














 , Ilaria Pappalardo 1, Anna Santarsiero 1, Swapnil Tripathi 2, Gyanendra Singh 2, Marcos Roberto de Oliveira 3,4,*
, Ilaria Pappalardo 1, Anna Santarsiero 1, Swapnil Tripathi 2, Gyanendra Singh 2, Marcos Roberto de Oliveira 3,4,*
1 Department of Science, University of Basilicata, 85100 Potenza, Italy
2 Division of Toxicology, ICMR-National Institute of Occupational Health, 380016 Ahmedabad, India
3 Department of Biochemistry, Institute of Basic Health Sciences, Federal University of Rio Grande do Sul (UFRGS), CEP 90035-000, Porto Alegre, Rio Grande do Sul, Brazil
4 Study Group on Neurochemistry and Neurobiology of Bioactive Molecules, Department of Chemistry, Federal University of Mato Grosso (UFMT), CEP 78060-900, Cuiaba, Mato Grosso, Brazil
Abstract
Carnosic acid (CA), a diterpene obtained mainly from Rosmarinus officinalis and Salvia officinalis, exerts antioxidant, anti-inflammatory, and anti-apoptotic effects in mammalian cells. At least in part, those benefits are associated with the ability that CA modulates mitochondrial physiology. CA attenuated bioenergetics collapse and redox impairments in the mitochondria obtained from brain cells exposed to several toxicants in both in vitro and in vivo experimental models. CA is a potent inducer of the major modulator of the redox biology in animal cells, the transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2), which controls the expression of a myriad of genes whose products are involved with cytoprotection in different contexts. Moreover, CA upregulates signaling pathways related to the degradation of damaged mitochondria (mitophagy) and with the synthesis of these organelles (mitochondrial biogenesis). Thus, CA may be considered an agent that induces mitochondrial renewal, depending on the circumstances. In this review, we discuss about the mechanisms of action by which CA promotes mitochondrial protection in brain cells.
Graphical Abstract
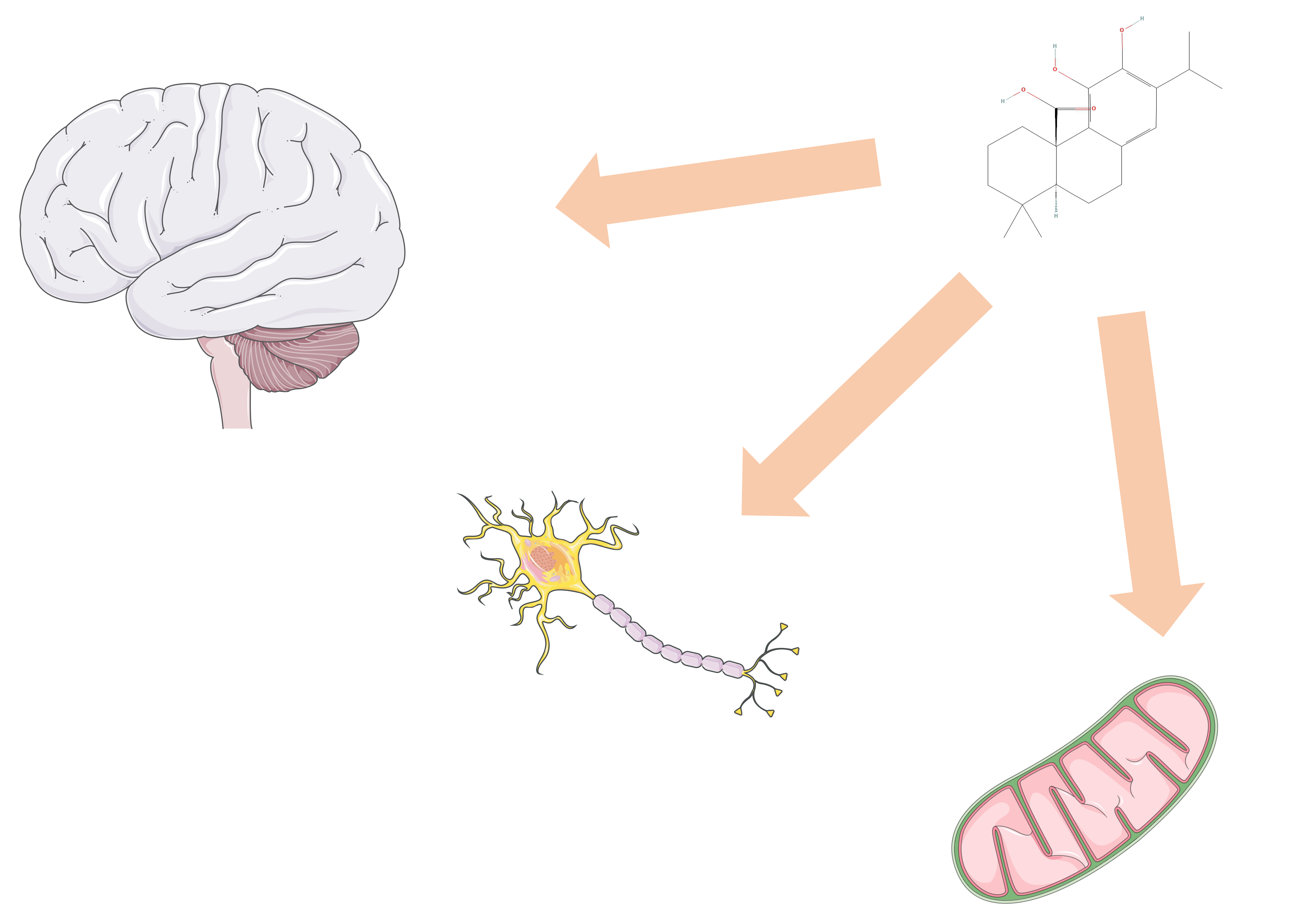
Keywords
- carnosic acid
- mitochondria
- neurons
- brain
- antioxidant
- Nrf2
Mitochondria are the cytoplasmic organelles of eukaryotic cells historically
recognized as the mainly responsible for the production of metabolic energy as
they house the oxidative phosphorylation (OXPHOS) [1]. Different metabolic
pathways, such as the tricarboxylic acid (TCA) cycle,
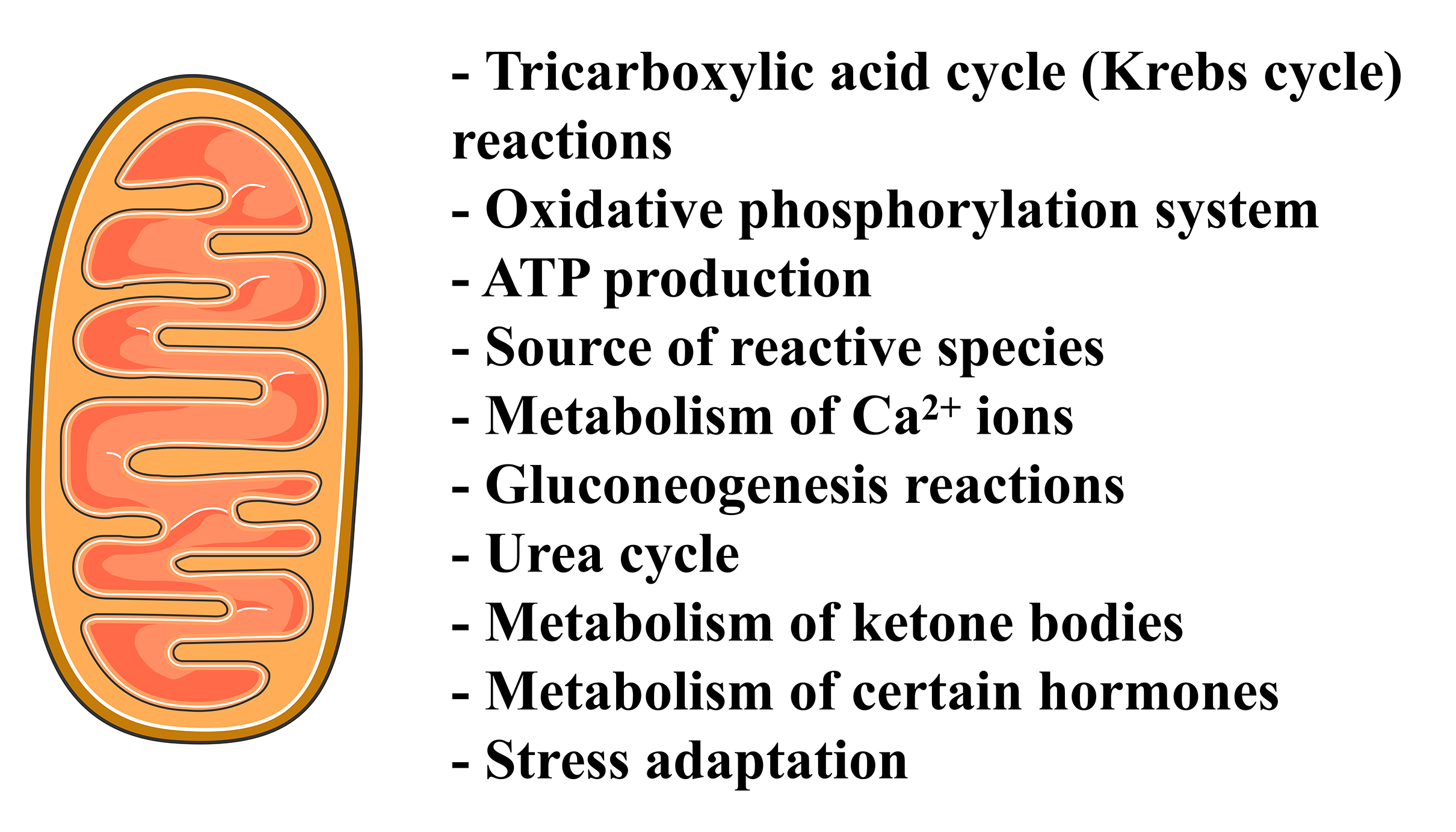 Fig. 1.
