





1 Genetics & Developmental Biology Laboratory, Department of Biotechnology & Bioengineering, Institute of Advanced Research (IAR), 382426 Gandhinagar, Gujarat, India
Abstract
Parkinson’s disease (PD) is a neurodegenerative disorder characterized by the
progressive loss of dopaminergic neurons in the substantia nigra pars compacta
region of the midbrain and the formation of intracellular protein aggregates
known as Lewy bodies, of which a major component is the protein
Keywords
- Parkinson's disease
- α-Synucleinopathies
- Lewy bodies
- mitochondria
- mitochondrial dynamics
- mitotherapy
Parkinson’s Disease (PD) is the second most common progressive neurodegenerative
disorder (NDD) after Alzheimer’s disease (AD). It was originally described by
James Parkinson in his article “An essay on the shaking Palsy” in 1817, after
which it was renamed PD [1, 2, 3, 4, 5, 6, 7, 8, 9]. PD is pathologically characterized by the loss of
dopaminergic (dopamine-producing) neurons in the substantia nigra pars compacts
(SNpc) of the midbrain and the formation of neuronal Lewy bodies (LBs) composed
of
PD is a multifactorial disease, in which genetic and non-genetic environmental factors are involved [24, 25, 26, 27]. Several biological processes are known to contribute to PD pathogenesis, such as accumulation of misfolded proteins [24, 28, 29, 30], oxidative stress [24, 31], mitochondrial dysfunction [24, 32], and neuroinflammation [33, 34]. However, the mechanistic details underlying these processes are not well understood. Eighteen genetic loci and twelve familial PD genes—ATP13A2, DNAJC6, DJ-1, EIF4G1, FBXO7, LRRK2, PARK2, PINK1, PLA2G6, SNCA, SYNJ1, and VPS35—have been identified to date [24, 30, 35].
It is widely reported that changes in several cellular processes play a pivotal role in the onset of PD. The details of some key processes are described below.
The accumulation of misfolded SNCA and other proteins are established primary
hallmarks of PD [36, 37, 38]. Several genetic, molecular, and biochemical studies
conducted on postmortem human brains from patients neuropathically diagnosed with
PD with dementia and DLB suggest that the accumulation of misfolded proteins such
as amyloid beta (A
The accumulation of hyper-phosphorylated tau can lead to the formation of paired helical filaments known as “neurofibrillary tangles” (NFTs), which are a hallmark of several neurodegenerative diseases such as AD, PSP, and frontotemporal dementia with parkinsonism (FTDP) [36]. FTDP is associated with a locus at chromosome 17 (FTDP-17), where p-tau aggregation occurs in the SNpc and cortex of the brain [36, 44]. P-tau can also be found in association with LBs and mutation of the LRRK2 gene, which are correlated with the development of sporadic PD [36, 44, 45]. Similarly, in FTDP cases, alteration in the gene coding for microtubule-associated protein (MPTP) leads to an increase in p-tau aggregation [36, 45]. Moreover, while NFTs are associated with AD, they can co-localize with SNCA in LBs and play a significant role in destabilizing DA neurons, ultimately resulting in their degeneration and death [36, 44, 46].
Oxidative stress (OS) plays a regulatory role in the aging process and directly affects the central nervous system (CNS) [47]. In addition, the oxidative stress theory is one of the most popular theories in PD and other neurodegenerative diseases such as AD, Huntington’s Disease (HD), and Amyloid Lateral Sclerosis (ALS) [36]. Under physiological conditions, reactive oxygen species (ROS) or free radicals are very important for gene transcription, regulation of synaptic plasticity, and apoptosis [47, 48]. Moreover, oxidative stress occurs when ROS increases the activity of cellular antioxidant enzymes. Due to the continuous increase of ROS, an aggregation of cytotoxic compounds occurs that results in lipid toxicity, protein collapse, failure of key enzymes activities, and induction of cell death in several neurons including DA-neuronal tissue [49]. Recently, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) has been proposed as a key ROS generator [50] and plays a pivotal role in the triggering of oxidative stress and inducing neurotoxicity [51], including within mitochondria [52]. Complex I and II of the electron transport chain (ETC) are thought to house most of the ROS produced in mitochondria [47]. Moreover, increased oxidative stress can reduce lysosomes and affect the lysosomal autophagy system, which is connected with oxidative stress to the build-up of SNCA [47]. Another hypothesis proposes that extra-cytosolic DA can be oxidized to generate DA-quinones. Further, DA-quinine-modified SNCA may partially inhibit chaperone-mediated autophagy and promote SNCA to self-assemble [47, 53, 54], while the accumulation of intracellular SNCA increases mitochondrial oxidative stress [55].
Mitochondrial dysfunction is known to be important in PD pathogenesis [47].
Moreover, several studies have shown that mitochondrial dysfunction induces
chronic ROS production and dopaminergic neurodegeneration. Further,
overexpression of SNCA in mice results in increased susceptibility to
toxins as compared with SNCA knockout mice, suggesting the toxicity of
mitochondrial SNCA [56, 57]. As mentioned above, PD affects mitochondrial complex
I activity, which is directly related to adenosine triphosphate (ATP) production
and, ultimately, leads to apoptosis [36, 58]. Further, dysregulation of
transcription factors causes changes in mitochondrial biogenesis, which leads to
mitochondrial dysfunction [47]. Peroxisome proliferator-activated receptor gamma
coactivator-1
Molecular and cellular analysis of the human brain has revealed
neuroinflammation-related damage in PD patients [47, 66]. Both innate and
adaptive immune responses are implicated in the progression of PD [47, 67, 68, 69].
