


 , Songyan Liu 1,*
, Songyan Liu 1,*1 Department of Neurology, China-Japan Union Hospital of Jilin University, 130033 Changchun, Jilin, China
2 Department of Anatomy, College of Basic Medicine Sciences, Jilin University, 130015 Changchun, Jilin, China
Abstract
Excessively high or synchronized neuronal activity in the brain is the underlying cause of epilepsy, a condition of the central nervous system. Epilepsy is caused mostly by an imbalance in the activity of inhibitory and excitatory neural networks. Recurrent or prolonged seizures lead to neuronal death, which in turn promotes epileptogenesis and epileptic seizures. Ferrous ion-mediated cell death is known as ferroptosis, which is due to the accumulation of lipid peroxidation products resulting from compromise of the glutathione (GSH)-dependent antioxidant system. The pathophysiology of epilepsy has been linked to anomalies in the glutathione peroxidase 4 (GPX4)/GSH redox pathway, lipid peroxidation, and iron metabolism. Studies have shown that inhibiting ferroptosis may alleviate cognitive impairment and decrease seizures, indicating that it is neuroprotective. With the hope of aiding the development of more novel approaches for the management of epilepsy, this research aimed to examine the role of ferroptosis in this disease.
Keywords
- ferroptosis
- iron
- epilepsy
- seizure
- epileptogenesis
Epilepsy is a long-term neurological condition characterized by repeated episodes of seizures. Short bursts of symptoms caused by excessive or synchronized brain activity are the hallmarks of an epileptic seizure. Electroencephalograms may reveal the abnormally excessive synchronization of dischargs [1]. Epilepsy affects more than 70 million individuals worldwide [2] and leads not only to impaired mental and physical functions and increased risk of accidents and injuries but also potentially to death; thus, it places a great economic burden on families and society and causes a series of social and psychological problems [3]. Treatment options include drugs, surgery, dietary changes and neurostimulation, but no ideal treatment has been identified [2]. Thus, further research into the pathophysiology of epilepsy is needed to provide a theoretical foundation for its diagnosis, prevention and treatment.
Dixon et al. [4] (2012) identified glutamate-induced cell death in organotypic hippocampal slices and erastin-induced death of tumour cells as a single cell death process distinct from other well-studied types of regulated cell death and subsequently named this cell death mechanism ferroptosis. Ferroptosis is a ferrous ion-dependent form of regulated cell death, and it is due to the accumulation of lipid peroxidation products resulting from compromise of the glutathione (GSH)-dependent antioxidant system [5]. While ferroptotic cell death primarily causes the shrinkage of dense mitochondria and a reduction in the number of mitochondrial cristae, it does not damage the nucleus, as has been demonstrated via electron microscopy, and the cell membrane ruptures during the late stage of ferroptosis [5, 6, 7].
A wide range of disorders affecting the central nervous system (CNS) have been associated with ferroptosis. These include Alzheimer’s disease and Parkinson’s disease as well as traumatic brain injury, stroke, brain tumours, and epilepsy [8, 9, 10, 11, 12, 13]. The mechanisms of epilepsy-related ferroptosis are poorly understood; however, research in animal models has shown a substantial association between the epilepsy and ferroptosis. To better understand ferroptosis and its function in epilepsy, this paper provides a comprehensive overview of the molecular pathways involved, as well as suggestions for further study.
There is a correlation between the natural increase in brain iron levels with age and an increased risk of ferroptosis with older ages [14, 15]. To improve tumour cell survival and proliferation, inhibiting ferroptosis is crucial. In order to develop drug resistance, tumour cells regulate the production and activity of ferroptosis-related proteins, which inhibits ferroptosis [16, 17]. This means that ferroptosis has connections to both physiological and pathological functions. The exact mechanism underlying ferroptosis is still being investigated; however, it is known to be the result of many simultaneous biological processes, including iron metabolism, lipid metabolism, and the glutathione peroxidase 4 (GPX4)/GSH redox pathway.