Fig. 1.A summary of the roles exerted by the mitochondria in mammalian
cells. The Figure was partly generated using Servier Medical Art, provided by
Servier, licensed under a Creative Commons Attribution 3.0 unported license. ATP,
adenosine triphosphate; Ca
Mitochondria represent a significant source of reactive oxygen species (ROS) and
are involved in calcium ions (Ca
Mitochondria are double-membrane surrounded organelles: the outer mitochondrial membrane (OMM), freely permeable due to the presence of a large number of porins [10], and the inner mitochondrial membrane (IMM), structured in cristae, so highly impermeable that almost all ions and molecules require membrane transporters to cross it [11]. The IMM is the primary site of adenosine triphosphate (ATP) synthesis, and it has a high percentage content of proteins involved in OXPHOS and transport of metabolites between the cytosol and the mitochondria [12]. The double-membrane system demarcates two independent compartments: the intermembrane space and the mitochondrial matrix. The internal matrix contains numerous enzymes involved in oxidative metabolism as well as the mitochondrial DNA (mtDNA) [13]. Mitochondria are unique among cytoplasmic organelles since their own maternally inherited, circular, and double-stranded DNA, closely resembles a prokaryotic genome [14]. The mtDNA encodes some proteins involved in the OXPHOS system, ribosomal RNAs and most of the transfer RNAs required for protein translation within mitochondria [15].
The TCA cycle is the central hub of metabolism where molecules derived from
different metabolic pathways, among which glycolysis and oxidation of fatty
acids, are oxidized and the energy obtained is stored in nicotinamide adenine
dinucleotide (NADH) and flavin adenine dinucleotide (FADH
Since O
Mitochondria regulate different types of cell death, with an essential role in apoptosis [34]. Various cellular stresses, among which DNA damage, hypoxia, viral infection, induce the intrinsic pathway of apoptosis initiated by the opening of the mitochondrial permeability transition pore (mPTP) located in the IMM. The aperture of the mPTP leads to the IMM swelling and the rupture of the OMM, followed by the leakage of cytochrome c, apoptosis-inducing factor (AIF) and endonuclease G [34]. Cytochrome c binds and activates apoptotic protease activating factor (Apaf-1), forming a complex known as apoptosome [35]. Apoptosome binds to and activates procaspase-9 to become the initiator caspase-9, which subsequently cleaves and activates the downstream effector caspases 3, 6, and 7, inducing apoptosis. Caspase-independent nuclear translocation of AIF and endonuclease G triggers chromatin condensation and degradation. Members of the B-cell lymphoma 2 (Bcl-2) family control these events exhibiting pro- and/or anti-apoptotic properties [36]. Mitochondria take part also in other cell death mechanisms [37], among which pyroptosis [38], necroptosis, and ferroptosis [39].
Mitochondrial biogenesis and mitochondria-selective autophagy (mitophagy), which fosters the elimination of damaged mitochondria, are two opposing processes that must be finely regulated and balanced to preserve mitochondrial homeostasis and ensure cellular adaptation to metabolic shifts, stress signals and environmental stimuli, such as aerobic exercise or caloric restriction [40]. Imbalance between mitochondrial biogenesis and mitophagy leads to development of countless pathologic conditions [41].
Mitochondrial biogenesis is the physiological response to adapt the number of
mitochondria to energy demands by which new organelles are formed by the growth
(increase in mitochondrial mass) and division of preexisting mitochondria [42, 43]. Mitochondrial biogenesis is controlled by both nuclear and mitochondrial
genomes [43]. The master regulator in mitochondrial biogenesis is peroxisome
proliferator-activated receptor-gamma coactivator-1 alpha (PGC-1
Mitophagy is the process of selective degradation of damaged and dysfunctional mitochondria through autophagy [53]. Mitochondria are enclosed by a membrane, forming autophagosomes which fuse with lysosomes for hydrolytic degradation. Phosphatase and tensin homolog (PTEN)-induced putative protein kinase 1 (PINK1)/Parkin pathway is the main molecular mechanisms regulating mitophagy [54]. PINK1 is a serine/threonine kinase usually present in low levels. When any damage occurs to mitochondria like mutations in mtDNA, accumulation of misfolded proteins, increased ROS, or depolarization, PINK1 accumulates at the OMM. PINK1 phosphorylates ubiquitin on Ser65 and recruits Parkin from the cytosol, phosphorylating and activating it. Parkin is an E3-ubiquitin ligase that, when activated, triggers the ubiquitination of a wide range of OMM proteins. These events result in the recruitment of autophagy receptors, such as p62/sequestosome 1 (SQSTM1), that facilitate the sequestering of damaged mitochondria within autophagosomes [55]. SQSTM1 comprises a domain interacting with polyubiquitinated proteins on the mitochondrial surface and a motif binding to microtubule-associated protein 1A/1B-light chain 3-II (LC3-II) present on autophagosomes [56]. Mitophagy can occur independently from functional PINK1, allowing ubiquitin-independent recognition of damaged mitochondria. In mammals the known receptors for receptor-mediated mitophagy include Bcl-2 interacting protein 3 (BNIP3), its analog NIX (BNIP3L) and FUN14 domain-containing protein 1 (FUNDC1) [54, 57]. These adapters detect mitochondrial damage and guide dysfunctional mitochondria to the autophagosome by changing their subcellular location or the proteins they interact with. FUNDC1 is a mitochondrial protein located in the OMM protein sensitive to hypoxia. The affinity of FUNDC1 for LC3 is regulated by phosphorylation at Ser17 and Ser13 under different stresses. While phosphorylation at Ser17 activates mitophagy, phosphorylation at Ser13 inhibits this process [55, 58, 59]. Mitophagy declines with aging determining accumulation of defective mitochondria, increased oxidative stress, and higher cell apoptosis [60].
In keeping with the multiple functions, mitochondria can change rapidly both form (size, shape, and position) and number to meet the physiological needs of cells undergoing the two opposing processes of fission and fusion [61, 62]. Mitochondrial homeostasis is strictly dependent on the equilibrium between fusion and fission dynamics which are important for mitochondrial inheritance and maintenance of functions (e.g., efficient OXPHOS, transport and regulation of mitophagy). Fission involves the separation of both OMM and IMM and their subsequent rejoining in the correct orientation, besides the properly partitioning of mitochondrial proteins and mtDNA so that each daughter organelle can function normally. In physiological condition, fission happens prior to cell division to guarantee that organelles are equally distributed between daughter cells [62]. Fission is also required as a preliminary event to mitophagy, to allow for segregation of damaged organelles, their inclusion in autophagosomes and finally their degradation [63, 64]. Mitochondrial dynamics require specialized nuclear-encoded protein, mainly represented by GTP-hydrolyzing proteins (GTPases) belonging to the dynamin superfamily. Three central players are (1) mitofusin 1 and mitofusin 2 (OMM fusion) [65], (2) optic atrophy 1 (OPA1) (IMM fusion) [66], and (3) dynamin-related protein 1 (Drp1) (division of IMM and OMM) [67]. Perturbations in mitochondrial dynamics have detrimental consequences on bioenergetic supply and represent a common feature of different neurogenerative disorders such as AD, PD, and HD [9].
AD usually affects people over the age of 65 and accounts for at least
two-thirds of dementia cases [68]. AD is characterized by the accumulation of
beta-amyloid (A
Many studies have been carried out to develop therapeutic strategies with the aim of attenuating mitochondrial dysfunction and restoring mitochondrial homeostasis in neurodegenerative diseases. Approaches target various mitochondrial processes such as energy metabolism, ROS generation, mitochondrial dynamics, mitochondrial biogenesis and mitochondrial protein synthesis. Different plant-derived natural products have been shown to be effective in delaying or treating neurodegenerative diseases via modulation of mitochondrial dysfunction [88]. Increasing evidence is establishing the relevance of carnosic acid (CA) as a promising neuroprotective agent.
Carnosic acid (CA; C
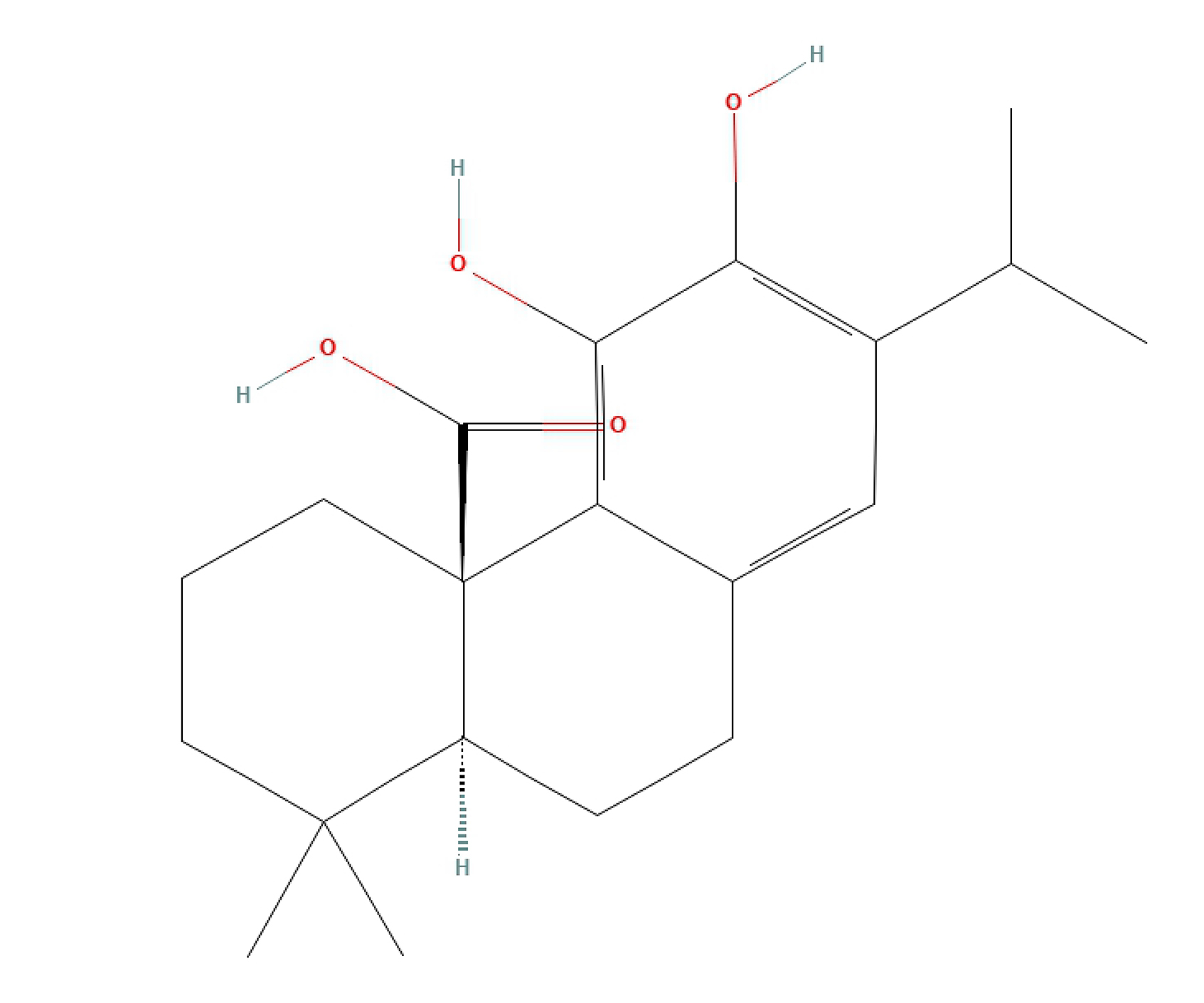 Fig. 2.