They regulate nuclear factor kappa-B (NF-
In an animal model of 6-Hydroxydopamine (6-OHDA)-induced neuronal degeneration,
microglial cells gradually shift from an anti-inflammatory M2 phenotype to a
proinflammatory M1 phenotype [47, 81]. Following this repolarization, NF-
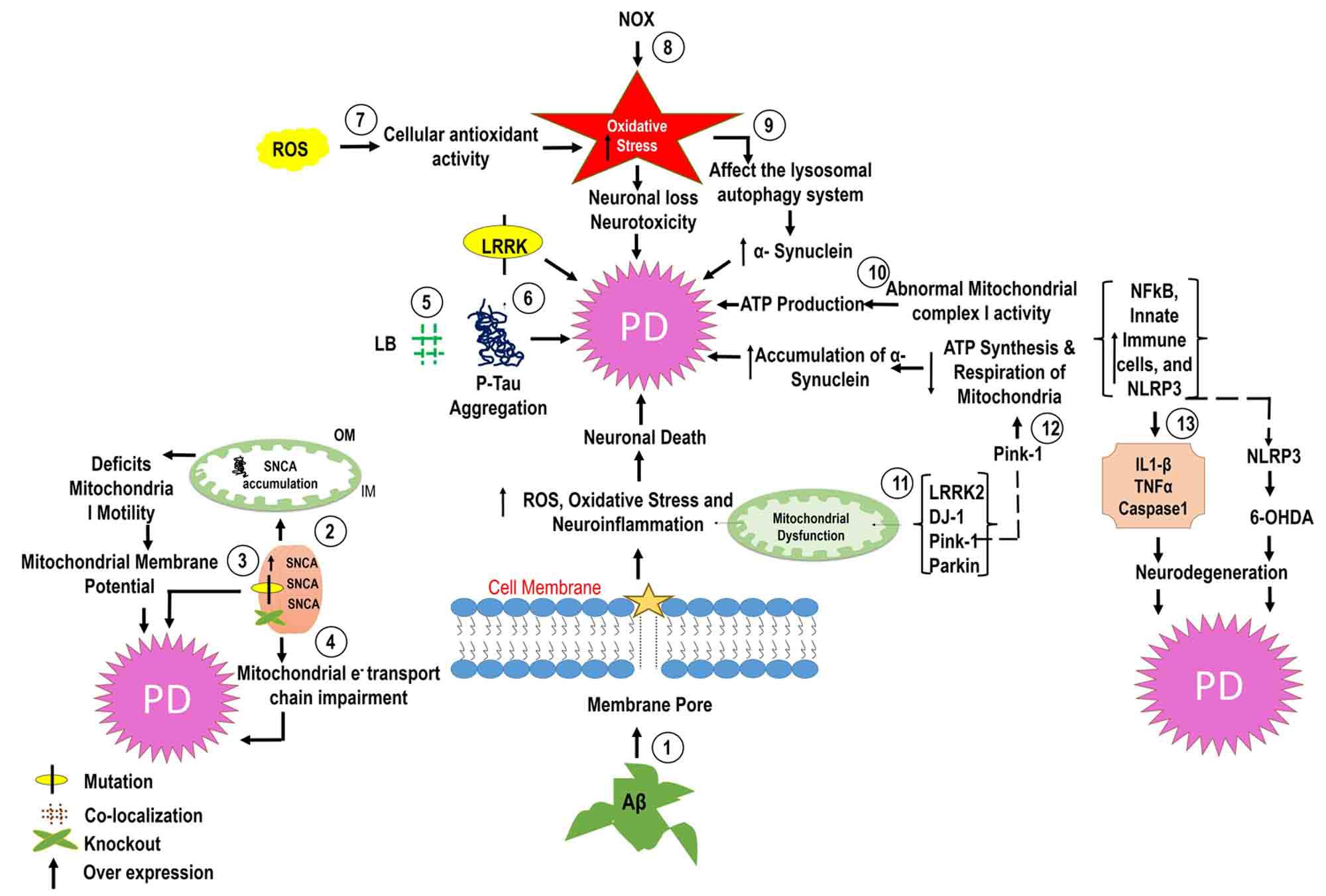 Fig. 1.
Fig. 1.Molecular mechanisms of PD. In PD, amyloid beta (A
Several studies have reported that SNCA binds with synaptic vesicle membranes and regulates neurotransmitter production [82]. In synucleinopathies, there is formation of SNCA aggregates in the brain that ultimately results in neuronal dysfunction [83, 84]. Further, pathological SNCA species lead to presynaptic deficits through their interference with the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex required for synaptic vesicle fusion and neurotransmitter release. SNCA oligomers have been shown to disturb SNARE-mediated vesicular fusion by binding to vesicle-associated membrane protein 2 (VAMP2) and inhibit SNARE complex formation, affecting neurotransmitter release and ultimately resulting in synaptic failure [85, 86]. Moreover, mitochondrial dysfunction is closely associated with presynaptic dysfunction caused by pathological SNCA. Importantly, SNCA can interact with mitochondria and induce mitochondrial fragmentation, impair mitochondrial fusion, and increase mitochondrial autophagy [39, 86]. Such abnormalities can impair energy production and the calcium homeostasis vital for synaptic transmission and vesicle fusion. Furthermore, dysfunctional mitochondria due to SNCA are also involved in oxidative stress leading to neuronal damage, worsening pre-synaptic deficits, and increasing synaptic dysfunction [39].
Tau, a microtubule-binding protein, is commonly found in axons, but in neurodegenerative conditions such as AD it is mislocalized to dendritic spines. Consequently, synaptic function and plasticity is disrupted, leading to postsynaptic deficits. In addition, tau mislocalization to dendritic spines affects the structural and functional integrity of synapses [39, 86, 87]. It also disrupts post-synaptic density organization and affects synaptic protein trafficking, which eventually results in the impairment of synaptic transmission and plasticity [86, 88].
Mitochondria are cytosolic free-floating organelles present in all eukaryotic cells and known as the “powerhouse” of the cell [6, 89, 90]. Mitochondria take care of cellular energy demands by generating cellular energy in the form of ATP by oxidative phosphorylation [6, 91] and are involved in cell death regulation via apoptosis, the formation of iron-sulfur clusters, calcium homeostasis, and the metabolism of amino acids and lipids (Fig. 2) [6, 92, 93]. Mitochondria regulate calcium homeostasis and play a key role in controlling programmed cell death and scavenging free radicals [6]. Structurally, mitochondria are double-membrane cell organelles composed of a lipid bilayer with a phospholipid, an inner membrane, and an outer membrane that surrounds the intracompartmental matrix [6]. The intermembrane space contains the major units of oxidative phosphorylation [6, 91] and the mitochondrial matrix contains 10–100 copies of mitochondrial DNA. Mitochondria are dynamic organelles that continuously undergo fission and fusion processes and regulate cell survival, cell growth, cell division, and cell differentiation [90, 94]. The balance between mitochondrial fusion and fission processes underpin the important roles of mitochondria, such as protecting mitochondrial DNA and controlling mitochondrial bioenergetics functioning [6, 95].
