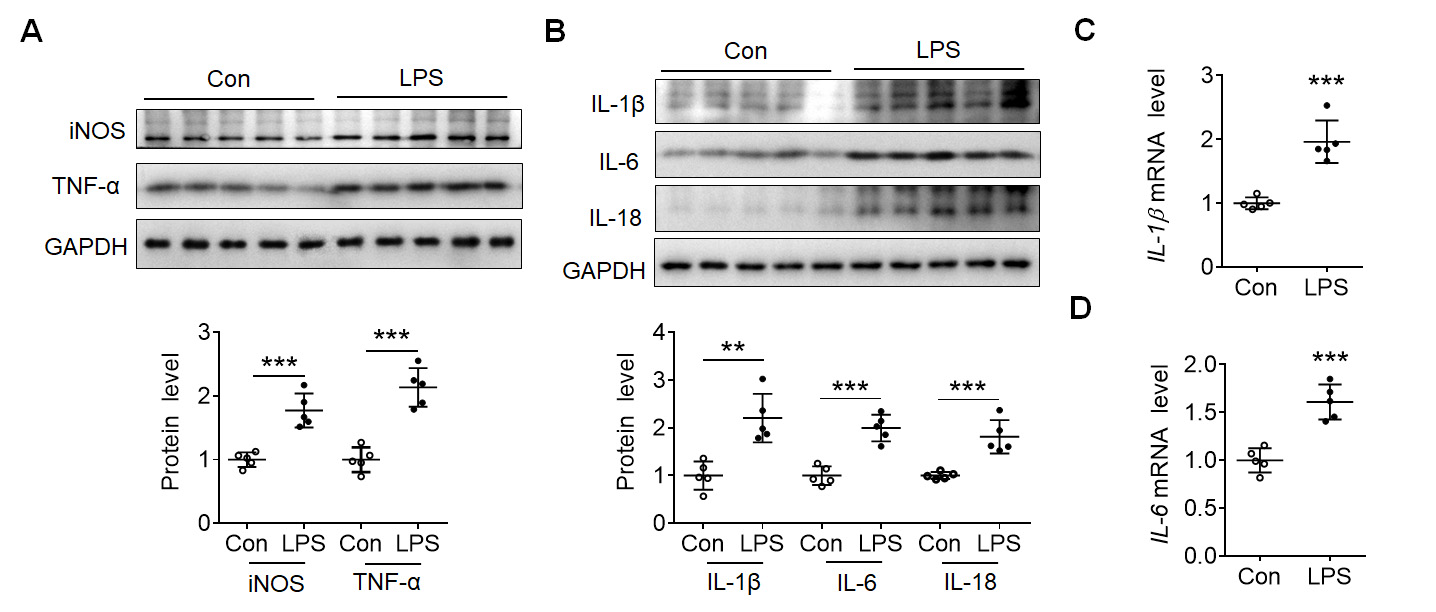
To investigate the mechanism of EndoMT in the inflammatory response, LPS was
administered intraperitoneally to mice several times to establish a chronic
inflammatory model. In comparison to the control group, the intraperitoneal