Fig. 2.Carnosic acid chemical structure. The chemical structure of carnosic acid was obtained from PubChem.
Also in planta, this molecule can give rise to various dehydrogenation derivatives such as carnosol, rosmanol and isorosmanol. If plants are subjected to high drought conditions or intense solar radiation, CA can be converted to methyl derivatives. It is clear that this molecule exerts a dual protective role in the rosemary plant against environmental stresses by capturing free radicals in the chloroplasts and preventing the breakdown of biological membranes [91, 92, 93]. As far as we know, CA has been identified in only one plant family, in only one out of 10 subfamilies of Lamiaceae (Nepetoideae), in only two out of 66 tribes (Mentheae, Ocimum), and in only nine out of 220 genera [89]. Most probably such a poor distribution in the plant kingdom is due to a very specific evolutionary strategy, but also to a scarcity of research, as many plant species still remain unknown and/or unstudied [89]. CA is not evenly diffused in plants, being present more in the aerial parts [94, 95] and its levels also depend on the phase of development. The biosynthesis and accumulation of CA occurs solely in young rosemary leaves at the branch apexes, where the diterpene molecule is partly employed during leaf formation and ageing [92, 96, 97, 98].
Bioavailability studies are few. It was reported that, in rats, 6 hours after
oral administration (64.3
Research over the past decade has revealed numerous bioactive abilities of CA including antioxidant, anti-inflammatory and anti-cancer activities, among others (Table 1, Ref. [92, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 119, 120, 121, 122, 123, 124, 125, 126, 127, 128, 129, 130, 131, 132, 133, 134, 135, 136]; Fig. 3). This fat-soluble compound is known for its documented antioxidant properties, which have enabled it to find various industrial applications in food and beverages, personal care, nutrition and health [89]. Most likely, the antioxidant properties of CA are linked to the presence of a catechol moiety and have been studied mainly in vitro in a wide variety of artificial and/or model systems [93, 102]. CA has been shown to protect fatty acids and triglycerides from oxidation in bulk and emulsified lipid systems [103, 104, 105]. Pearson et al. [106] showed how CA prevents oxidation of low-density lipoproteins in human aortic endothelial cells. Wijeratne and Cuppett [107] observed CA-mediated lowering of oxidative stress occurring by lipid hydroperoxide in Caco-2 cells. Moreover, it has been reported that CA reduces lipid peroxidation in rat liver microsomes and brain [100, 108]. Foods such as raw or cooked meat and oil have been protected from oxidation by CA, generally with greater efficiency than synthetic antioxidants [109, 110, 111, 112]. CA has been shown to have a very high reactivity towards ROS, being quickly oxidized and converted to a variety of metabolites in the process. Acting as a scavenger of ROS, CA eliminates both singlet oxygen, an excited form of oxygen, and free radicals. Furthermore, oxidized CA derivatives were found in rosemary leaves under both control and stress conditions, and prolonged exposure to darkness resulted in a striking reduction in their concentrations. These data indicate the chronic oxidation of CA in plants by light and suggest a protective role of CA even under low light conditions [92].
| Bioactive properties | Mechanism of action | References |
| Antioxidant | - Protection of fatty acids and triglycerides from oxidation | [103, 104, 105, 106, 107] |
| - Inhibition of lipid peroxidation | [100, 108, 109, 110, 111, 112] | |
| - H |
[92] | |
| Antimicrobial | - Antilisterial, antibacterial effect against Gram-positive and Gram-negative bacteria | [113, 114, 115, 116, 117, 118, 119, 120] |
| - Modulation of the bacterial membrane permeability | [121, 122, 123] | |
| Anti-cancer | - Proapoptotic effect | [124] |
| - Antiproliferative activity | [125] | |
| - Anti-angiogenic action | [126] | |
| - Chemoprotective function | [127] | |
| - Antiplatelet properties | [128] | |
| Anti-inflammatory | - Interleukin suppression | [129] |
| - Blocking of proto-oncogene tyrosine-protein kinase Src pathway | ||
| - Increasing of PGE |
[130] | |
| - Inhibition of NO release | [102] | |
| - TNF- |
[131] | |
| - NF- |
[132] | |
| - PPAR |
[133, 134] | |
| Anti-adipogenic | - Fasting glycaemia and plasma cholesterol levels reduction | [135, 136] |
| - Inhibition of pancreatic lipase activity | ||
| - PPAR | ||
| - Hepatocyte lipid accumulation reduction | [101] | |
| - Reduction of inflammatory cytokines expression (IL-1 | ||
| - Increasing of ATP, acetyl CoA, NAD(P)(+), and NAD(P)H | ||
| - EGFR, MAPK, AMPK, and ACC activation | ||
| - Inhibition of palmitate-induced cellular lipid accumulation | ||
| - Stimulation of EGFR and MAPK phosphorylation |
CA, carnosic acid; H
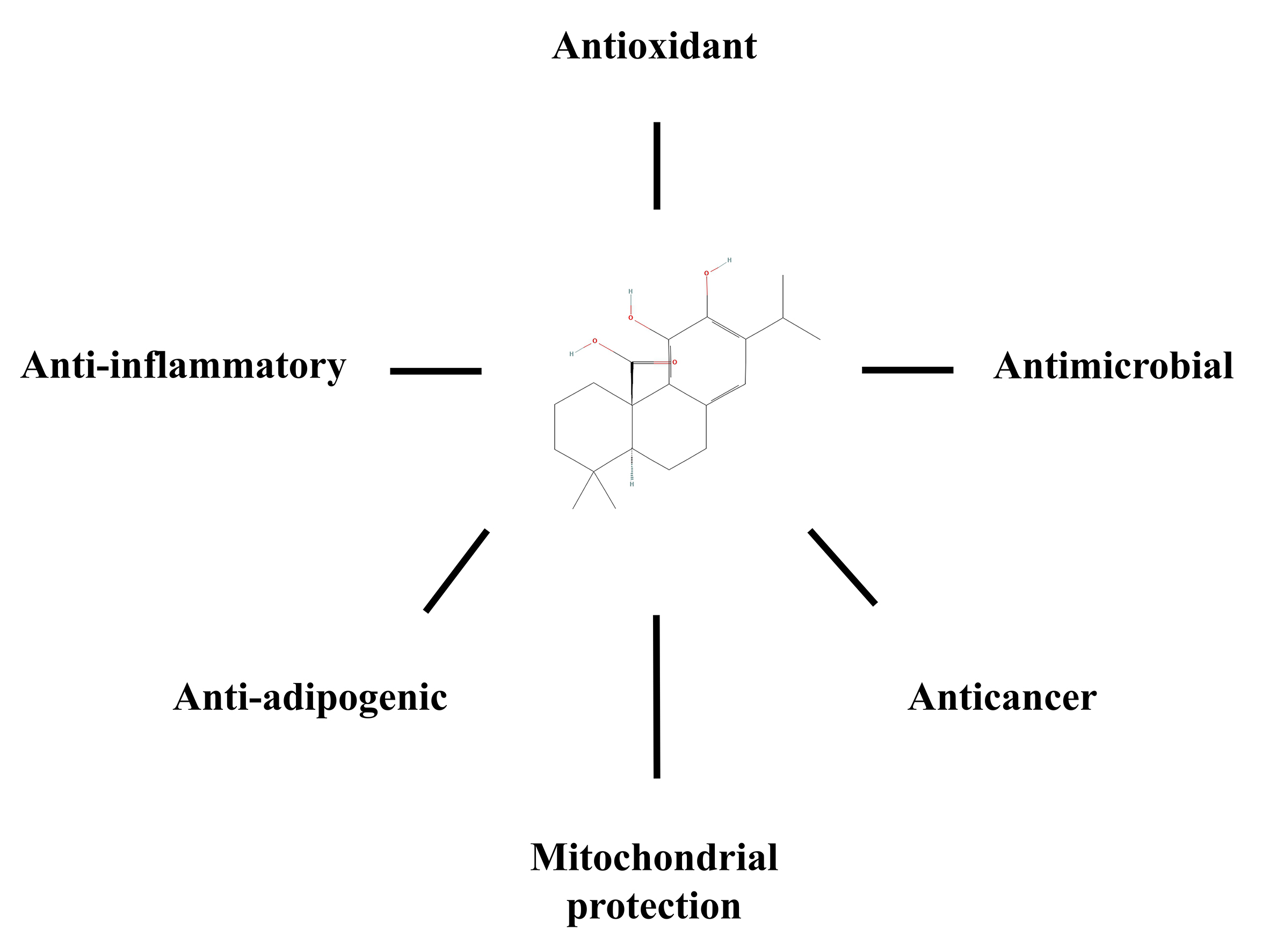 Fig. 3.