Metabolic processes, energy production, and DNA synthesis all rely on iron, which is a protein cofactor [18]. Cellular ferrous ions stimulate the generation of lipid peroxides and, via the Fenton reaction with peroxide, generate lethal hydroxyl radicals [5]. Thus, ferroptosis is primarily driven by ferrous ions. Iron intake, transport, utilization, storage, and translocation are all parts of iron metabolism that may be altered ferroptosis. Iron bound to transferrin (TF) is the most common type of dietary iron carried throughout the body through the blood after absorption in the duodenum. Iron must be efficiently carried across the blood-brain barrier (BBB) before it can reach the brain parenchyma. The passage of iron through the BBB in adult animals is mediated by the transferrin receptor (TFRC) complex on the surface of the anterior lumen of cerebral microvascular endothelial cells [19, 20]. After the iron-TF-TFRC complex is internalized into the cytoplasm by endocytosis, ferric ions are reduced to ferrous ions by the six transmembrane epithelial antigen of prostate 3 (STEAP3) in the endosome [21]. Knocking down the TFRC gene in Ras-mutant cells inhibits erastin-induced ferroptosis and cysteine deficiency-induced ferroptosis [22]. Accordingly, TFRC is now thought to be a biomarker for ferroptosis [23]. An integral part of iron metabolism, the labile iron pool (LIP) is formed when ferrous ions are released into the cytoplasm via divalent metal transporter 1 (DMT1) on the endosomal membrane [24]. Iron is transported to ferritin by cytoplasmic poly-(RC)-binding protein 1 (PCBP1), which binds directly to ferrous ions in the LIP [25]. The protein ferritin is composed of two parts: ferritin heavy chain 1 (FTH1) and ferritin light chain (FTL). Within the ferritin mineral core, FTH1 has the ability to transform ferrous ions into ferric ions. The iron loading capacity of ferritin is decreased and the cytoplasmic LIP content is increased by PCBP1 degradation [26]. Nuclear receptor coactivator 4 (NCOA4) facilitates ferritinophagy, the last step in ferritin breakdown. NCOA4 overexpression leads to ferritin degradation, which in turn induces ferroptosis; however, knocking down NCOA4 can prevent ferritin degradation and erastin-induced ferroptosis [27]. Prominin-2 (PROM2)-mediated exosomes may mediate the extracellular release of ferritin, which in turn diminishes ferroptosis sensitivity [28]. The only way nonheme iron is transported is through ferroportin 1 (FPN1) [29]. Ferrous ions that are transported out of the cell are oxidized to ferric ions by ferroxidases such as ceruloplasmin (CP) [30]. Poly-(RC)-binding protein 2 (PCBP2) acts as a regulator of FPN1 by binding to it and receiving ferrous ions. Silencing PCBP2 can inhibit the iron uptake function of FPN1, leading to a decrease in iron efflux [26]. An imbalance in iron homeostasis can change the amount of intracellular free ferrous ions, thus affecting sensitivity to ferroptosis.
Ferroptosis induction requires membrane damage and lipid peroxidation [5]. Polyunsaturated fatty acids (PUFAs)-containing cell membranes are especially vulnerable to lipid peroxidation, and this process is significantly accelerated when ferrous ions are present [31]. The levels of cellular lipid peroxidation and ferroptosis are controlled by the level and location of PUFAs [32]. There is evidence that monounsaturated fatty acids (MUFAs) may prevent ferroptosis [33, 34]. Among PUFAs, the essential fatty acids for ferroptosis are arachidonic acid (AA) and its derivative, adrenergic acid (AdA) [35, 36]. Acyl-CoA synthetase long-chain member 4 (ACSL4) catalyses the synthesis of AA or AdA acyl-CoA derivatives from AA or AdA [35]. Moreover, ferroptosis sensitivity increases when ACSL4 is expressed and ACSL4 is a driving factor of ferroptosis [37, 38]. In the presence of lysophosphatidylcholine acyltransferase 3 (LPCAT3), AA-phosphatidylethanolamines (PEs) or AdA-PEs may be produced from AA or AdA acyl-CoA derivatives [39]. It was found that 12/15-lipoxygenase (12/15-LOX) oxidize AA or AdA-PEs to lipid peroxides, which enhance ferroptosis [34, 35, 36, 40].