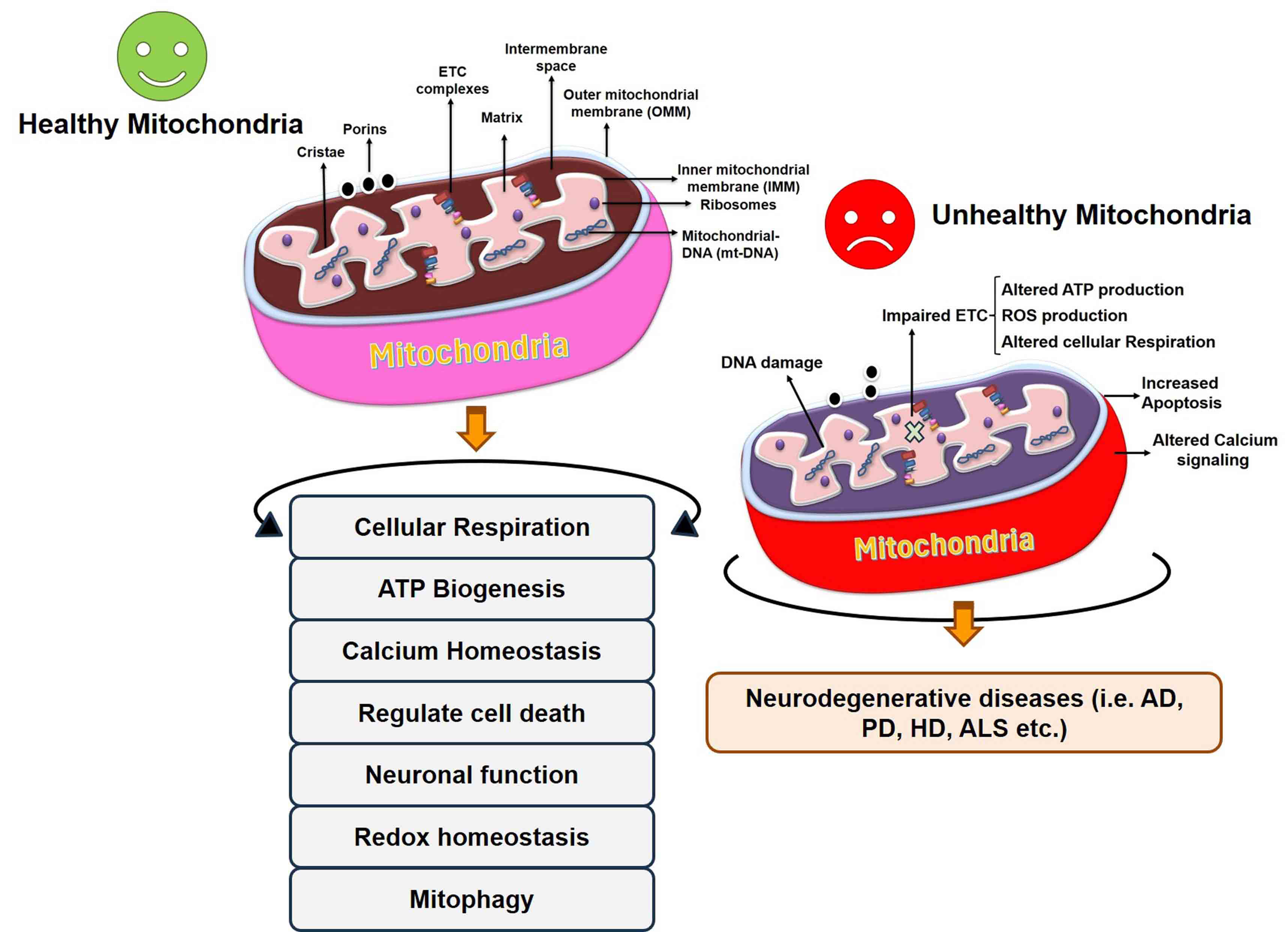
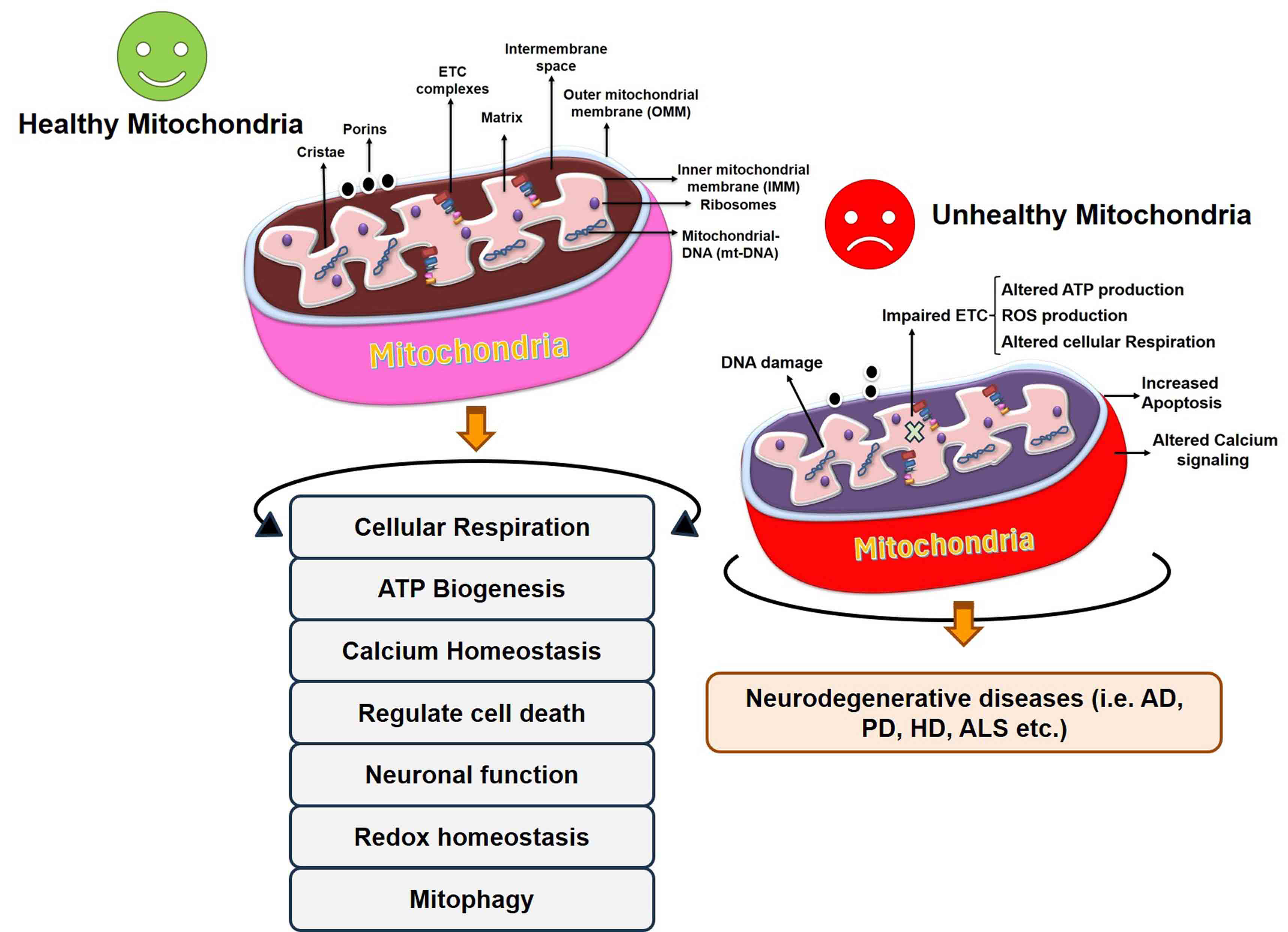 Fig. 2.
Fig. 2.Regulation of cellular processes by healthy mitochondria and alteration in mitochondrial function in the diseased condition. Unhealthy mitochondria result in abnormalities in different processes and ultimately the onset of neurodegenerative disease. AD, Alzheimer’s disease; HD, Huntington’s Disease; ALS, Amyloid Lateral Sclerosis; ETC, electron transport chain.