Fig. 3.A summary of the biological effects promoted by carnosic acid in mammals. The chemical structure of carnosic acid was obtained from PubChem.
Antimicrobial activities have been described for CA [93, 102, 113, 114]. This property was determined by evaluating the antilisterial effect of a series of rosemary phenolic compounds and it was found that CA exhibited the strongest antilisterial activity [115] when tested for 24 h at 30 °C after inoculation. Moreover, this activity was further enhanced at low pH and high NaCl content. Other Gram-positive bacteria, such as Bacillus, Enterococcus, Streptococcus and Staphylococcus, and Gram-negative bacteria, such as Escherichia, Salmonella and Campylobacter, were also sensitive to CA [113, 116, 117, 118, 119, 120]. The proposed antimicrobial action mechanism provides that, thanks to its lipophilic structure, CA can be inserted into the bacterial membrane [123], where the hydrogen binding donor group could interact with phosphorylated membrane groups [121] or that CA modules the release of ethyl bromide responsible for membrane permeability [122].
Anti-cancer effects of CA have also been reported due to its proapoptotic [124], antiproliferative [125], anti-angiogenic [126], chemoprotective [127], antitumor [137, 138] and antiplatelet properties [128]. Some in vitro studies have highlighted the preventive [139, 140] and inhibitory [102, 135, 141, 142] action against cancer of plant extracts containing CA.
Numerous other investigations have shown the anti-inflammatory properties of
plant extracts rich in CA. These effects include interleukin suppression,
blocking of the proto-oncogene tyrosine-protein kinase Src pathway [129],
regulation of prostaglandin E
Furthermore, CA shows the capacity to promote mitochondrial protection in neural cells, so it could be used as a therapeutic agent for the treatment of neurodegenerative disorders such as AD, PD and HD, in which mitochondrial dysfunction plays a central role in initiation and development [144]. This review summarizes the in vitro and in vivo studies performed so far concerning the effect of CA on mitochondrial function in neurodegenerative diseases.
CA exerts cytoprotection in several in vitro experimental models
involving brain cells [89, 144, 145]. CA induces antioxidant effects mainly by a
mechanism associated with the upregulation of both non-enzymatic and enzymatic
antioxidant defenses in neurons [144] (Fig. 4). Satoh et al. [146]
reported that CA at 10 µM for 6–48 h stimulated the
antioxidant-responsive element (ARE)/nuclear
factor erythroid 2-related factor 2 (Nrf2), upregulating the enzymes heme
oxygenase-1 (HO-1), NAD(P)H quinone oxidoreductase-1 (NQO-1), and also the
catalytic (GCLC) and modifier (GCLM) subunits of
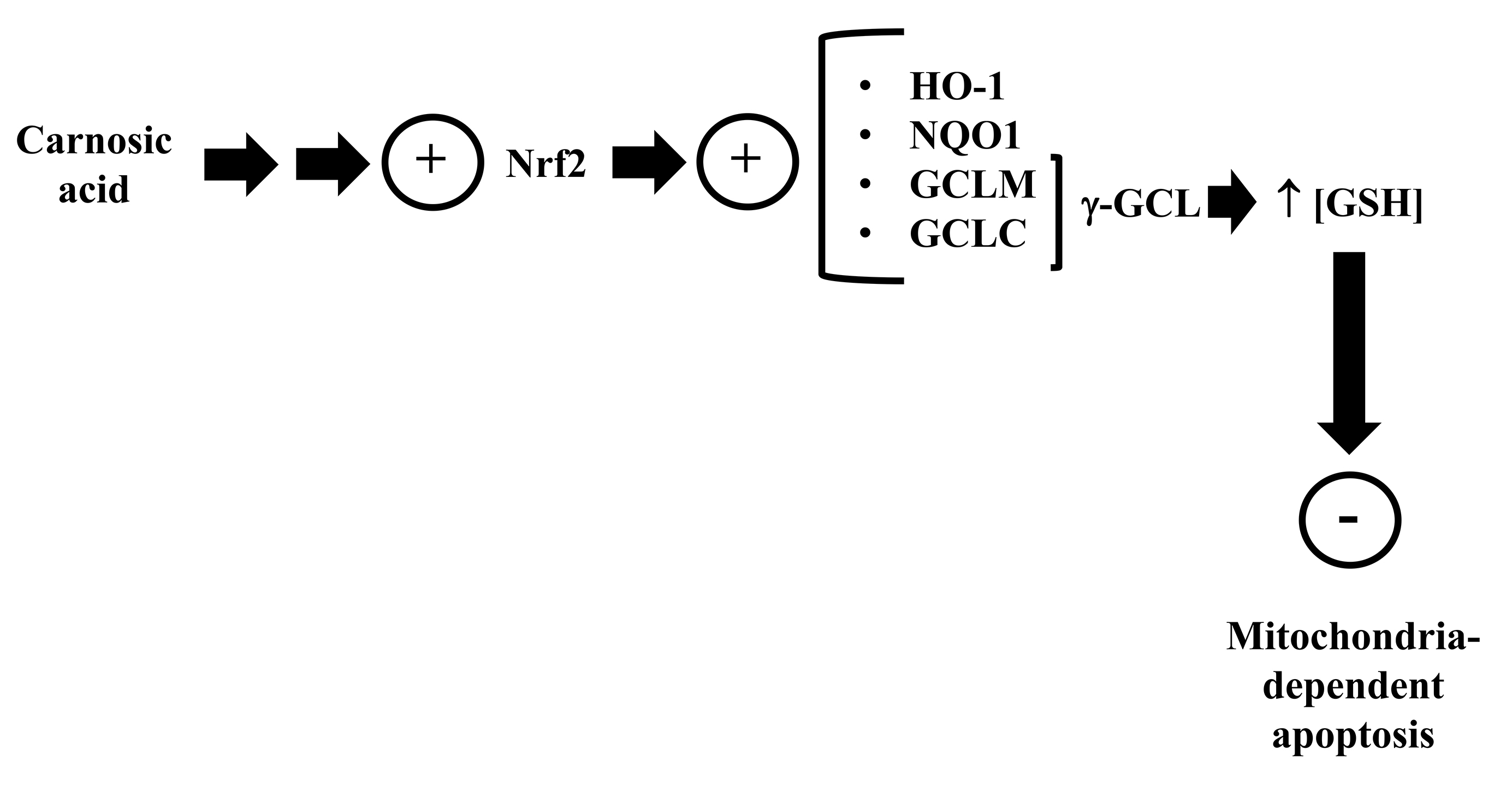 Fig. 4.
Fig. 4.Carnosic acid stimulates the transcription factor Nrf2, leading to an upregulated expression and
synthesis of several cytoprotective enzymes, including those involved with the
metabolism of GSH. Please, read the text for detailed information.
HO-1, heme oxygenase-1; NQO1, NAD(P)H quinone oxidoreductase-1; GCLC,
glutamate-cysteine ligase catalytic subunit; GCLM, glutamate-cysteine ligase
modifier subunit;
Even though the existence of findings indicating that CA would be capable to promote neuroprotective actions, it was not demonstrated whether those effects would be linked to the ability of this diterpene in modulating mitochondrial physiology (which involves bioenergetics, redox biology, dynamics, and biogenesis of the organelles). Furthermore, it was not clearly demonstrated whether Nrf2 (or its downstream targets) would present a role in the mechanism promoted by CA with regard to the mitochondria. Thus, it was investigated whether CA would prevent mitochondrial dysfunction in different in vitro experimental designs related to neurotoxicity. We found that CA at 1 µM for 12 h attenuated the mitochondrial dysfunction induced by the agrochemical paraquat (a mitochondrial toxicant) on the human dopaminergic SH-SY5Y cells [153]. CA blocked the Bax upregulation induced by paraquat, consequently decreasing the release of cytochrome c from the mitochondria to the cytosol. Moreover, CA attenuated the paraquat-induced upregulation in the activity of the pro-apoptotic caspases-9 and -3, resulting in a decrease in the fragmentation of DNA. CA significantly reduced the impact caused by paraquat on the activity of the Complexes I and V, as well as on the mitochondrial membrane potential (MMP) and production of ATP, indicating a role for CA in rescuing mitochondrial function (Fig. 5). In the same work, we demonstrated that CA prevented the paraquat-induced increase in the mitochondrial levels of malondialdehyde (an index of lipid peroxidation) and protein carbonylation. These effects may be linked to the ability of CA in upregulating the levels of GSH and Mn-SOD in the mitochondria of the SH-SY5Y cells (Fig. 6). Nonetheless, it was not examined in that work. It was found that blockade of PI3K/Akt or knocking down of the transcription factor Nrf2 suppressed the mitochondria-related cytoprotection caused by CA in that experimental model, suggesting that the PI3K/Akt/Nrf2 axis would be involved in mediating the effects promoted by CA in the SH-SY5Y cells. Similarly, de Oliveira et al. [154] reported that blockade of the PI3K/Akt/Nrf2 axis attenuated the effects caused by CA on the mitochondria of the SH-SY5Y cells challenged with methylglyoxal, a dicarbonyl commonly associated with diabetes mellitus and neurodegeneration [155] (Fig. 5).