injection of LPS significantly enhanced the expressions of iNOS and TNF-
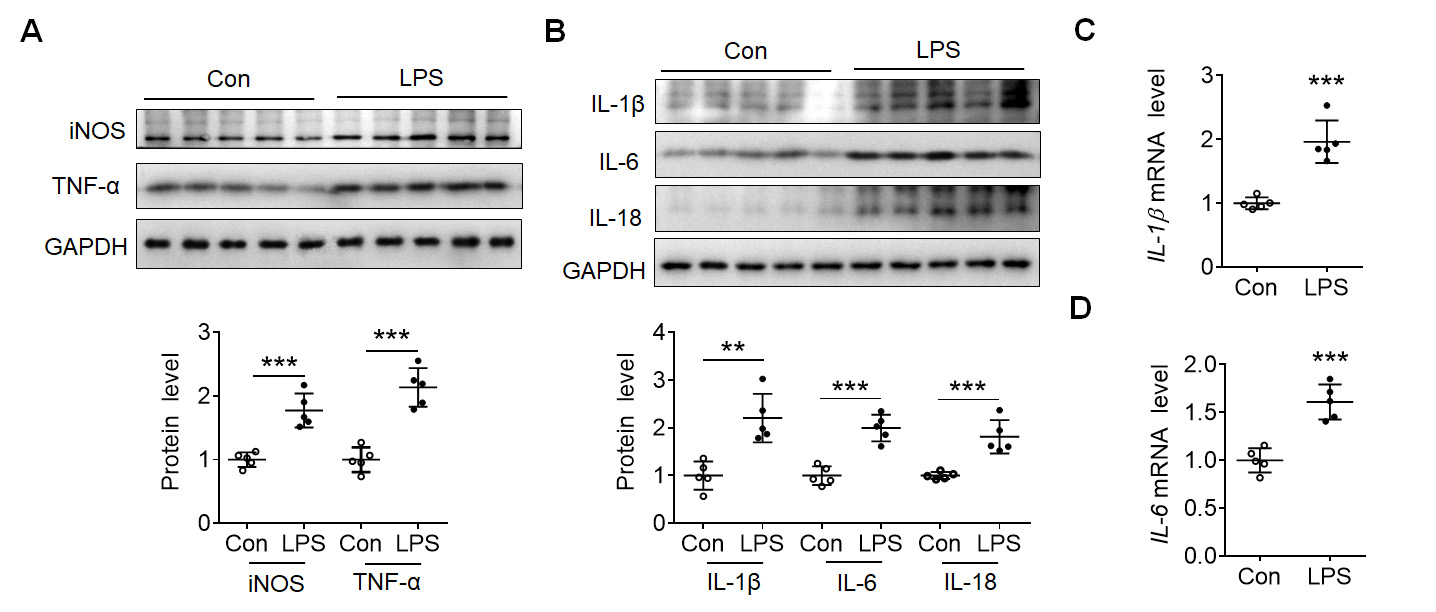 Fig. 1.
Fig. 1.Lipolysaccharide (LPS) induced inflammatory reaction in mouse
cortex. (A) Representative blots showed the expression of inducible nitric oxide