The GPX4/GSH redox pathway, a significant antioxidant pathway, is a crucial
pathway involved in ferroptosis. However, glycine, glutamate, and cysteine
generate tripeptide reduced GSH, with cysteine acting as the
rate-limiting precursor. The majority of cells take up cystine mostly via the
cystine-glutamate antiporter System Xc
In addition to the GPX4/GSH pathway, intracellular lipid peroxidation levels can also be regulated via the ferroptosis suppressor protein 1 (FSP1)-coenzyme Q10 (CoQ10)-NADPH pathway. FSP1 is an enzyme that may regenerate CoQ10 with NADPH, allowing the capture of lipid peroxide free radicals and providing protection against GPX4 deletion-mediated ferroptosis [52]. CoQ10 inhibits lipid peroxidation by neutralizing free radical intermediates [53]. Notably, isopentenyl pyrophosphate (IPP) is the precursor of CoQ10 and a limiting substrate for the enzymatic isopentenylation of selenocysteine-tRNA (Sec-tRNA). Therefore, IPP affects the expression of GPX4 [54].
In addition to FSP1-CoQ10-NADPH pathway, the guanosine triphosphate cyclohydrolase 1 (GCH1)/tetrahydrobiopterin (BH4) and dihydroorotate dehydrogenase (DHODH) pathways are also important for ferroptosis [55]. GCH1 is the rate-limiting enzyme for BH4. BH4 can promote the synthesis of CoQ10. BH4 may inhibit ferroptosis by inducing lipid remodelling of the plasma membrane [56]. DHODH is an enzyme located on the outer surface of the inner mitochondrial membrane [57]. In mitochondria, DHODH can reduce CoQ to coenzyme QH2 (CoQH2), reducing oxygen free radicals, inhibiting phospholipid peroxidation, thereby inhibiting ferroptosis [55].
As the global population has aged, CNS diseases such as Parkinson’s disease, Alzheimer’s disease, stroke and epilepsy have become serious economic and mental burdens. The majority of research on ferroptosis’s function in CNS diseases has focused on neoplasms and some neurodegenerative disorders. An increasing number of studies examining the mechanisms of ferroptosis have shown a substantial association between ferroptosis and the pathophysiological features of epilepsy.
One of the reasons for the increased incidence of epileptogenesis in some
patients after haemorrhagic stroke or traumatic brain injury is that locally
extravasated blood and iron released from haemoglobin-containing blood cells leak
through the destroyed BBB into the brain parenchyma, leading to an elevated iron
concentration and iron accumulation within local brain tissues [58, 59]. After
nanoscale intracortical iron injection (100 mM, 350 nL), the number of
Ikeda [63] reported that the serum iron concentration and TF saturation of epileptic patients are greater than those of healthy individuals, indicating that there is iron overload in the blood of epileptic patients. In an animal model of temporal lobe epilepsy (TLE), ferritin expression is upregulated within areas with cell death, indicating that iron easily accumulates in the lesion [64]. After seizures, BBB damage and changes in brain cell-related iron metabolism promote iron accumulation within areas responsible for the generation of seizures. After stimulating hippocampal brain slices with the epileptiform stimulant ferric ammonium citrate, neuronal iron uptake increases, and the accumulated iron increases neuronal excitability by impairing astrocyte function and stimulating neuroinflammation [65]. The absorption of iron by neurons is enhanced during epileptiform activity due to either the direct promotion of iron absorption by n-methyl-d-aspartic acid (NMDA) or its dependence on DMT1 [66, 67]. Studies have shown that under circumstances that enhance cellular iron overload-related toxicity, voltage-gated calcium channels provide alternative routes for iron to enter neuronal cells [68]. Epileptogenic brain tissue from individuals with focal cortical dysplasia type Ⅱb (FCD Ⅱb) and tuberous sclerosis complex (TSC) was investigated by Zimmer et al. [69]. It was found that increased expression of 4-hydroxynonenal (4-HNE), nuclear factor erythroid 2-like 2 (NRF2), haem oxygenase 1 (HO1), and iron regulatory genes in the epileptogenic brain tissue and increased levels of unstable iron ions generated due to the high HO1 expression may aggravate oxidative stress by generating additional reactive free radicals through the Fenton reaction. The authors suggested that oxidative stress and iron metabolism are closely related and exert synergistic effects and that both exacerbate cellular dysfunction and damage in epilepsy [69].