Mitochondria are interconnected entities that fulfill cellular energy requirements by changing their morphology and mobilizing within the cell [96, 97]. To ensure cellular energy demands, mitochondria pass through several processes such as the fission and fusion processes, axonal transport, and mitophagy [97, 98, 99]. There are several proteins available on the mitochondrial outer and inner membrane that regulate the mitochondrial fusion/fission process and mitochondrial transport. The fusion protein optic atrophy protein 1 (OPA1) is located on the inner mitochondrial membrane (IMM) while GTPase mitofusin1 and 2 (Mfn1/2) are located on the outer mitochondrial membrane (OMM) [97, 100, 101, 102, 103, 104]. In addition to OPA1 and Mfn1/2, several other proteins such as dynamin-related protein 1 (Drp1), mitochondrial fission 1 protein (Fis1), mitochondrial dynamics protein of 49 kDa (MiD49), mitochondrial dynamics protein of 52 kDa (MiD52), and mitochondrial fission factor (MFF) participate in the fission process.
Intracellular mitochondrial dynamics involve mitochondrial movement from one
cell compartment to another and are important in neuronal cells that are
connected with long axons and dendrites [105]. The movement of mitochondria in
axonal cells occurs by a process known as mitochondrial axonal transport, during
which mitochondria travel through the axon to fulfill the neuronal energy
requirements [106]. There are two types of mitochondrial axonal transport:
anterograde and retrograde. During anterograde mitochondrial movement,
mitochondria move from the cell body to the axon toward the microtubule (MT) plus
end, mediated by the kinesin motor protein to provide energy at the synapses. In
retrograde transport, mitochondria move from the axon to the cell body toward the
MT minus end, mediated by the dynein motor protein to eliminate the damaged
mitochondria [106, 107, 108, 109]. The movement of mitochondria is mediated by a protein
complex between the OMM protein mitochondrial rho GTPase (Miro), MT-bound motor
proteins, and adaptor proteins such as trafficking kinesin protein 1 and 2 (TRAK1
and 2) [97, 110]. Further, Miro1 also works with Drp1/Fis1-independent
mitochondrial shape transition protein (Mist) for mitophagy [97, 111]. MTs are
polar
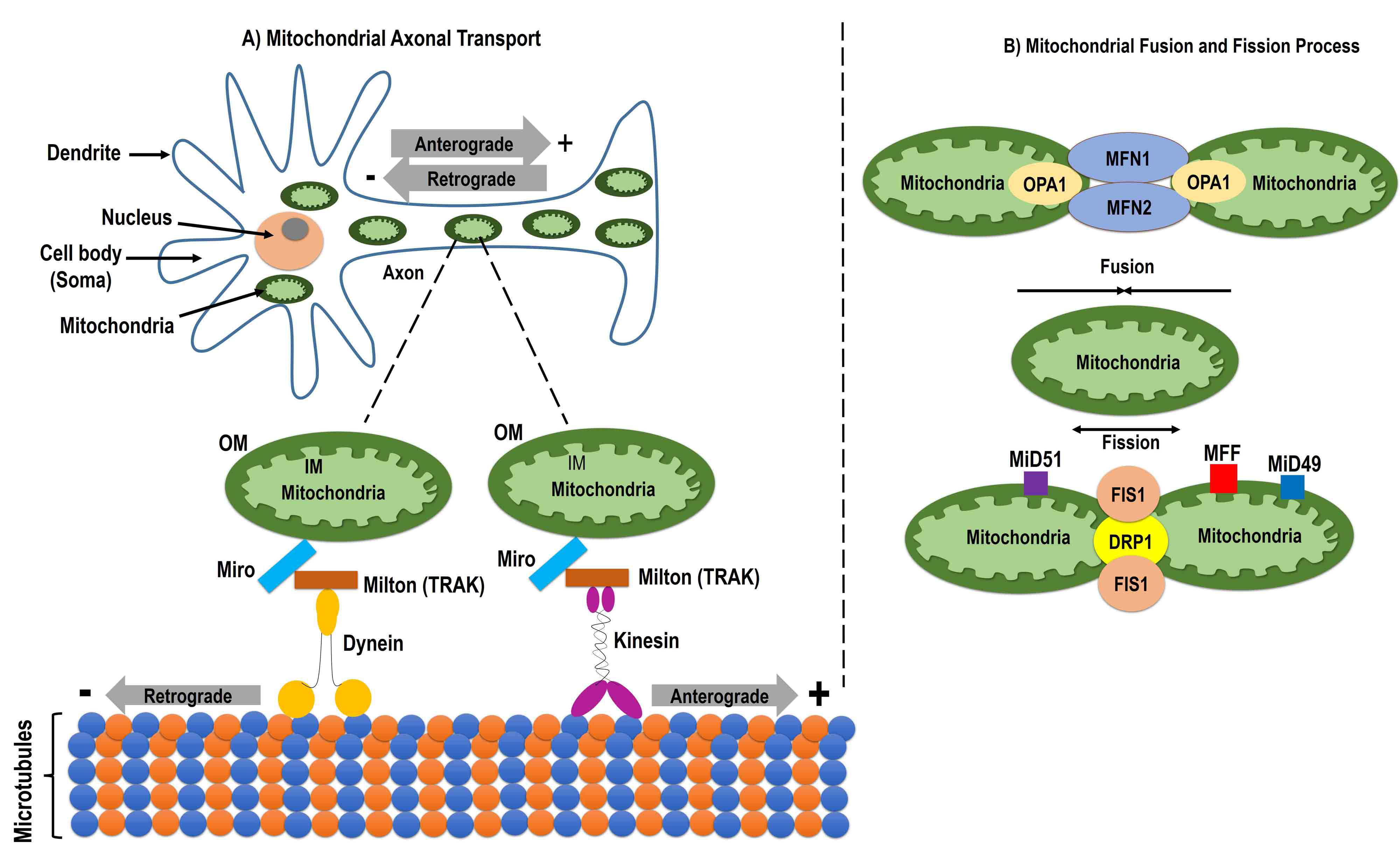
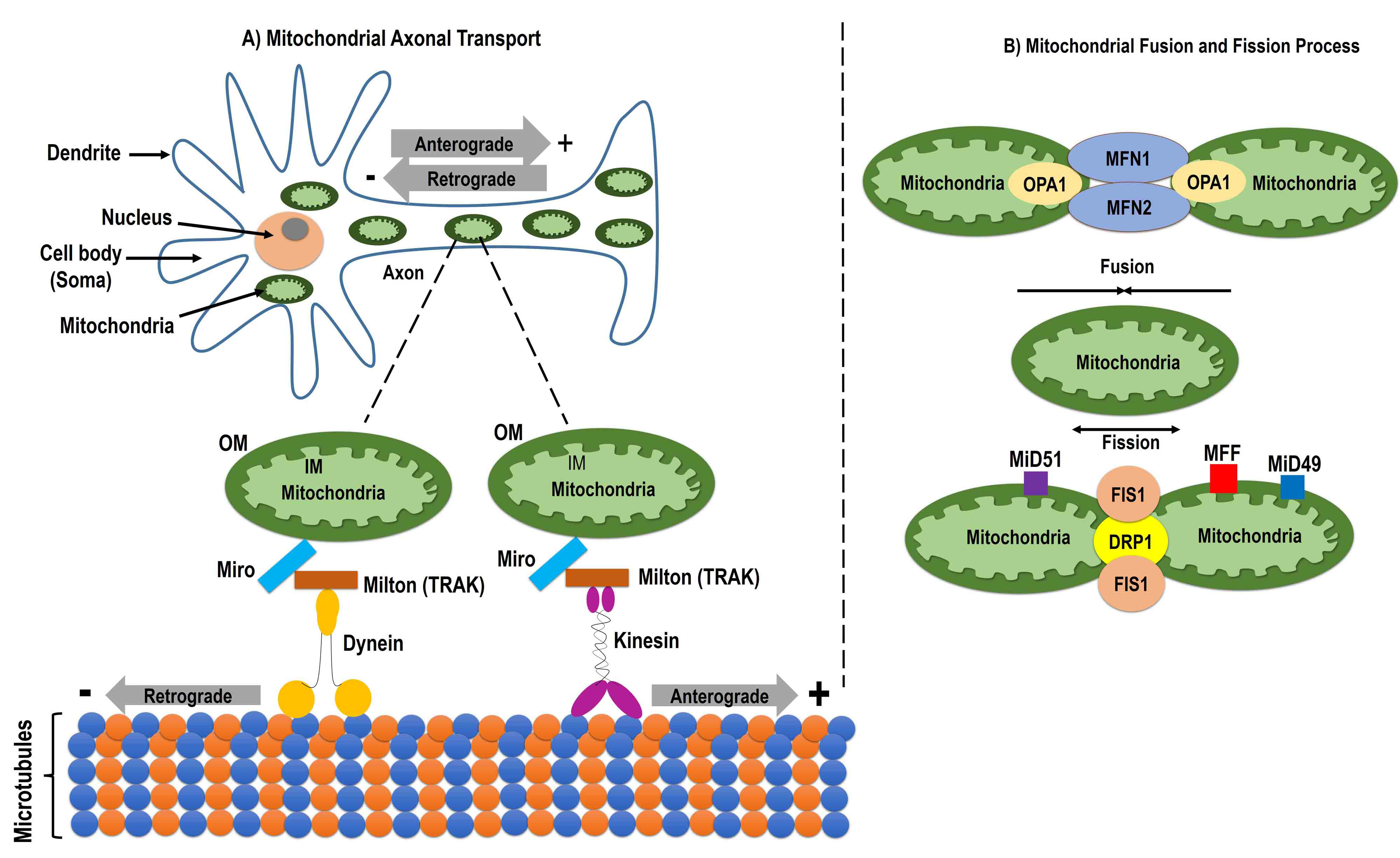 Fig. 3.
Fig. 3.Mitochondrial dynamics. Schematic image showing mitochondrial dynamics including the mitochondrial axonal transport, and fusion and fission process. Mitochondrial axonal transport uses the mitochondrial outer membrane protein Miro, trafficking kinesin-binding protein (TRAK), an adaptor protein milton, and the motor proteins kinesin and dynein. Miro makes a protein complex with motor proteins with the help of milton (TRAK). Kinesin and dynein help in making the connection between microtubules and mitochondria and facilitates axonal transport. Anterograde transport occurs