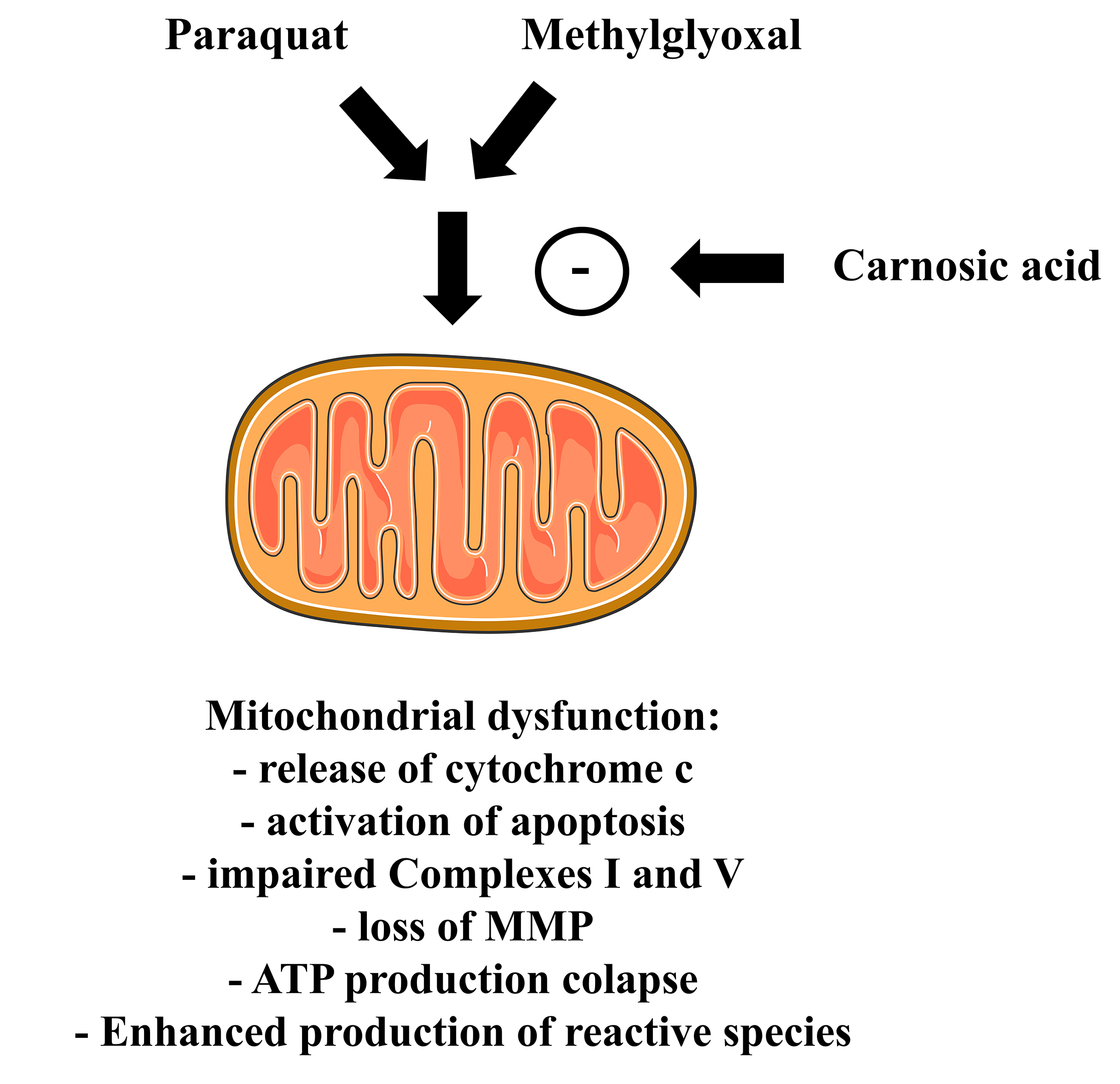 Fig. 5.
Fig. 5.Carnosic acid attenuates the effects caused by either paraquat or methylglyoxal on the mitochondria of cultured cells. Please, read the text for detailed information. The Figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license. MMP, mitochondrial membrane potential.
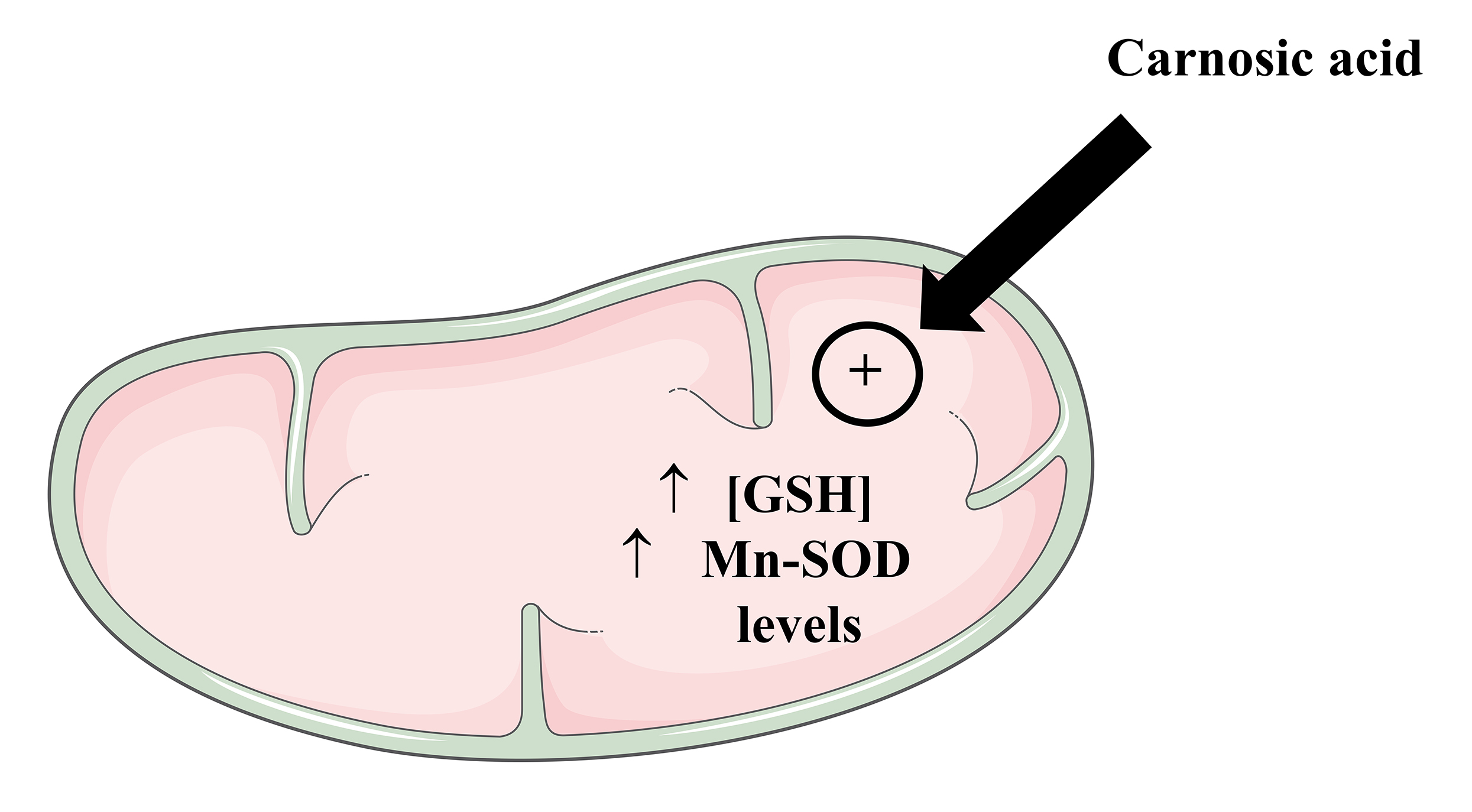 Fig. 6.
Fig. 6.Carnosic acid upregulates the levels of GSH and Mn-SOD in the mitochondria of cultured cells. The Figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license. Mn-SOD, manganese-dependent superoxide dismutase.
CA is a potent activator of Nrf2, leading to the expression of several enzymes associated with cytoprotection in different cell types [146, 149]. The enzyme HO-1, one of the most studied downstream targets of Nrf2, mediates heme degradation generating biliverdin, carbon monoxide (CO), and free iron and promoting cytoprotection, mainly by antioxidant and anti-inflammatory-related mechanisms, in different human cell types [156]. Biliverdin generated by HO-1 is further converted into bilirubin by the enzyme biliverdin reductase (BVR) [157]. Since it was showed that CA upregulated HO-1 in previous works [146, 153], we investigated whether this enzyme would exert a role in mediating the protection of mitochondria in CA-treated SH-SY5Y cells exposed to paraquat [158]. It was found that the inhibition of HO-1 by using zinc protoporphyrin-IX (ZnPP-IX) blocked the preventive effects caused by CA on the mitochondrial function and redox parameters (please, see Table 2 (Ref. [149, 150, 152, 153, 154, 158, 159, 160, 161, 162, 163, 164, 165, 166]) for detailed information). Similarly, inhibition of HO-1 abrogated the effects stimulated by CA in SH-SY5Y cells challenged with glutamate [167]. The authors also found that a treatment with either bilirubin or carbon monoxide-releasing molecule-2 (CORM-2, which generates CO) alleviated the alterations caused by glutamate in the mitochondria, suggesting a link between the products generated by the HO-1/BVR axis in mediating the protection of the organelles (Fig. 7).