synthase (iNOS) and tumor necrosis factor
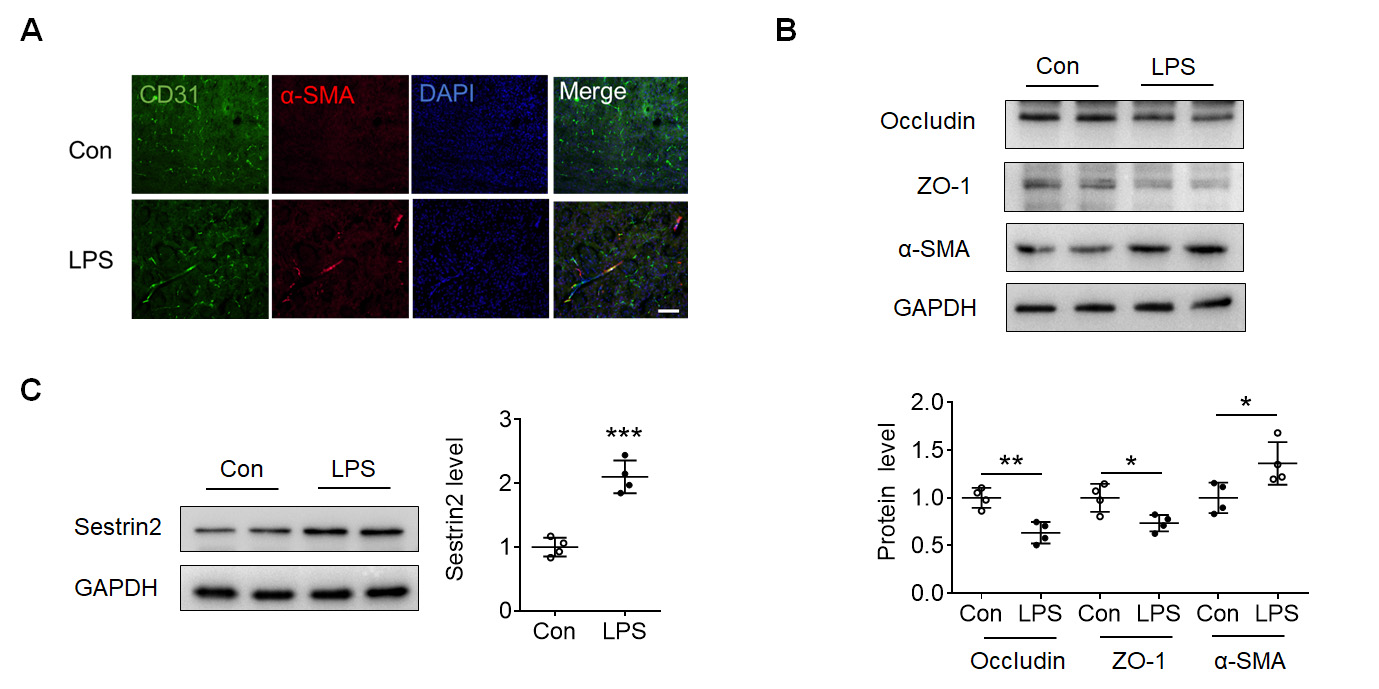
After determining the inflammation induced by LPS, immunofluorescence staining
of CD31 and
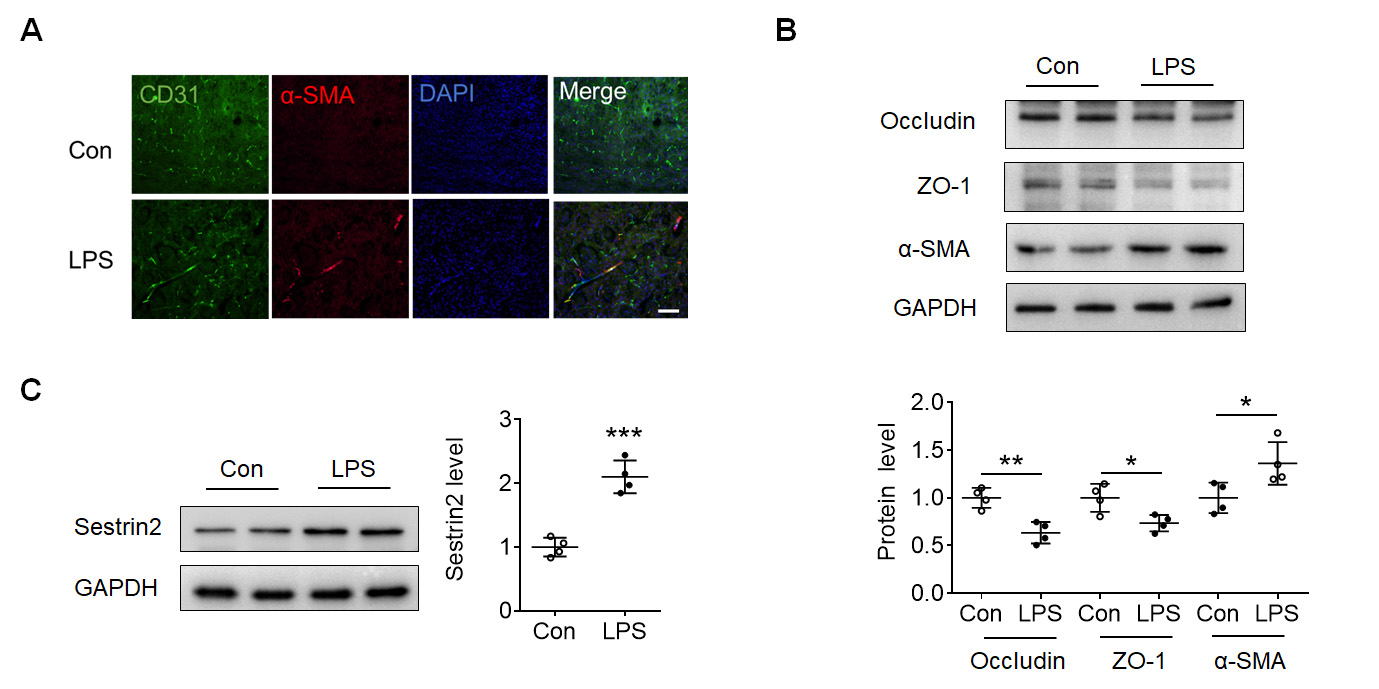 Fig. 2.