through kinesin from the cell body to the axon and retrograde transport is mediated by dynein from the axon to the cell body (A). The mitochondrial fusion process is induced by the mitochondrial outer membrane protein Mfn and the mitochondrial inner membrane protein optic atrophy 1 (OPA1), which help in the fusion of the outer and inner membrane, respectively. Mitochondrial fission occurs by the mitochondrial outer membrane protein mitochondrial fission 1 protein (Fis1) and the cytosolic protein dynamin-related protein 1 (Drp1). During fission, Drp1 forms high molecular weight oligomers with Fis1 on the mitochondrial membrane. Once Fis1 is detached from the mitochondria, the fission process is complete (B). Mfn, mitofusin1; MiD49, mitochondrial dynamics protein of 49 kDa; MiD52, mitochondrial dynamics protein of 52 kDa; MFF, mitochondrial fission factor.
Further, triplication or mutation of SNCA; chemicals such as rotenone,
maneb, and 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydrodropyridine (MPTP); and
mutations in PINK1, LRRK2,
Parkinson disease protein 7 (DJ1), and vacuolar protein sorting-associated protein 35 (VPS35) can induce
inhibition of complex 1 [6, 119, 120]. Inhibition of complex 1 induces apoptosis
and produces ROS, resulting in the activation of intrinsic pro-apoptotic
pathways, translocation of Bax into the mitochondria, and release of cytochrome c
(cyt c) [6]. Phosphorylation of tumor necrosis factor receptor-associated protein
1 (TRAP1) or high-temperature requirement A2 (HTRA2), the substrate of PINK1,
results in attenuation of cell death by mitochondrial PINK1 [6]. Mutations in
PINK1 and Parkin are correlated with PD and can induce abnormal
mitophagy resulting in the accumulation of damaged mitochondria, which eventually
leads to cellular dysfunction and ultimately cell death [6]. SNCA has been shown
to influence mitochondrial size, in both a dependent and independent manner in
fission/fusion proteins, and transport and mitophagy processes [97, 121, 122, 123, 124, 125].
Further, SNCA oligomers can bind to lipids in the OMM and distress the membrane
bend which leads to reduced mitochondrial fusion [22, 97]. Furthermore,
upregulation of SNCA in a transgenic mouse model reduces the level of Mfn1/2 and
is associated with a decrease in mitochondrial fusion rate [97, 126], while
down-regulation of SNCA was shown to be associated with mitochondrial elongation
[97]. An increased concentration of SNCA was reported in a mitochondrial traffic
jam well before the axonal degeneration that affects mitochondrial mobility
(anterograde and retrograde) in the axon [22, 123]. In anterograde transport,
SNCA oligomers are disrupted by direct interaction between MTs and kinesin, which
leads to an increased expression of tau and eventually affects MT
structure [97, 127, 128]. SNCA induces fragmentation of MTs directly as well,
hampering the movement of mitochondria from the distal cell area [129].