| Cell line | Experimental model | Mitochondria-related main effects | Interpretation | References |
| SH-SY5Y cells | CA at 1 µM for 12 h before exposure of the cells to 6-hydroxydopamine (6-OHDA) | - CA upregulated ARE/Nrf2, GCLC), GCLM, and GSH | CA depends on GSH to suppress the mitochondria-related pro-apoptotic effect triggered by 6-OHDA | [149] |
| - CA prevented the 6-OHDA-dependent activation of caspase-3 and the cleavage of PARP | ||||
| - L-buthionine sulfoximine (BSO) at 100 µM blocked the mitochondria-related anti-apoptotic effects induced by CA | ||||
| SH-SY5Y cells | CA at 1 µM for 18 h before the administration of 6-OHDA for 12 or 18 h | - CA upregulated the PI3K/Akt/NF- |
CA depends on the PI3K/Akt/NF- |
[150] |
| - CA increased the immunocontent and activity of the enzyme GSTP | ||||
| - CA blocked the activation of caspase-3 by a mechanism dependent on the PI3K/Akt/GSTP axis | ||||
| SH-SY5Y cells | CA at 1 µM for 8 h prior to the exposure of the cells to 6-OHDA for additional 18 h | - CA prevented the 6-OHDA-induced downregulation of GCLC, GCLM, glutathione reductase (GR), and superoxide dismutase (SOD) | The blockade of the mitochondria-related pro-apoptotic signal is an important part of the anti-apoptotic action induced by CA | [152] |
| - A pretreatment with CA blocked the 6-OHDA-induced alterations on the levels of Bax, Bcl-2, caspase-3, and cleaved PARP | ||||
| - BAM7, an activator of Bax, suppressed the mitochondria-related anti-apoptotic effects elicited by CA | ||||
| SH-SY5Y cells | CA at 1 µM for 12 h prior to the exposure of the cells to paraquat at 100 µM for different periods | - The pretreatment with CA attenuated the paraquat-induced increase in the levels of Bax, the release of cytochrome c to the cytosol and the activation of the caspases-9 and -3 | CA depended on the PI3K/Akt/Nrf2 axis to promote mitochondrial protection in the cells exposed to paraquat (a mitochondrial toxicant) | [153] |
| - CA promoted mitochondrial protection by attenuating the effects of paraquat on the activity of the Complexes I and V and on the levels of ATP and MMP | ||||
| - CA prevented the paraquat-induced oxidation of lipids and proteins in the mitochondria | ||||
| - CA induced an increase in the levels of mitochondrial GSH and Mn-SOD | ||||
| - Inhibition of the PI3K/Akt/Nrf2 signaling pathway suppressed the mitochondrial and cellular protection promoted by CA in the paraquat-treated cells | ||||
| SH-SY5Y cells | CA at 1 µM for 12 h before the administration of methylglyoxal at 500 µM for different periods | - CA upregulated Mn-SOD | CA depended on the PI3K/Akt/Nrf2 axis to prevent the mitochondrial and cellular collapses caused by methylglyoxal | [154] |
| - CA prevented the methylglyoxal-induced decline in the levels of ATP | ||||
| - Inhibition of the PI3K/Akt/Nrf2 signaling pathway abrogated the mitochondria-related protection induced by CA | ||||
| SH-SY5Y cells | CA at 1 µM for 12 h prior to the exposure of the cells to paraquat at 100 µM for different periods | - CA prevented the paraquat-induced loss of MMP and decline in the levels of ATP | CA depended on the Nrf2/HO-1 axis to promote mitochondrial protection in the cells exposed to paraquat | [158] |
| - CA prevented the redox impairment (lipid peroxidation and protein carbonylation and nitration) induced by paraquat on the membranes of mitochondria | ||||
| - The pretreatment with CA attenuated the paraquat-induced decrease in the activity of the Complexes I and V | ||||
| - CA prevented the mitochondria-related pro-apoptotic action triggered by paraquat | ||||
| - Silencing of Nrf2 or inhibition of HO-1 suppressed the mitochondrial and cellular protection induced by CA | ||||
| SH-SY5Y cells | CA at 1 µM for 12 h prior to the exposure of the cells to methylglyoxal at 500 µM for different periods | - BSO suppressed the antioxidant effects elicited by CA in the membranes of mitochondria isolated from the cells exposed to methylglyoxal | CA relies on GSH to prevent mitochondrial collapse caused by methylglyoxal | [159] |
| - CA failed to prevent the mitochondrial dysfunction caused by methylglyoxal in the cells exposed to BSO | ||||
| - BSO abrogated the effect caused by CA on the production of the radical anion superoxide (O |
||||
| SH-SY5Y cells | CA at 1 µM for 8 h before the administration of 6-OHDA at 100 µM for further 18 h | - CA prevented the 6-OHDA-induced inhibition of the chymotrypsin-like proteasome activity | CA stimulated the PINK1/parkin axis leading to a mitochondria-related anti-apoptotic action in the cells exposed to 6-OHDA | [160] |
| - CA upregulated PINK1 and parkin | ||||
| - CA prevented the 6-OHDA-induced downregulation in PINK1 and parkin | ||||
| - Silencing of parkin or administration of MG132 (an inhibitor of the proteasome) suppressed the mitochondria-related anti-apoptotic effects induced by CA in the cells exposed to 6-OHDA | ||||
| SH-SY5Y cells | CA at 1 µM for 18 h before the administration of 6-OHDA at 100 µM for additional 12 h | - CA prevented the 6-OHDA-induced decrease in the levels of VDAC1 | CA induced a mitochondria-related anti-apoptotic action by a mechanism dependent on the PINK1/parkin/mitophagy axis | [161] |
| - CA prevented the 6-OHDA reduction in the levels of ubiquitinated VDAC1 | ||||
| - Silencing of PINK1 suppressed the CA-induced increase in the levels of parkin, VDAC1, and LC3-II | ||||
| - Silencing of PINK1 abrogated the CA-induced decrease in the activation of caspase-3 and cleavage of PARP | ||||
| SH-SY5Y cells | CA at 1 µM was administrated to the cells for 18 h prior to the challenge with 100 µM 6-OHDA for further 9 h | - CA upregulated Mic60 and citrate synthase | CA treatment favored the interaction between Mic60 and PINK1 leading to neuronal survival by a mechanism dependent on mitophagy | [162] |
| - Silencing of Mic60 suppressed the CA-induced blockade on the release of cytochrome c | ||||
| - CA upregulated PINK1 | ||||
| - Silencing of PINK1 abrogated the CA-induced effect on the phosphorylation of Mic60 | ||||
| - CA downregulated PKA | ||||
| - CA promoted mitophagy | ||||
| SH-SY5Y cells | CA at 1 µM for 18 h before the administration of 6-OHDA at 100 µM for further 12 h | - CA upregulated PI3K p100, Beclin1, Atg7, and the conversion of LC3-I into LC3-II | CA depended on the induction of autophagy to block the 6-OHDA-elicited mitochondria-related apoptosis in the cells exposed to 6-OHDA | [163] |
| - CA stimulated the phosphorylation of mTOR | ||||
| - CA upregulated parkin | ||||
| - Silencing of parkin abrogated the effects of CA on PI3K p100, Beclin1, and Atg7, as well as on the conversion of LC3-I into LC3-II | ||||
| - Inhibition of autophagy suppressed the mitochondria-related antiapoptotic effects caused by CA in the cells challenged with 6-OHDA | ||||
| SH-SY5Y cells | CA at 1 µM for 18 h before the administration of 6-OHDA at 100 µM for further 12 h | - CA prevented 6-OHDA-induced upregulation of ARTS and downregulation of XIAP | CA modulated the parkin/ARTS/XIAP signaling to prevent the 6-OHDA-induced apoptosis in the SH-SY5Y cells | [164] |
| - CA attenuated the activation of the caspases-9 and -7 | ||||
| - Silencing of parkin suppressed the CA-induced ARTS downregulation and XIAP upregulation in the 6-OHDA-treated cells | ||||
| - Silencing of XIAP abrogated the Ca-induced attenuation of the activation of the caspases-9, -7, and -3 and the cleavage of PARP | ||||
| SH-SY5Y cells | CA at 1 µM for 24 h before the administration of 6-OHDA at 100 µM for further 12 or 18 h | - CA upregulated parkin | CA depended on the parkin/IKK/NF- |
[165] |
| - CA stimulated the translocation of p65 to the cell nucleus | ||||
| - CA upregulated OPA1 | ||||
| - Silencing of parkin suppressed the effects of CA on p65 and OPA1 | ||||
| - Silencing of OPA1 abrogated the anti-apoptotic effects caused by 6-OHDA in the cells | ||||
| SH-SY5Y cells | CA at 1 µM for 18 h prior to the administration of 6-OHDA for additional 6–12 h | - CA prevented the 6-OHDA-induced downregulation of NRF1 and TFAM | CA depended on the parkin/PARIS/PGC-1 |
[166] |
| - CA upregulated PGC-1 |
||||
| - CA prevented the 6-OHDA-induced PARIS upregulation | ||||
| - Silencing of parkin suppressed the effects induced by CA on PARIS and PGC-1 |
||||
| - Silencing of PGC-1 |
||||
| - Silencing of PGC-1 |
ARE, antioxidant-responsive element; PARP, poly (ADP-ribose) polymerase; PI3K,
phosphatidylinositol 3-kinase; GSTP, glutathione-S-transferase; PINK1, PTEN-induced putative protein kinase 1;
PKA, protein kinase A; XIAP, X-liked inhibitor of apoptosis protein; ARTS,
apoptosis-related protein in the TGF-
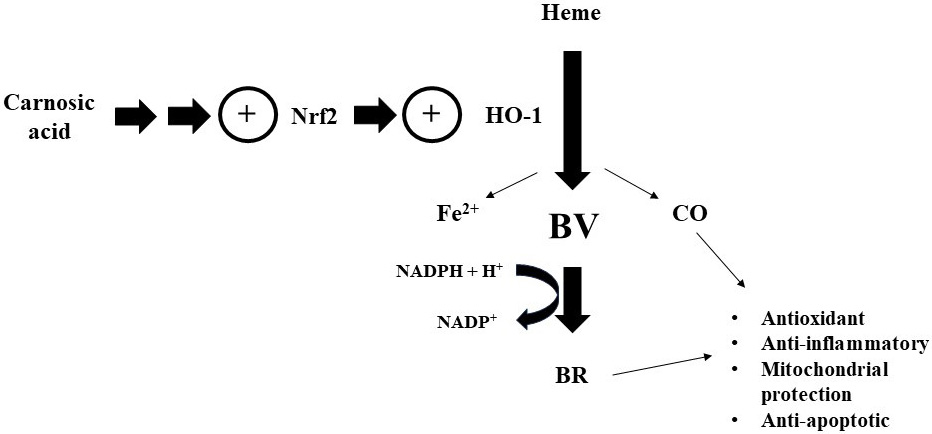 Fig. 7.
Fig. 7.Carnosic acid upregulates the enzyme HO-1 by a mechanism dependent on the transcription factor Nrf2, causing several beneficial effects related to the mitochondria in cultured cells. Please, read the text for detailed information. CO, carbon monoxide; BV, biliverdin; BR, bilirubin; NADPH, reduced nicotinamide-adenine dinucleotide phosphate; NADP, nicotinamide-adenine dinucleotide phosphate (oxidized form).