Fig. 2.LPS induced Endothelial-to-mesenchymal transition (EndoMT) in
mouse cortex. (A) Immunofluorescence staining of CD31 (green), alpha smooth
muscle actin (
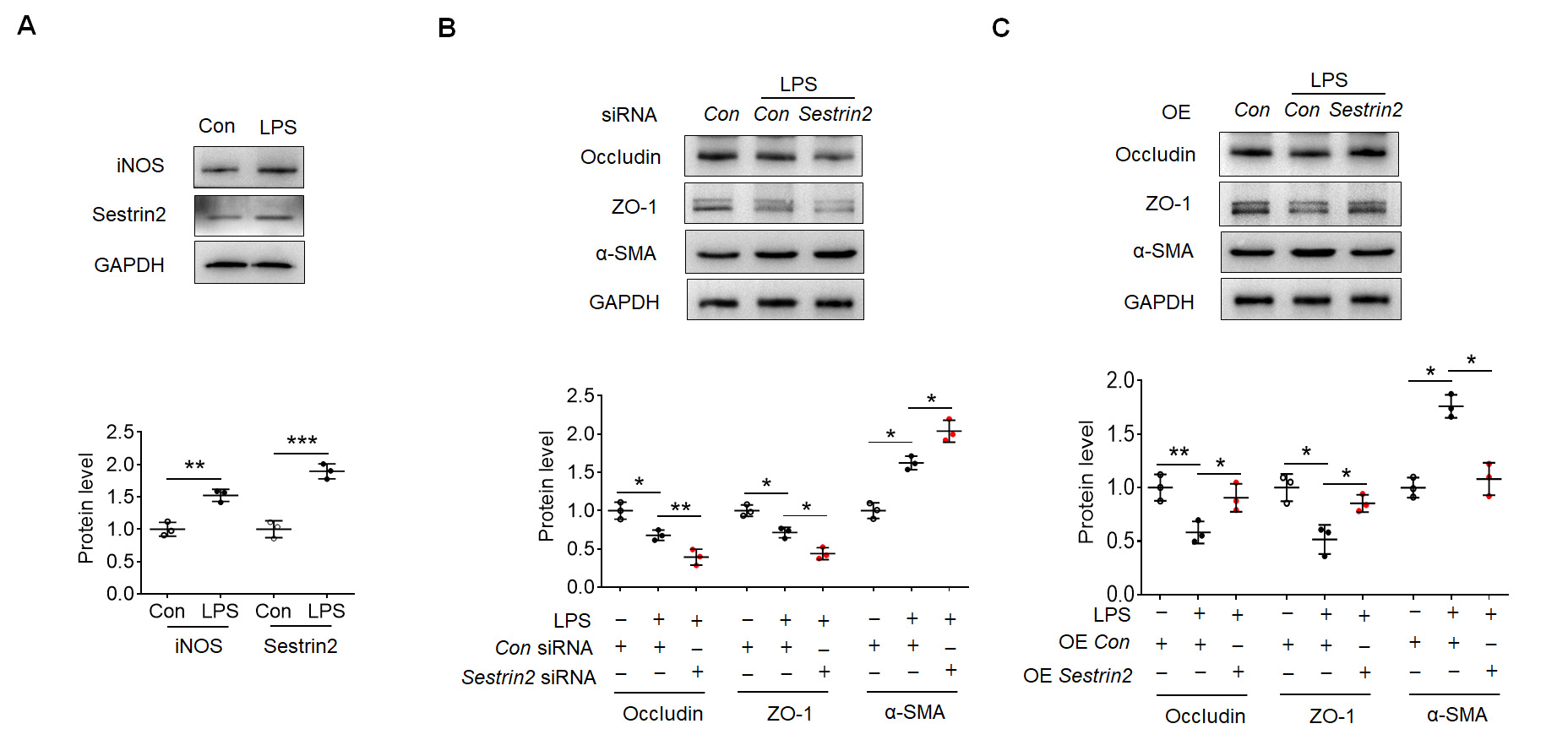
Subsequently, HBMECs were subjected to LPS exposure in vitro. This
exposure led to an upregulation of iNOS and Sestrin2 expression in HBMECs, as
depicted in Fig. 3A. To investigate the impact of Sestrin2 on EndoMT, specific
Sestrin2 siRNAs were designed and transfected into HBMECs. Among these