Experiments have shown that GSH levels decrease and that redox haemostasis is disrupted in epilepsy. Ryan et al. [70] reported that GSH levels in the hippocampi of rats with TLE decrease during several stages of epilepsy, i.e., the acute, latent, and chronic stages. After 8 and 24 hours of kainite treatment, GSSG levels increase, and by 48 hours, they are considerably elevated, increasing throughout the latent stage. From 24 hours to the sixth week, there is a significant decrease in the GSH to GSSG ratio [70]. Smeland et al. [71] reported that hippocampal GSH levels in epileptic mice decrease 3.5 weeks after pilocarpine treatment. According to the findings of Shin et al. [72], genetically epilepsy-prone rats (GEPRs) exhibit elevated lipid peroxidation and decreased GSH/GSSG ratios and glutathione peroxidase (GPX) activity in the hippocampus. This finding suggests that epileptogenesis in GEPRs is caused by antioxidant system damage mediated by GPX, as well as GSH [72]. Acute exposure to trimethyltin results in behavioural symptoms in adult rats, including epilepsy susceptibility, hyperactivity and aggressiveness, similar to those observed in certain humans with epilepsy [73]. Trimethyltin decreases the GSH/GSSG ratio, GPX activity and GSH protein expression in the hippocampus of rats [74]. Decreased antioxidant enzyme activity is observed after seizures, and the number of seizures can be reduced if antioxidant enzyme activity is restored [75]. GSH levels in the parietal and occipital lobes of both hemispheres are markedly lower in epileptic patients than in normal individuals [76]. Hazany et al. [77] showed that GSH levels in grey matter and white matter were significantly increased after a ketogenic diet was given to 2 patients with intractable epilepsy, and alleviation of seizure symptoms was observed, indicating that GSH may have anticonvulsant effects in the brain. Consumption of a ketogenic diet stimulates new GSH synthesis and may improve brain redox status by promoting mitochondrial function [77]. Increased posttranslational modification of the biosynthetic enzyme glutamate cysteine ligase increases intracellular GSH levels, which in turn decreases neuronal hyperexcitability in primary cerebrocortical neuronal-glial cultures; this is the mechanism by which the dithiol-containing compound dimercaprol can increase the intracellular GSH concentration. Treating Dravet syndrome model zebrafish larvae with dimercaprol increases their GSH levels and preventes convulsive, abrupt “seizure-like” swimming behaviour [78].
Low levels of selenium and GPX in epileptic patients suggest that damage to the
antioxidant system is associated with the frequent occurrence of seizures [79, 80]. Seizures among children with reduced GPX activity may be ameliorated by the
addition of selenium to their diet [81]. Behavioural symptoms, oxidative damage,
and neuronal death in mice with pentylenetetrazole-induced epileptic episodes can
be reversed by pretreatment with selenium nanoparticles [82]. Seizure phenotypes
can be induced in mice and humans by the genetic suppression of GPX4 activity.
Mutations in the GPX4 gene cause a rare paediatric syndrome known as
sedaghatian-type spondylometaphyseal dysplasia, which is characterized by
seizures, cerebellar hypoplasia and severe neurological deficits [83].
Neuron-specific GPX4 knockout mice exhibit a distinct hyperexcitability
phenotype at postnatal day 12; in these mice, seizures are likely to occur when
the pups are touched by littermates [84]. The loss of inhibitory interneurons
that are positive for parvalbumin in the hippocampus causes severe convulsions in
mice that express cysteine at the GPX4 active site instead of selenocysteine
[50]. According to Brigelius-Flohé and Maiorino [85] (2013), NRF2 controls GPX4
transcription. The binding of NRF2 to antioxidant response components in the
nucleus promotes the transcription of many antioxidant genes [86, 87]. NRF2
activation alters the development of epilepsy following seizures and mediates
antioxidant responses in a cell type- and space-dependent manner [88].