Furthermore, the PD-linked protein LRRK2 alters the MT polymerization cycles,
affecting mitochondrial trafficking [130, 131]. The parkinsonian toxin 1-methyl-4-phenylpyridinium (MPP
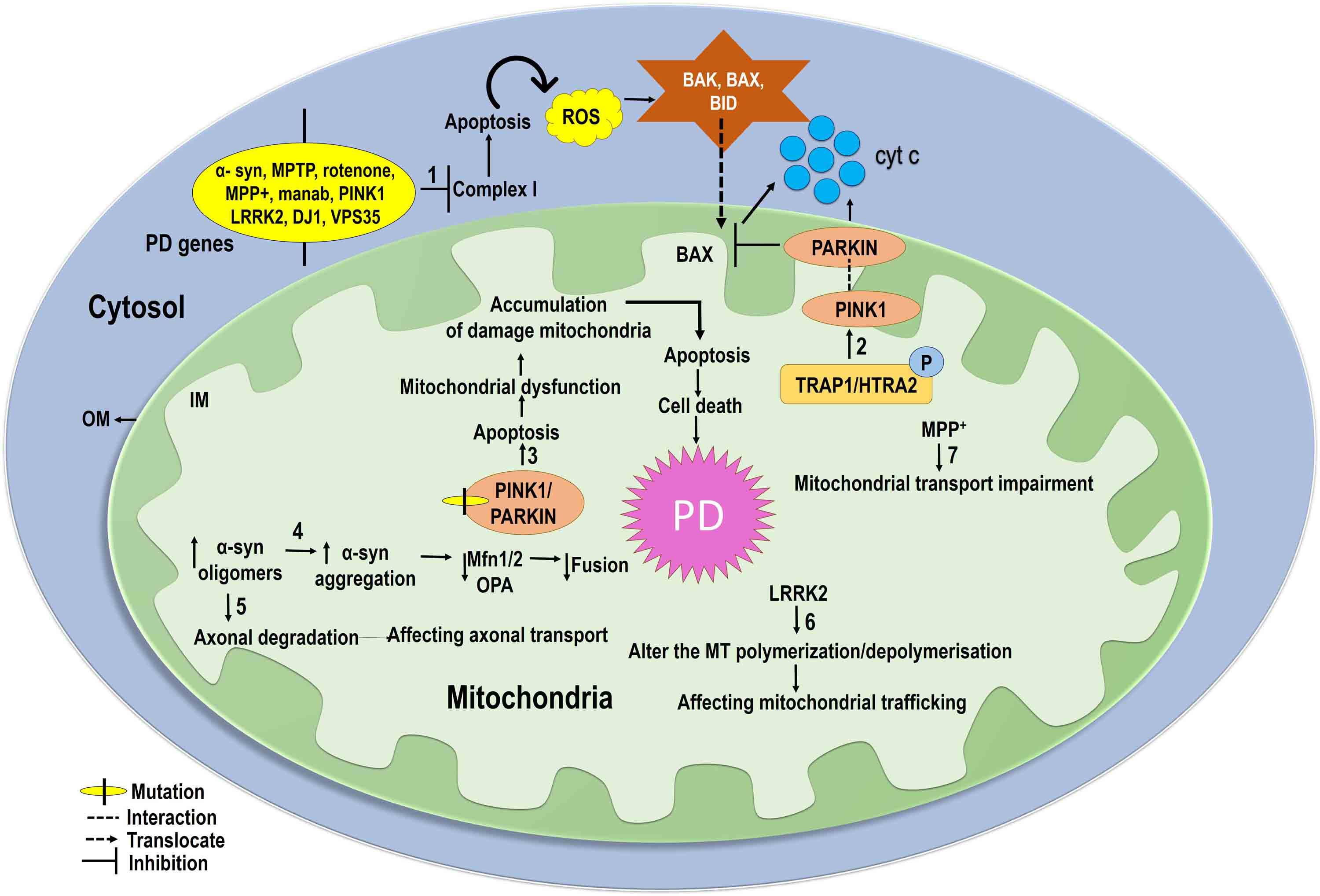 Fig. 4.
Fig. 4.Mitochondrial dynamics in PD: Mutations in SNCA, MPTP, PINK1, LRRK2, DJ1, and VPS35 inhibit mitochondrial complex I activity and produce ROS, leading to intrinsic pro-apoptotic pathway activation. Bax translocates to the mitochondrial inner membrane and releases cyt c in the mitochondria, eventually leading to apoptotic cell death (1). PINK1 reduces apoptotic cell death through phosphorylation of tumor necrosis factor receptor-associated protein 1 (TRAP1) and high-temperature requirement A2 (HTRA2) (2). Mutations in PINK1 and PARKIN reduce mitochondrial dysfunction and accumulation of damaged mitochondria induce apoptosis in PD (3). Increased SNCA oligomers increase the accumulation of SNCA and reduce levels of Mfn1/2 and OPA1, and decrease mitochondrial fusion (4). Increased SNCA oligomers hamper axonal degradation, which affects axonal transport (5). Furthermore, the PD-linked LRRK2 seems to affect microtubule (MT) polymerization, altering mitochondrial trafficking (6). The parkinsonian toxin 1-methyl-4-phenylpyridinium (MPP+) impairs mitochondrial transport and also inhibits kinesin-1-mediated anterograde transport (7).
As mentioned above, mitochondrial dysfunction results in decreased oxidative
phosphorylation affecting cellular energy production and increased production of
ROS, one of the key factors associated with aging and age-related diseases [135, 136]. Thus, it is unequivocally accepted that mitochondrial quality control or
mitochondrial health plays a vital role in the onset of several neurodegenerative
disorders, and abnormal mitochondrial health/abnormal mitochondrial function has
been documented in PD patients and various
In the past decade, mitochondria have been examined as a potential target for identifying possible therapeutic strategies for several conditions including neurological, metabolic, genetic, cancer, and viral diseases [140]. Mitochondria, the powerhouse of cells through generating ATP via the electron transfer from complex I to IV, become nonfunctional in several diseases which makes them a target for reviving cells affected by pathological conditions. Mitotherapy (mitochondrial therapy) utilizes approaches that can be used to improve mitochondrial health and subsequently helps to restore neuronal dysfunction and energy metabolism in neurons, promoting their capacity for energy regeneration [140, 147].
Several strategies have been examined for their efficacy in improving mitochondrial health and disease conditions. These include the use of several molecules, antioxidants, nanoparticles, genetic therapy-based approaches, plant secondary metabolites, mitochondria-targeted drug molecules, efficient drug delivery methods, and mitochondrial transplantation. The details of some of these potential strategies are outlined in Fig. 5.