siRNAs, Sestrin2 siRNA 1 exhibited the most effective knockdown
efficiency in terms of Sestrin2 expression, as shown in Supplementary
Fig. 1A. Consequently, Sestrin2 siRNA 1 was employed in subsequent
experiments. Notably, knockdown of Sestrin2 exacerbated the reduced expression
levels of Occludin and ZO-1 in the LPS-treated group (Fig. 3B). Simultaneously,
knockdown of Sestrin2 facilitated the enhanced expression of
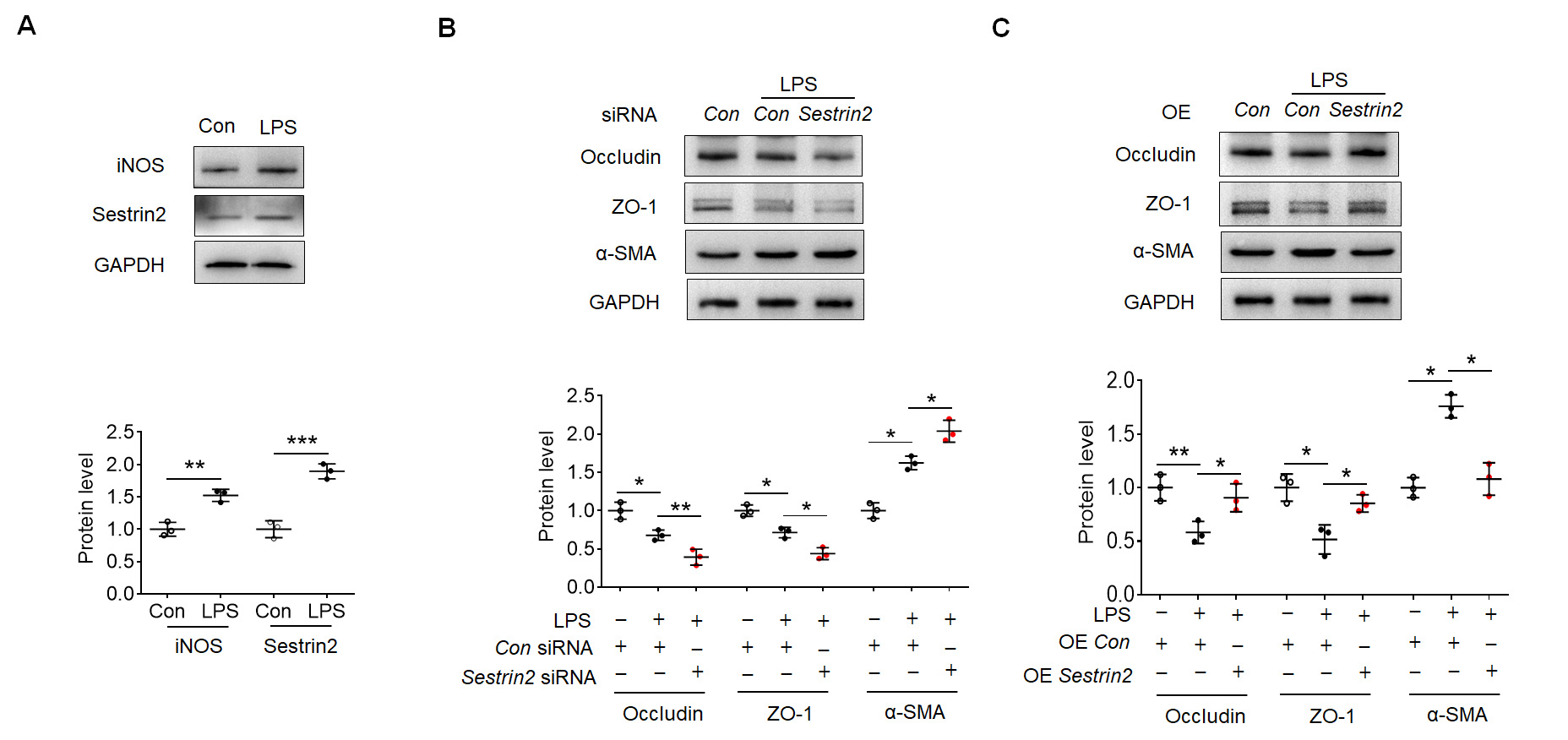 Fig. 3.