Additionally, in rat models of pilocarpine-induced epilepsy, peroxisome
proliferator-activated receptor
Seizures cause an increase in reactive oxygen species (ROS) levels due to mitochondrial malfunction and increased activities of NADPH oxidase and xanthine oxidase [91, 92]. Moreover, neurons may die via damage to DNA, peroxidation of lipids, damage to mitochondria, and inhibition of enzymes caused by excess ROS [93]. Most enzymes attached to membranes, the function of the calcium pump, and membrane permeability are all impacted by lipid peroxidation [94, 95]. Łukawski and Czuczwar [96] summarized the changing trends of oxidative parameters such as lipid peroxidation (LPO), superoxide dismutase (SOD), catalase (CAT) and GSH activities in various animal models of acquired epilepsy. These changes are associated with convulsive activity in animal models, elevate immediately after seizures, persist during seizures, and still detectable during chronic periods [96]. According to Petrillo et al. [97], who studied ferroptosis indicators in epileptic children’s blood, excess ROS levels and increased lipid peroxidation result from NADPH oxidase 2 activation. Lipid alcohols are unable to effectively neutralize lipid peroxidation due to decreased levels of GSH and GPX4. This leads to ferroptosis, and oxidative damage mediated by ferroptosis is a particular process that contributes to epilepsy pathogenesis [97]. Inhibiting lipid peroxidation contributes to seizure control, which in turn contributes to the development of epilepsy [91]. The link between epilepsy and elevated lipid peroxidation was validated in a systematic review by Martinc et al. [98], who also found that some antiepileptic medications can potentially further elevate lipid peroxidation levels.
Seizures are associated with elevated levels of ROS and oxidative stress in neurons, with ROS being mostly produced by mitochondria [99, 100, 101]. Recurrent epileptic seizures may result from mitochondrial malfunction in neurons, which decrease adenosine-triphosphate (ATP) generation and alters brain metabolism [102, 103, 104]. Therefore, impaired mitochondrial function is one of the pathophysiological manifestations of various types of epilepsy [105]. There is increasing clinical data indicating that mutations in nuclear coding DNA or mitochondrial DNA might underlie epilepsy [106]. Mitochondria in ferroptotic cells are often smaller, and the cristae around them are either less prominent or nonexistent [4, 5, 107]. The mitochondria are the main site for utilizing and storing iron, and excessive ferrous ions can accumulate in mitochondria [108]. Labile ferrous ions are the key factor triggering ferroptosis; therefore, mitochondria are involved in ferroptosis [109, 110, 111]. Mitochondrial encephalopathy known as pontocerebellar hypoplasia type 6 (PCH6), characterized by refractory epilepsy, may result from mitochondrial arginyl-tRNA synthetase 2 (RARS2) mutations [112, 113]. A study involving immunohistochemical analysis of patient’s brain tissue revealed mitochondrial respiratory chain abnormalities [114]. Patients treated with vatiquinone in clinical trials for PCH6 reported fewer seizures and fewer related complications. By regulating the activity of two enzymes, vatiquinone inhibits ferroptosis in tissues from patients. One enzyme, 15-LOX, decreases lipid peroxidation, while the other, GPX4, increases lipid peroxide clearance [13]. Hence, ferroptosis occurs in mitochondrial disorder-related epilepsy [115] (Fig. 1). Inhibition of ferroptosis may be effective in treating this type of epilepsy [13].