Activation of Nrf2 by CA also can lead to increased production of GSH, as previously reported by different research groups [146, 148, 149]. Thus, we investigated whether the CA-induced upregulation in the Nrf2/GSH axis would be associated with the benefits caused by CA in the mitochondria of SH-SY5Y cells exposed to methylglyoxal, whose biotransformation depends on the availability of GSH [155]. It was found that GSH production blockade, by using BSO, suppressed the actions promoted by CA on the mitochondria of the methylglyoxal-treated cells [159]. CA failed to prevent the redox impairment in the membranes of mitochondria extracted from the methylglyoxal-treated cells exposed to BSO. BSO also suppressed the CA-stimulated effects on the function of mitochondria (which was assessed through the quantification of the activity of the Complexes I and V, of MMP and of the levels of ATP) in the cells challenged with methylglyoxal. Using a different approach, Tamaki et al. [148] showed that CA activated the metabolism of GSH in the HT22 cells, leading to cytoprotection against glutamate. Similarly, Nishimoto et al. [168] reported that upregulation of GSH attenuated the pro-apoptotic stimuli caused by methylglyoxal in the SH-SY5Y cells. Since GSH also exerts signaling functions in mammalian cells [169], it should be further examined whether other mechanisms, in addition to those related to antioxidant and detoxifying defenses, would be associated with the mitochondria-related benefits promoted by GSH in brain cells (Fig. 8).
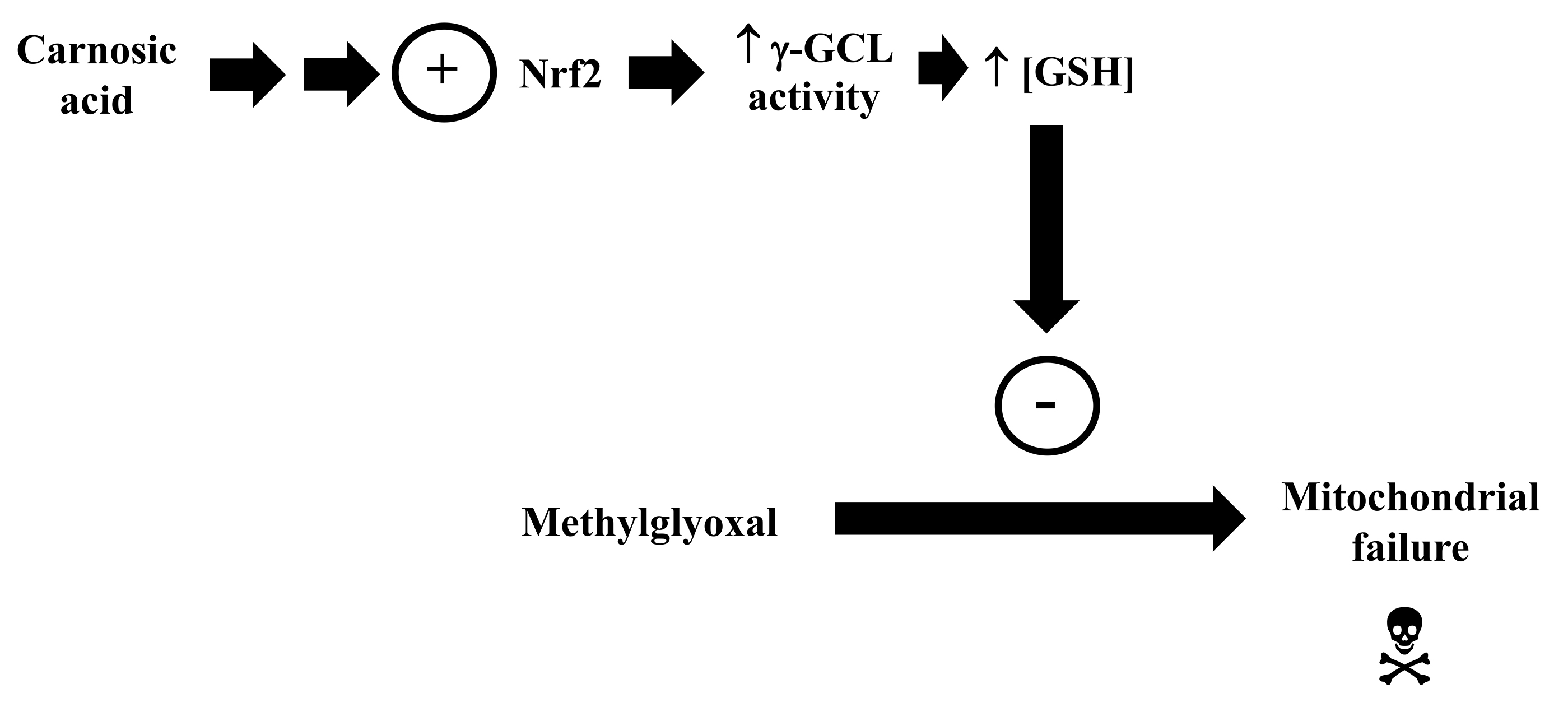 Fig. 8.
Fig. 8.Carnosic acid stimulates the transcription factor Nrf2, causing an increase in the activity of the
enzyme
In addition to promoting antioxidant and detoxifying defenses that lead to mitochondrial protection, CA also affords cytoprotection by modulating other aspects related to mitochondrial physiology. Lin et al. [160] published that CA prevented the pro-apoptotic effects caused by 6-OHDA in SH-SY5Y cells by a mechanism involving upregulation of PINK1 and parkin. The authors also found that silencing of parkin suppressed the mitochondria-related anti-apoptotic effects caused by CA in the 6-OHDA-challegned cells. PINK1, a serine/threonine kinase associated with the IMM, is responsible for recruiting parkin for damaged mitochondria leading to the removal of the dysfunctional organelles from the cells by mitophagy [170]. Proteins linked to ubiquitin are digested by the ubiquitin-proteasome system (UPS), allowing clearance of damaged proteins [171]. Mutations in PINK1 or parkin may lead to parkinsonism due to a failure to maintain mitophagy, among other consequences, such as redox impairment and bioenergetics collapse [172]. Lin and Tsai [161] found that CA stimulated mitophagy by activation of the PINK1/parkin axis, preventing the mitochondria-related apoptosis in the 6-OHDA-treated SH-SY5Y cells (Fig. 9). Silencing of PINK1 abrogated the upregulation of parkin and of voltage-dependent anion channel 1 (VDAC1, which is present in the outer membrane of mitochondria and serve as a target of parkin after mitochondrial depolarization) observed in the cells pretreated with CA and challenged with 6-OHDA. Thus, CA promoted neuroprotection by depending on the stimulation of the PINK1/parkin-dependent mitophagy.
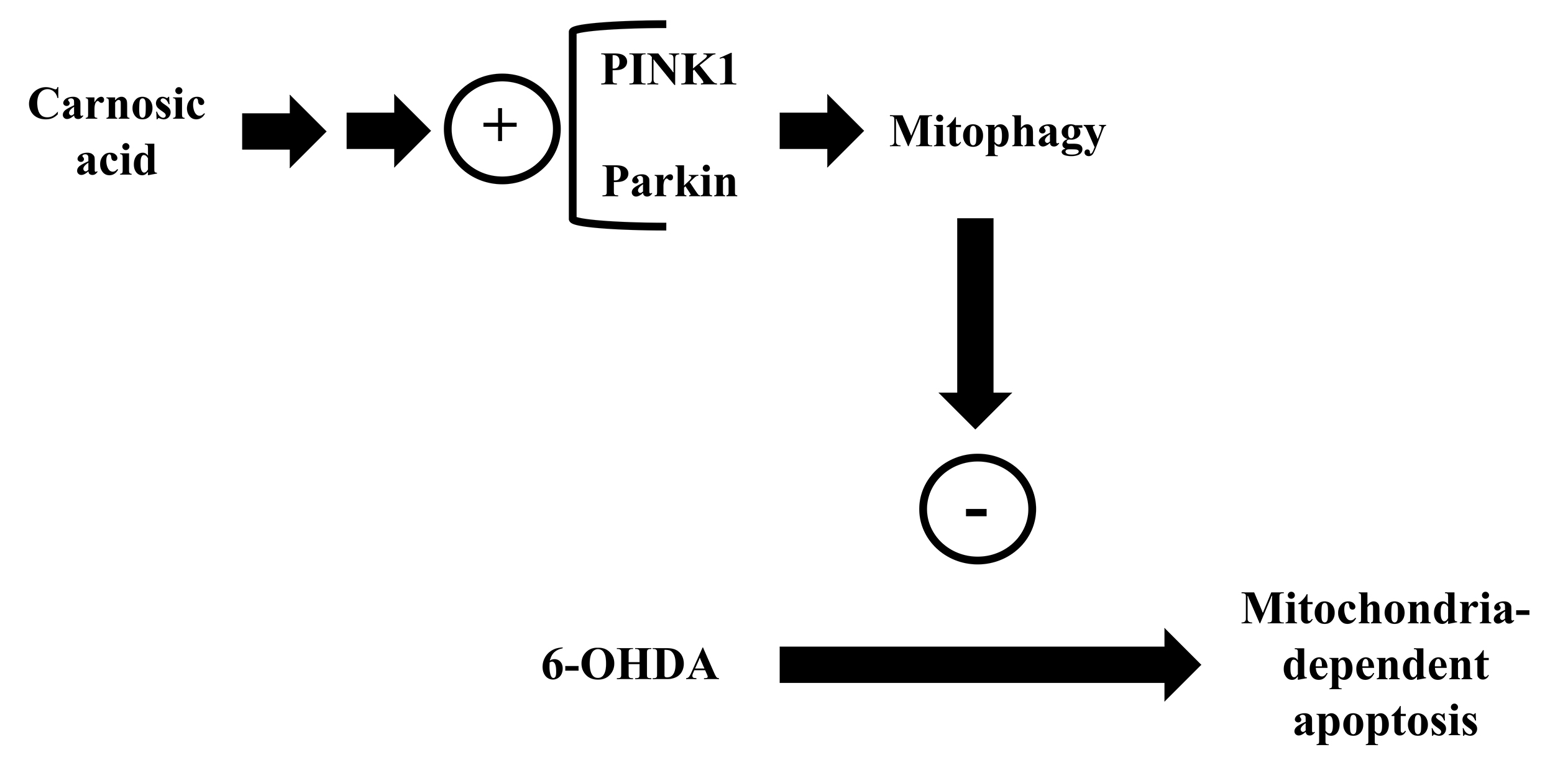 Fig. 9.