Fig. 3.Sestrin2 overexpression attenuated EndoMT in human brain
microvascular endothelial cells (HBMECs). (A) Representative blots showed iNOS
and Sestrin2 expression in the control and LPS-exposed HBMECs. (B) Representative
blots showed Occludin, ZO-1 and
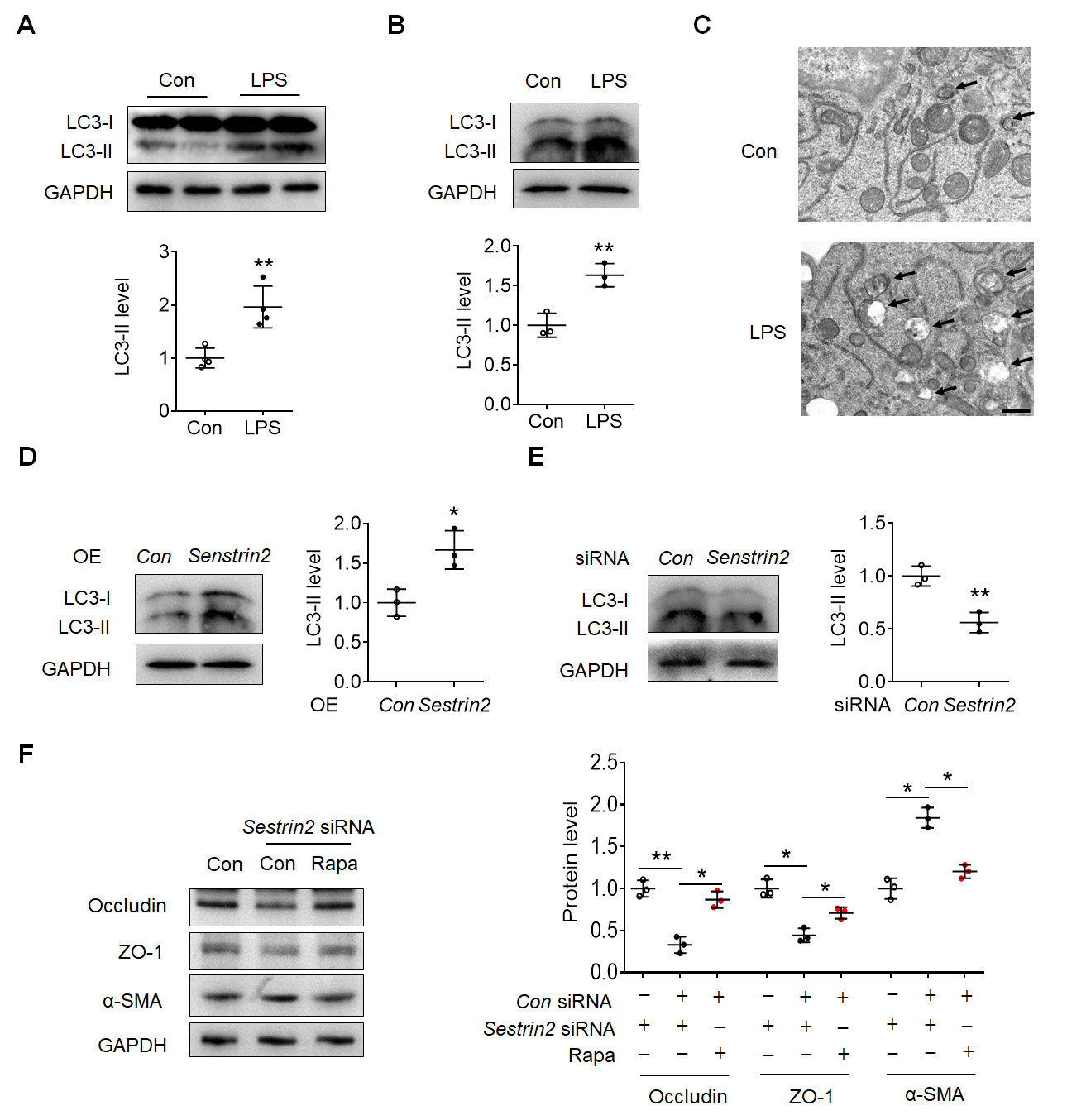
Subsequently, we aimed to investigate the downstream effects of Sestrin2 in the
LPS-induced EndoMT process. As shown in Fig. 4A, the expression of
microtubule-associated protein1 LC3-II was observed to be
elevated in the cortex of mice treated with LPS. Similarly, an increase in
LC3B-II expression was observed in LPS-exposed HBMECs (Fig. 4B). LPS treatment
increased the autolysosomes (black arrows) in HBMECs (Fig. 4C). Overexpression of
Sestrin2 facilitated the LC3-II expression in HBMECs (Fig. 4D), whereas knockdown
of Sestrin2 suppressed the expression of LC3-II in HBMECs (Fig. 4E). Furthermore,
knockdown of Sestrin2 resulted in reduced expression levels of Occludin and ZO-1,
which were initially inhibited by treatment with the autophagy inducer rapamycin
(Fig. 4F). Additionally, the increased expression of
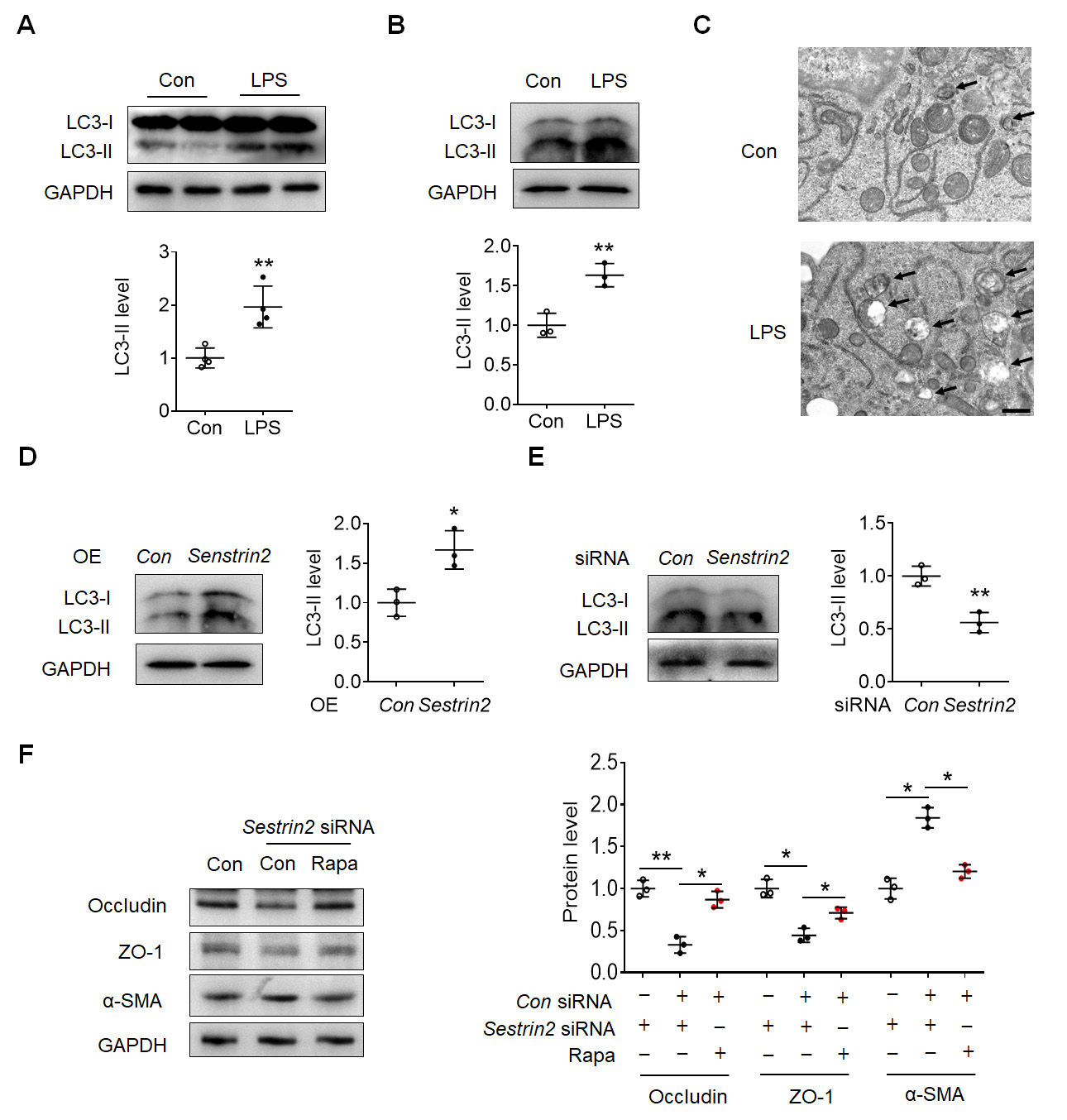 Fig. 4.
Fig. 4.Sestrin2 regulated EndoMT depending on autophagy in HBMECs. (A)
Representative blots showed the LC3-II expression in the cortex of control and
LPS-injected mouse. (B) Representative blots showed the expression of LC3-II in
the control and LPS-exposed HBMECs. (C) Transmission electron microscope