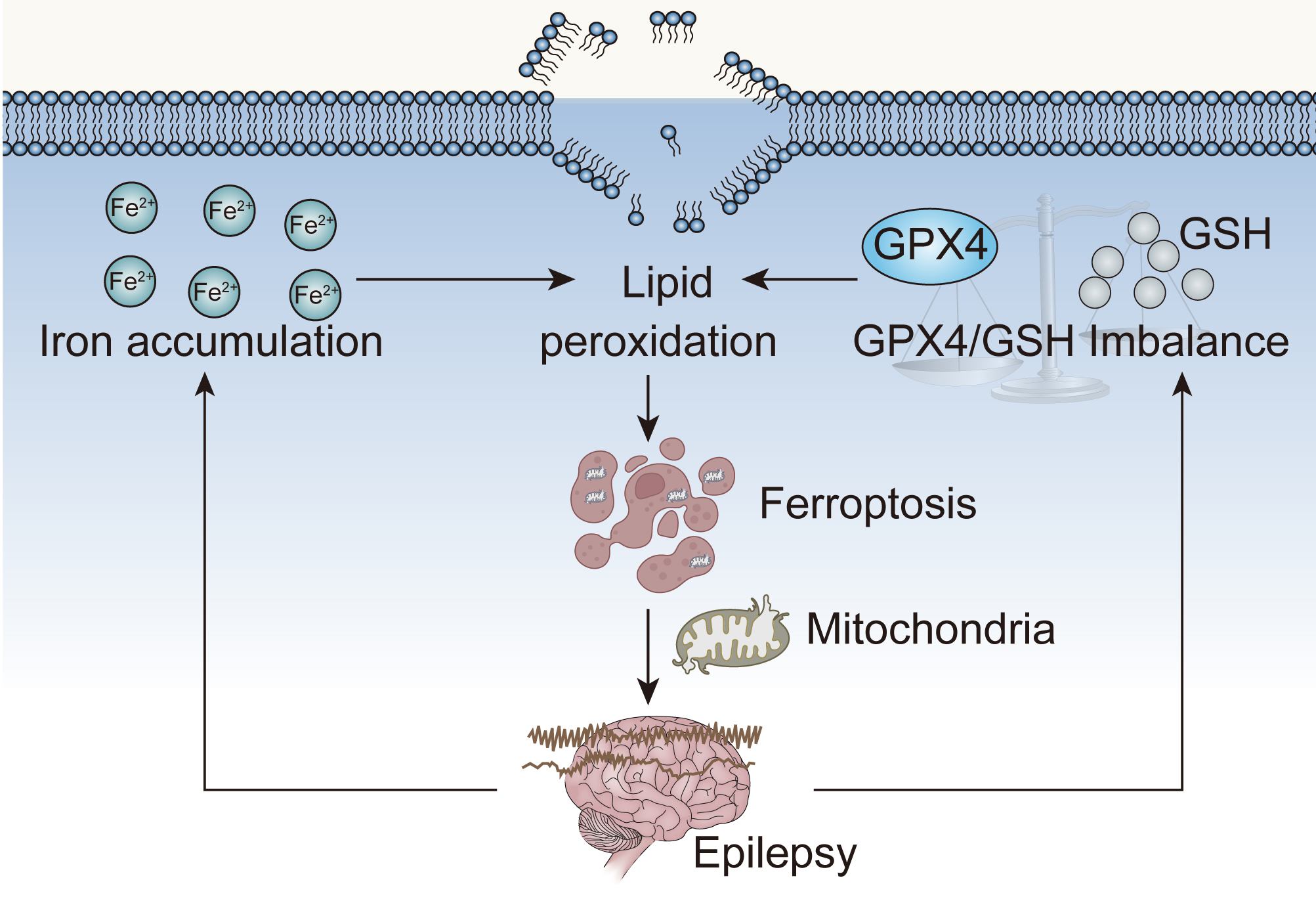 Fig. 1.
Fig. 1.Epilepsy, ferroptosis and mitochondrial dysfunction. GPX4, glutathione peroxidase 4; GSH, glutathione.
Ferroptosis may play a role in the development of epilepsy, according to the aforementioned findings; therefore, targeting ferroptosis-related molecules to treat epilepsy is a feasible approach (Fig. 2). Sodium valproate, an antiepileptic drug, can promote the expression of the FPN1 protein in cerebral capillary endothelial cells at the BBB and increase iron ion output, thus inhibiting ferroptosis [116].