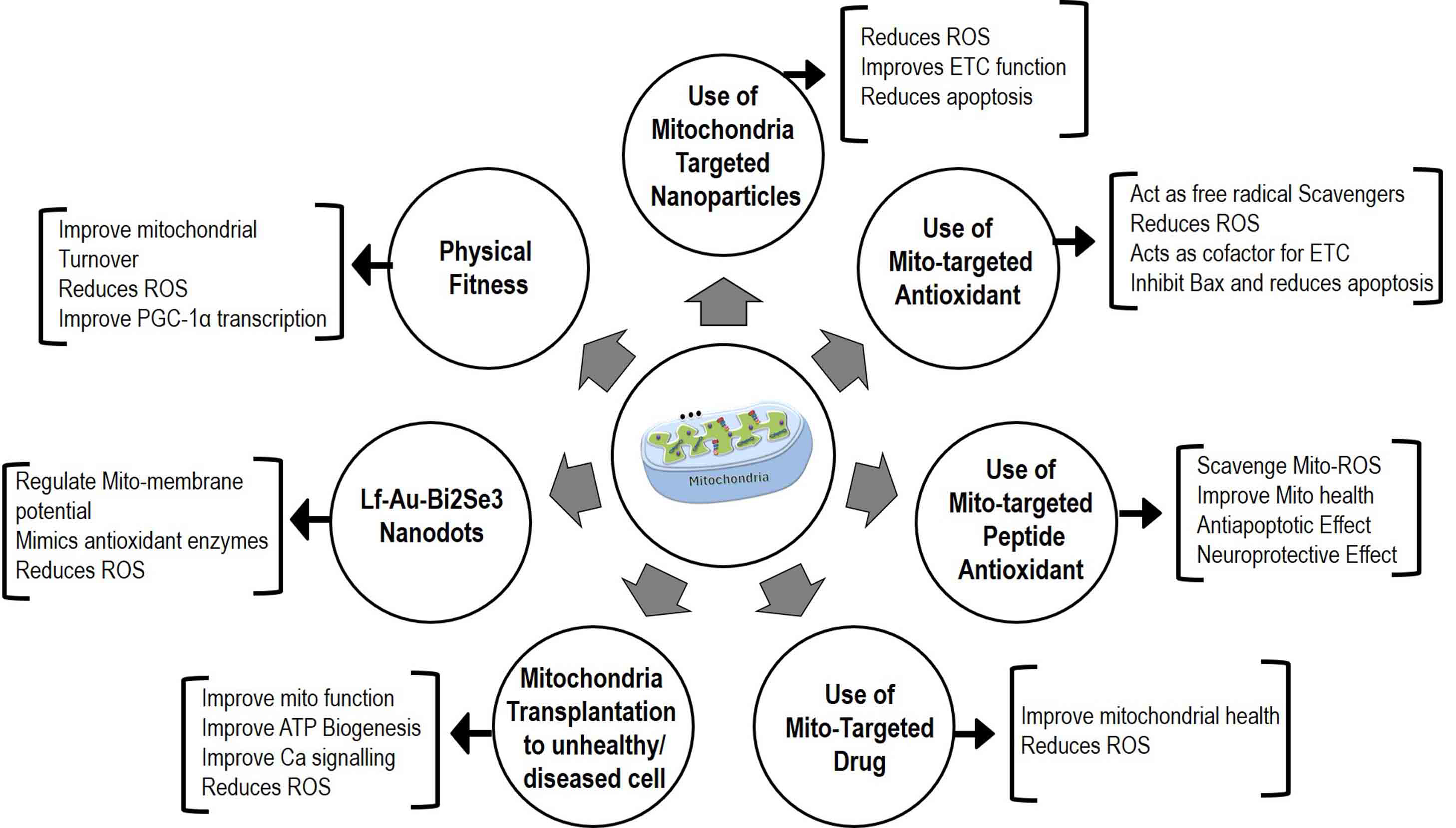 Fig. 5.
Fig. 5.Mitotherapy-based strategies for
Coenzyme Q10 (CoQ10) is a potential target and has been used in several
clinical trials for PD. CoQ10 acts as a lipid-soluble endogenous compound and
serves as a cofactor for the ETC by accepting electrons from complexes I, II, and
III [141, 148]. In microsomal lipid membranes and inner mitochondrial membranes,
CoQ10 acts as a free radical scavenger by regenerating
MitoQ (mitochondrial quinoline derivative), a mitochondria-targeted
antioxidant, has shown potential for improving mitochondrial health in humans and
animal models. MitoQ helps in the conversion of hydrogen peroxide
(H
Mitochondria-targeted neuroprotective peptides are synthetic peptides of less than 10 amino acid residues in length. They are also known as Szeto-Schiller (SS) peptides as they were developed by Hazel H. Szeto and Peter W. Schiller at CRI, Quebec, Canada. SS peptides (SS02, SS31, SS20) are well studied and can easily transfer to mitochondria and scavenge the mitochondrial ROS, helping to improve mitochondrial health [154, 158, 159, 160]. The use of SS peptides in different animal models of neurodegenerative diseases suggests that SS peptides (SS-02, SS-31) possess anti-apoptotic and necrosis effects [160]. Further, SS20 and SS30 have been reported to play a neuroprotective role by improving mitochondrial function in a PD model [160].
A study by Sekhar, 2022 [161] demonstrated that supplementation of Glycine and N-Acetylcysteine (GlyNAC) to mice and type 2 diabetic patients improves mitochondrial function by lowering oxidative stress [161].
Lactoferrin-functionalized Au-Bi2Se3 nanodots (Lf-Au-Bi2Se3 NDs) is one of the promising molecules that have the potential for the treatment of PD by targeting the mitochondria and attenuating ROS [153, 162, 163]. Lf-Au-Bi2Se3 NDs exhibit strong blood-brain barrier (BBB) permeability and can efficiently cross BBB to reach the brain [154, 155, 156]. Within the brain, these nanodots (NDs) function as different enzymes, including superoxide dismutase, catalase, glutathione peroxidase, and reductase [164]. They help to regulate the level of cellular ROS and maintain mitochondrial membrane potential. In an in vivo study using a PD mouse model, it has been shown that use of Lactoferrin-functionalized Au-Bi2Se3 nanodots (Lf-Au-Bi2Se3 NDs) improves memory and motility, protected mitochondria, and reduced loss of dopaminergic neurons in the SNpc region of the brain [162, 165]. Further, it was demonstrated that Lf-Au-Bi2Se3 NDs mimic antioxidant enzymes to reduce ROS levels and protect cells from toxicity. Furthermore, Lf modification enhances receptor adsorption-mediated transcytosis in the BBB [156], thus increasing therapeutic specificity in PD. NDs were also found to be close to mitochondria, further supporting their mitochondria-protective effect [60, 166, 167]. Overall, Lf-Au-Bi2Se3 NDs show promise as a potential therapeutic target for PD [52, 162].
Several studies have reported the utilization of gold nanoparticles (GNPs) conjugated with lipoic acid (LA) for the treatment of PD through the mitigation of oxidative stress [168, 169, 170, 171]. Oxidative stress, caused by the excessive generation of ROS, is intricately linked to the gradual degeneration of neuronal cells in PD [171, 172, 173]. The study establishes the capacity of GNP-LA nanoconjugates to effectively shield SH-SY5Y cells (a cellular model for PD) from the detrimental effects induced by ROS [174, 175, 176, 177]. In addition, it also emphasizes the biocompatibility of GNPs-LA nanoconjugates and their capacity to alleviate cellular damage caused by heightened levels of ROS [177, 178]. This promising treatment for PD takes advantage of the antioxidant attributes of LA, coupled with the drug delivery capabilities of GNPs. Furthermore, it highlights the use of atomic force microscopy to gain insights into the mechanical changes of living cells in response to physiological and pathological changes. This technique provides valuable insights into the effectiveness of GNP-LA nanoconjugates in protecting cells against oxidative stress [168, 177, 179, 180].
Schlichtmann et al. (2022) [181] and others discussed the use of nanoparticles as a targeted drug delivery system by using polyanhydride nanoparticles (NP) functionalized with a triphenylphosphonium derivative named (3-carboxypropyl) triphenylphosphonium (CPTP) [182, 183, 184, 185] to improve the delivery of a neuroprotective drug called mito-metformin [186, 187, 188, 189, 190, 191]. The objective of the study was to treat mitochondrial dysfunction, which is a significant factor in the progression of PD, by focusing on dopaminergic neuronal atrophy [192, 193, 194, 195]. First, the researchers tested the internalization of nonfunctional and functionalized CPTP NPs by neurons [196, 197, 198]. They observed that functionalization of CPTP significantly enhanced the cellular internalization of NPs. After that, it was determined whether nanoformulas were effective in treating mitochondrial dysfunction caused by rotenone [189, 190, 192, 199, 200]. The results showed that mito-metformin-coated NPs, functionalized by CPTP, confer significant protection against rotenone-induced toxicity, while non-functional and soluble mito-metformin NPs did not have a similar protective effect [201, 202, 203]. The mechanism of action involves the activation of adenosine monophosphate-activated protein kinase (AMPK) by mito-metformin. AMPK plays an important role in maintaining cellular metabolism and is attenuated in PD [192, 193, 200, 204, 205]. By activating AMPK, mito-metformin helps restore cellular function and protect against dopaminergic-induced cell death [206]. Targeted delivery of mito-metformin using a CPTP-functionalized NP improves its efficacy and reduces the required therapeutic dose, thereby minimizing the risk of toxicity. Overall, this study demonstrates the potential of targeted nanocarriers, especially CPTP-functionalized polyanhydride NPs, in the treatment of PD. This approach could help slow the progression of the disease by enhancing drug delivery and mito-metformin’s neuroprotective effects and improving the quality of life for people with PD [57, 182].