photograph showed the autolysosomes (black arrows). Scale bar: 5 µm. (D)
Representative blots showed the LC3-II expression in the control and LPS-exposed
HBMECs with/without Sestrin2 overexpressive lentivirus infection. (E)
Representative blots showed the LC3-II expression in the control and LPS-exposed
HBMECs with/without Sestrin2 siRNA treatment. (F) Representative blots
showed the LC3-II expression in the control and rapamycin-exposed HBMECs
with/without Sestrin2 siRNA treatment. *p
Vascular disease is thought to begin with endothelial dysfunction, which occurs
at a preclinical stage, and is prone to complications [13]. As a result of
endothelial maladaptation to mechanical, metabolic, or oxidative stress, it
causes impaired vasodilator response, altered angiogenesis, and increased
expression of pro-inflammatory and pro-thrombotic factors [14]. EndoMT is a cell
phenotypic transition driven by inflammation and an important cytopathological
manifestation of endothelial dysfunction, which plays an important role in the
occurrence and development of chronic cardiovascular diseases [15]. EndoMT is
regulated by several signaling pathways, such as basic fibroblast growth
factor/fibroblast growth factor receptor 1, transforming growth factor
(TGF)-
Oxidative stress is a prominent determinant of EndoMT, and its significance in
the development of vascular pathogenesis has been extensively investigated.