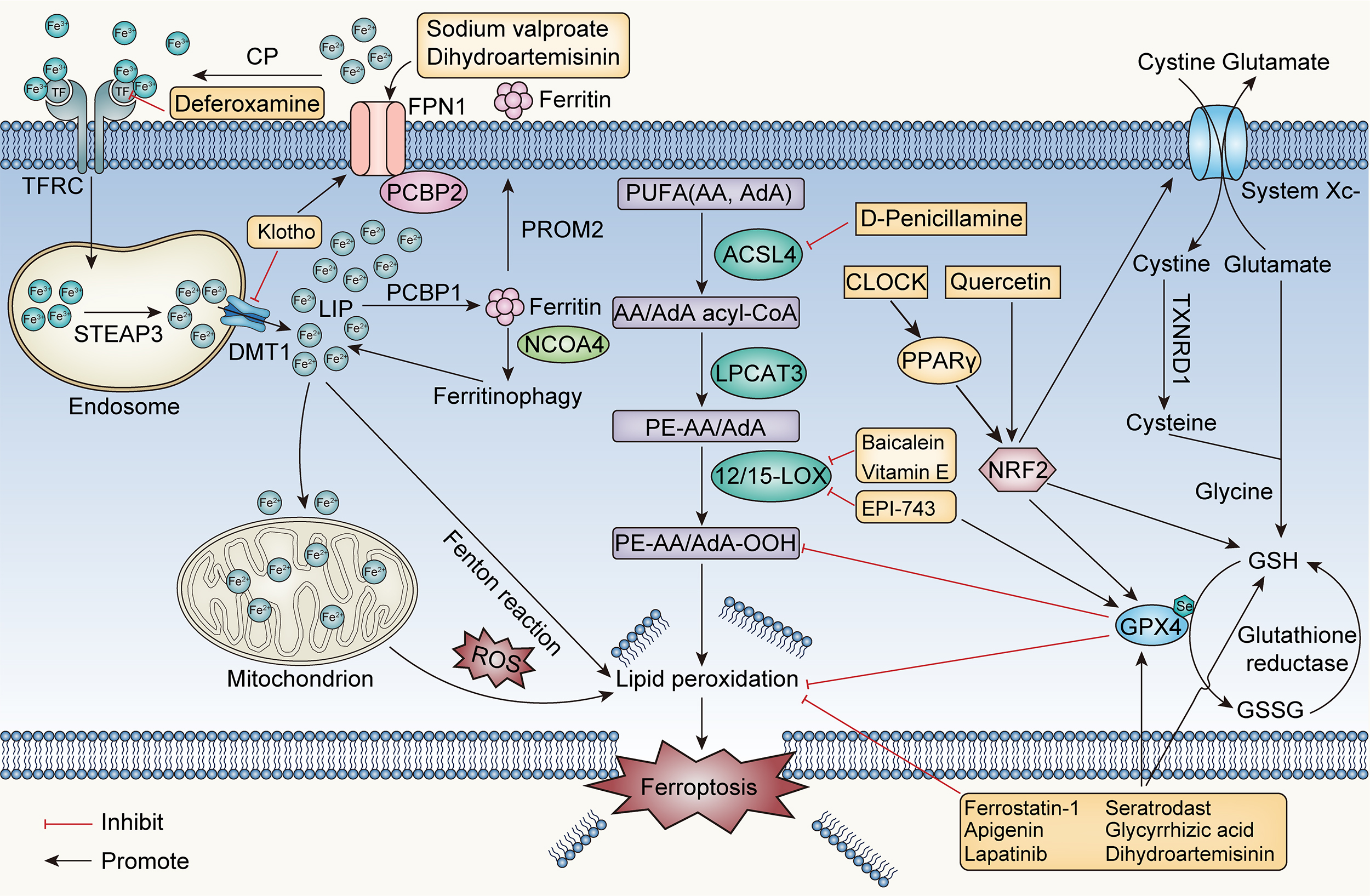 Fig. 2.