Plant secondary metabolites modulate the oxidative stress and
neuroinflammation in PD through numerous mechanisms [89, 181, 207, 208, 209, 210, 211]. They
have antioxidant outcomes and modulate the complexes/enzymes of nerve cells,
stimulating mitochondrial biosynthesis through pathways consisting of sirtuin 1 (SIRT1),
peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1
Apocynin, a non-poisonous plant molecule possesses neuroprotective effects and has the potential to treat PD [192]. Apocynin functions as an antioxidant, is centered on mitochondria, and allows less oxidative harm, which is an essential pathological mechanism that causes neurodegeneration in PD [177, 179, 192]. A study by Brenza et al. (2017) [176] in a preclinical animal model of PD demonstrated that the orally active apocynin spinoff decreased neuroinflammation and guarded toward dopaminergic neurodegeneration, thus playing a neuroprotective role [192, 219, 220]. Several other studies have also shown that the use of apocynin derivatives to target mitochondria plays a protective role in dopaminergic neurodegeneration in a preclinical animal model of PD [192, 219, 220, 221]. Further, nano-formulated, mitochondrial-focused apocynin provides extraordinary safety in opposition to oxidative stress-caused mitochondrial disorder and neuronal harm in a variety of neurons, including the dopaminergic neuronal lineage [158, 159, 160, 161]. Thus, available evidence suggests that apocynin can be a useful remedy for ameliorating PD with the aid of stopping neuronal damage by reducing oxidative stress [219], specifically when used in conjunction with nanoparticles. Detailed research is vital to evaluate the effectiveness of nanoparticle-mediated apocynin transport and its potential to cross the BBB [163, 220].
Mitochondrial transplantation/mitochondrial transfer a therapeutic modality that aims to enhance cellular function and mitigate damage by replacing damaged mitochondria with healthy ones [222, 223, 224]. This approach entails the isolation of functional mitochondria, which are then introduced into the damaged tissue. The transplanted mitochondria have the potential to restore bioenergetic function, augment ATP production [225, 226], improve calcium regulation, and reduce oxidative stress, thereby promoting cellular survival and function [227, 228, 229]. However, mitochondrial transplantation as a therapeutic strategy poses potential risks and challenges. One of the challenges is the efficient delivery of mitochondria to the target tissue [230, 231, 232, 233]. Various methods have been developed, including direct injection into the damaged organ or systemic administration of a bolus of mitochondria [137]. Direct injection allows for a focal concentration of mitochondria but is invasive and may require multiple injections. Systemic administration allows for global distribution but accessing certain organs such as the brain may be challenging [234]. Another challenge is understanding the incorporation and protective mechanisms of transplanted mitochondria. It is imperative to determine how the transplanted mitochondria integrate into the recipient cells’ mitochondrial network and whether they can maintain their functionality in the long term [227, 235, 236]. Additionally, there is a need to assess the potential risks associated with mitochondrial transplantation, such as immune responses, potential adverse effects, and the risk of introducing dysfunctional mitochondria [237, 238, 239, 240]. In order to fully comprehend the efficacy, safety, and long-term effects of mitochondrial transplantation as a therapeutic strategy, in-depth research is required to address these challenges and fully comprehend their effects.
Several studies have reported that with aging and in neurodegenerative diseases
there is a progressive decline in motor function due to reduced mitochondrial
function and ATP biogenesis. Further, regular exercise and physical activity
increase mitochondrial turnover and ATP biogenesis by reducing ROS [241, 242, 243, 244, 245]. It
has been demonstrated that exercise restores aging-induced reduced
PGC-1
SkQ1, consisting of plastoquinone in conjugation with tetraphenylphosphonium (TPP+), targets
mitochondria and within mitochondria it helps to reduce reduced skulachev plastoquinone 1 (SkQ1H
Creatine is guanidine compound and acts as a crucial energy reservoir for ATP. Creatine is found in skeletal muscle and several organs, such as the brain, is transported by specific creatine transporters, and serves as a substrate for cytosolic and mitochondrial creatine kinase [152, 167]. Creatine blocks apoptosis by inhibiting mitochondrial permeability transition pores [192]. Free radicals are induced by oxidative stress and promote the conversion of an octameric form of creatine kinase to a dimeric state, resulting in the opening of permeability transition pores to mediate cell death [152, 219]. Moreover, creatine/phosphocreatine possess neuroprotective functions through the creatine kinase-mitochondrial permeability transition pore system, suggesting that during oxidative stress induction, creatine and phosphocreatine enhances cytosolic, high energy phosphates, maintain the level of ATP and induce neurodegeneration [152, 220, 221].
In addition, other molecules for mitochondrial therapy such as exosomes, which are an emerging branch of drug delivery, and medicinal molecules such as curcumin have been shown to decrease neurodegeneration in traumatic brain injury and stroke, thereby improving mitochondrial function [140].
There are various genetic therapies for improving mitochondrial function, such
as mitochondria-based transcription activator-like effector nucleases (TALENs),
clustered regularly interspaced short palindromic repeat (CRISPR) approaches with
mitochondria-specific mitochondrially targeted Cas9 (mitoCas9), mtTALENs, and gene therapy by targeting Cytochrome P450 Family 46 Subfamily A Member 1 (CYP46A1)
in AD. Several drugs that facilitate the clearance of A
Neurodegenerative diseases are challenging issues worldwide, with no effective drugs available to treat them. Thus, researchers are trying hard to understand the molecular details of disease pathology, the cellular interactions of disease genes, and ways to positively alter the gene function to improve health. Mitochondria are vital cell organelles, responsible for fulfilling cellular energy demands and are affected in several neurological conditions including PD. Thus, mitotherapy, aimed at improving mitochondrial health, has emerged as a potential therapeutic strategy. It is interesting to examine how mitochondrial dynamics and PD are linked, with the aim of determining possible approaches for curing PD. Our review highlights recent possible approaches to treating PD, focusing on mitochondria and the fascinating studies exploring neuroprotective therapies in PD that serve as potential targets for restoring mitochondrial function.
AKT: conceptualized and designed the review, VB, HS & AKT: collected the information and drafted the manuscript, AKT: edited and approved the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
The authors are thankful to the peer reviewers for their opinions and suggestions.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.