Suppression of EndoMT has been shown to diminish renal ROS levels and mitigate
renal fibrosis [18]. Sestrin2, a widely recognized antioxidant protein, assumes a
critical function in curtailing the generation and buildup of ROS, thereby
safeguarding cells against oxidative harm [19]. Consequently, we hypothesized
that Sestrin2 might be implicated in the induction of EndoMT through LPS
treatment. In our study, it was revealed that Sestrin2 was upregulated in the
presence of LPS in the mouse cortex and HBMECs. Sestrin2 siRNA was utilized in
the rescue experiment conducted on HBMECs. Surprisingly, the knockdown of
Sestrin2 did not suppress the EndoMT induced by LPS; rather, it facilitated its
occurrence. Subsequent experiments corroborated that the overexpression of
Sestrin2 safeguarded HBMECs against LPS-induced EndoMT. In summary, Sestrin2
exhibited a favorable impact on the preservation of endothelial cell function.
The augmentation of Sestrin2 expression in response to LPS stimulation may
represent an intrinsic protective mechanism initiated by the cells themselves.
Nevertheless, the elevation of endogenous Sestrin2 expression was inadequate to
manifest its protective influence. Other studies have found the similar result
[20, 21]. LPS treatment significantly induced the upregulation of Sestrin2 in
dendritic cells, and Sestrin2 overexpression suppressed the ferroptosis of
dendritic cells in sepsis by inhibiting the activating transcription factor
4-CCAAT enhancer binding protein homologous protein signaling pathway [20].
Similarly, Sestrin2 expression was induced in retinal ganglion cells exposed to
H
Autophagy is a self-degrading process that is crucial for balancing energy
sources in response to nutritional stress and during vital periods of development
[22]. It serves as a crucial mechanism for cellular maintenance by eliminating
misfolded or aggregated proteins, clearing damaged organelles such as
mitochondria, endoplasmic reticulum, and peroxisome, and intracellular pathogens.
Consequently, autophagy is commonly acknowledged as a survival mechanism, albeit
excessive autophagy may also trigger cell death [23]. Beyond its role in removing
intracellular damaged or aggregated organelles, autophagy facilitates cell aging
and cell surface antigen presentation, and prevents genomic instability and cell
necrosis, thereby assuming a pivotal function in various disease processes. The
available literatures indicate that autophagy plays a role in the regulation of
EndoMT [3]. However, the specific mechanism by which autophagy operates and the
means by which it modulates EndoMT remain unclear. In human umbilical vein
endothelial cells, the autophagy inhibitor 3-Methyladenine was found to suppress
the EndoMT induced by SiO
There is a limitation to our experiment as we were unable to definitively establish that the increased in Sestrin2 and inflammatory response in the mouse cortex induced by LPS were exclusive to endothelial cells. It is possible that these changes are a result of a mixed reaction involving multiple cell types in mouse brain. Subsequent cellular experiment confirmed the increased Sestrin2 and inflammatory response in HBMECs. In conclusion, the findings from both the animal and cell experiments provide support for our conclusion.
Our findings demonstrated that Sestrin2 effectively suppressed endothelial inflammation and EndoMT by promoting autophagy, thereby presenting a promising therapeutic target for cerebrovascular inflammatory injury.
All data in this study were provided in the manuscript.
Investigation: RH, KS, and CD. Statistical quantification: RH, LL and KS. Drafting of the paper: RH, LL and KS. Study concept, design, review and editing: CD and ZF. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The animal experiments conducted in this study received approval and strictly adhered to the regulations established by the Animal Ethics Committee of Nantong University (S20230315-005).
Not applicable.
This work was supported by the National Natural Science Foundation of China (82101593, 32101027), the Natural Science Foundation of Jiangsu Province (BK20210844, BK20210838).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.