Fig. 2.Core mechanisms of ferroptosis and ferroptosis-based epilepsy
management. CP, ceruloplasmin; TFRC, transferrin receptor; STEAP3, six
transmembrane epithelial antigen of prostate 3; DMT1, divalent metal transporter
1; FPN1, ferroportin 1; PCBP, poly-(RC)-binding protein; LIP, labile iron
pool; PROM2, prominin-2; NCOA4, nuclear receptor coactivator 4; PUFA,
polyunsaturated fatty acids; AA, arachidonic acid; AdA, adrenergic acid; ACSL4,
acyl-CoA synthetase long-chain member 4; LPCAT3, lysophosphatidylcholine
acyltransferase 3; ROS, reactive oxygen species; NRF2, nuclear factor erythroid
2-like 2; PPAR-
Studies on the effects of ferroptosis inhibitors in several epilepsy models have shown that the therapeutic effects of these agents vary. Deferoxamine, an iron chelator, can reduce the epilepsy symptoms induced by ferric chloride by reducing TF levels and eliminating iron ions to inhibit ferroptosis [117]. Ferroptosis is induced in the hippocampal neurons of epileptic mice by pentylenetetrazole and pilocarpine, and the number of mitochondria in these neurons decreases, as determined via electron microscopy. Seizures in mice can be alleviated by ferrostatin-1 therapy, which increases GPX4 and GSH levels, decreases malondialdehyde (MDA) and 4-HNE levels, and inhibits iron accumulation and prostaglandin-endoperoxide synthase 2 (Ptgs2) mRNA expression [90]. One study revealed that ferrostatin-1 alleviates cognitive impairment in a rat model of TLE by blocking P38 mitogen-activated protein kinase, another study revealed that ferrostatin-1 does not reduce spontaneous seizures [118, 119]. In post traumatic epilepsy models induced by ferric chloride, ferrostatin-1 does not affect the occurrence of epilepsy but alleviates the severity of acute phase seizure behaviour and chronic-phase cognitive impairment [120]. Vitamin E prevents PUFA peroxidation and interrupts free radical chain reactions [121]. In a rat model of epilepsy, vitamin E inhibits 15-LOX expression, which in turn decreases neuronal ferroptosis and alleviates seizures [122].
Most flavonoids have strong antioxidant effects and inhibit ferroptosis. Moreover, via GPX4 and sirtuin 1 (SIRT1) activation, apigenin reduces oxidative stress mediated by myeloperoxidase and prevents neuronal ferroptosis in the hippocampus of epileptic mice treated with kainic acid [123]. Quercetin inhibits hippocampal neuronal ferroptosis, alleviates seizures and protects cognitive function by enhancing NRF2 expression in a mouse model of kainic acid induced epilepsy [124]. Baicalein suppresses ferroptosis by halting the expression of 12-LOX and 15-LOX and has neuroprotective effects against posttraumatic epileptic seizures [125].
The transcription factor PPAR-
With the exploration of the relationship between the pathophysiological process
of epilepsy and ferroptosis combined with the clinical application of drugs,
researchers have found that inhibiting the ferroptotic pathway can play a role in
treating epilepsy, laying the foundation for the use of ferroptosis inhibitors as
drugs for epilepsy. Lapatinib, a breast cancer drug, also exerts neuroprotective
effects against seizures by inhibiting GPX4-mediated ferroptosis [129].
Glycyrrhizic acid has antiviral, immunomodulatory, and anti-inflammatory effects
[130]. Glycyrrhizin-treated rats with TLE exhibit reduced neuronal ferroptosis
and neuronal damage [131]. The asthma medication seratrodast is an antagonist of
the thromboxane A2 receptor [132]. Seratrodast was shown to shorten seizure
latency and seizure duration by inhibiting ferroptosis in a
pentylenetetrazole-induced model. It controls the redox pathway involving System
Xc
Inhibiting ferroptosis is an effective therapeutic approach for preventing seizures and protecting against cognitive dysfunction, but related studies are at the basic stage (animal and cellular experiments), and extensive clinical studies have not yet been carried out. Although studies on the role of ferroptosis in epilepsy have focused on hippocampal neurons, little is known about how ferroptosis coordinates various processes in the pathophysiology of epilepsy. New treatment targets may be discovered by delving into the function of ferroptosis in epilepsy.
HL conceptualized the manuscript and acquired financial support; LH wrote the original manuscript and designed the figures; SL reviewed and edited the manuscript, performed the literature searches. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This work was financially supported by the Research cooperation platform project of Sino-Japanese Friendship Hospital of Jilin University and Basic Medical School of Jilin University (Grant NO.KYXZ2022JC04).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.