Fig. 9.Carnosic acid stimulates the PINK1/ E3 ubiquitin ligase (parkin) axis causing cytoprotection by a mechanism dependent on mitophagy. Please, read the text for detailed information.
Lin et al. [162] reported that CA stimulated PINK1 leading to Mic 60 phosphorylation at a threonine residue in SH-SY5Y cells administrated with 6-OHDA. Mic60 is part of the mitochondrial contact site and cristae junction organizing system (MICOS), which is responsible for the maintenance of mitochondrial architecture regarding cristae plasticity [173]. Moreover, Mic60 also binds to PINK1 and stabilizes this protein on the surface of damaged mitochondria, favoring the binding to parkin and the triggering of mitophagy [174]. Blockade of this interaction leads to mitochondrial impairment and cell death [174]. Furthermore, Mic60 is a target for protein kinase A (PKA), that phosphorylates Mic60 at a serine residue, affecting the interaction of this protein with PINK1 and promoting several negative consequences, including the suppression of the translocation of parkin to the injured mitochondria and the triggering of cytochrome c release to the cytosol [175]. The effects seen by the authors in the cells administrated with CA indicate that this diterpene downregulated PKA, stimulating the Mic60/PINK1-mediated mitophagy and rescuing neurons from the mitochondria-dependent cell death triggered by 6-OHDA (Fig. 10).
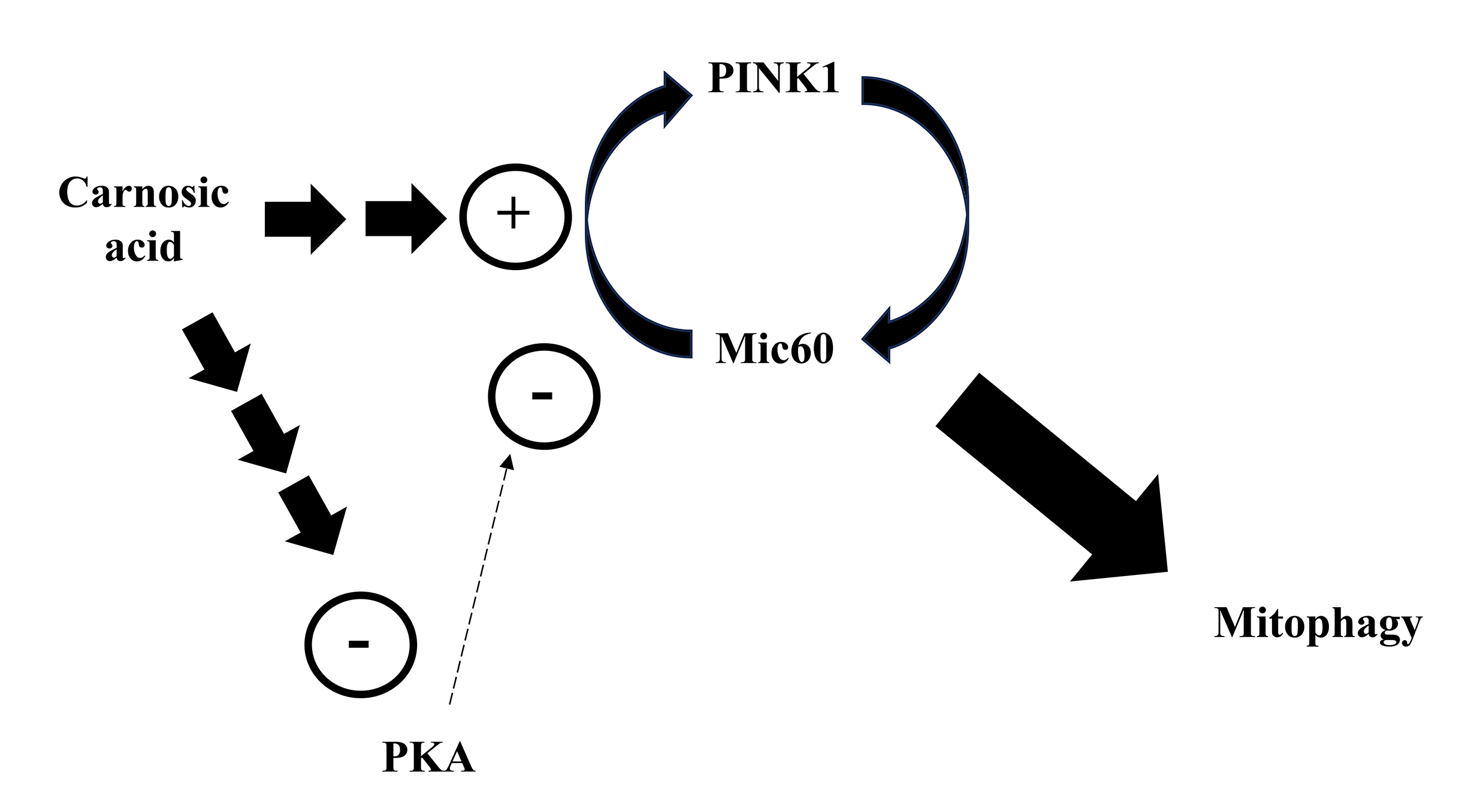 Fig. 10.
Fig. 10.Carnosic acid stimulates the PINK1 and Mic60 interaction by a mechanism associated with the downregulation of PKA, causing mitophagy and cytoprotection in cultured cells. Please, read the text for detailed information.
Parkin participates in the control of autophagy also by interacting with
Beclin1, which is central for the formation of the autophagosome [176]. For
example, silencing of Beclin1 blocked the degradation and consequent removal of
Interestingly, Sun et al. [178] demonstrated that Beclin1 can improve
mitochondria-associated membranes (MAMs) in the heart of mice submitted to an
experimental model of endotoxemia. MAMs may be characterized as regions in which
it is observed a close physical connection involving the outer mitochondrial
membrane and other biomembranas intracellularly [179]. MAMs are important to the
maintenance of communication between the mitochondria and the endoplasmic
reticulum necessary to the transport of Ca
Parkin has been considered as a cytoprotective protein also due to its ability
to control the apoptosis-related protein in the transforming growth factor-
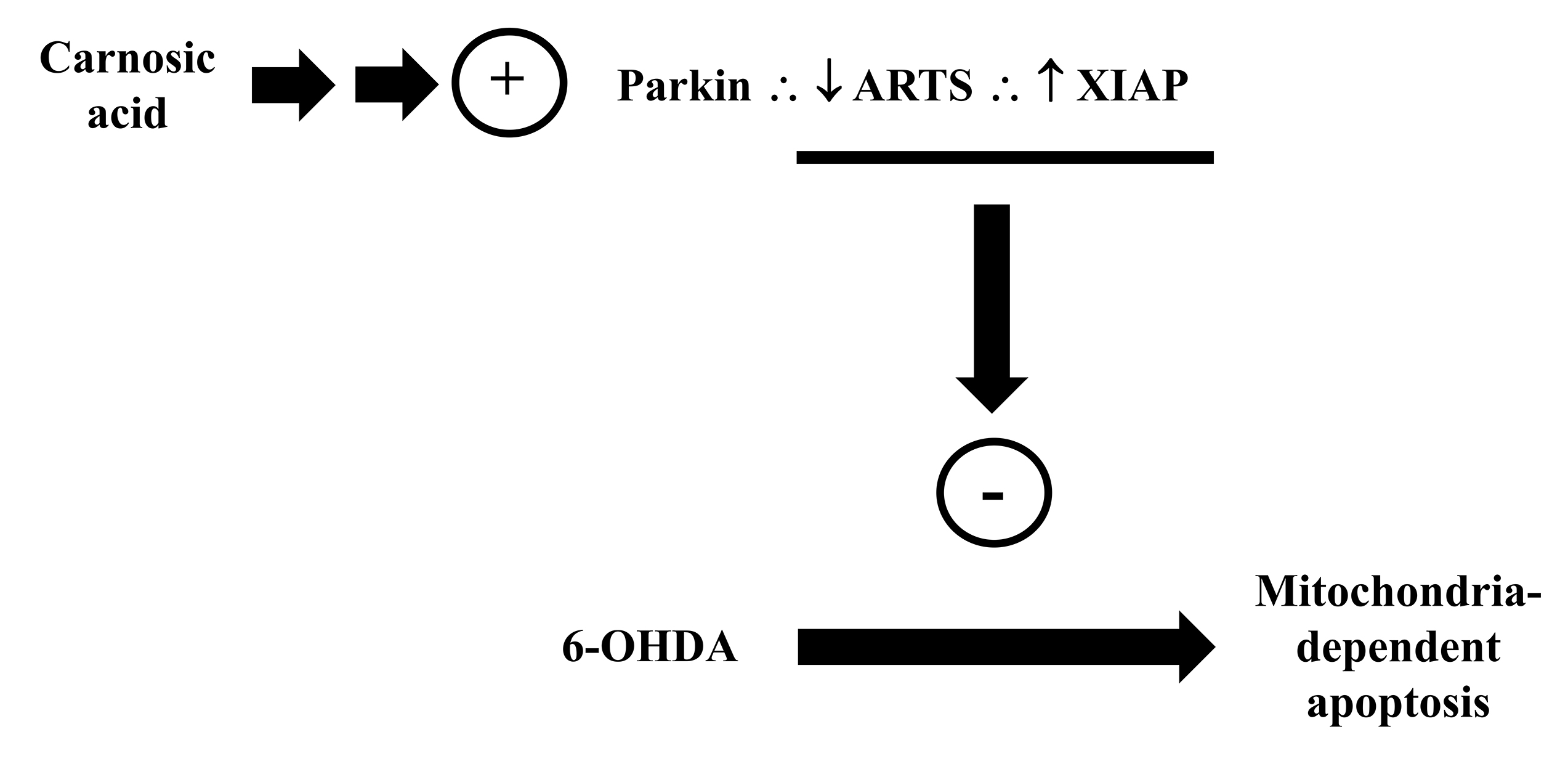 Fig. 11.
Fig. 11.Carnosic acid modulates the parkin/ARTS/ XIAP signaling pathway blocking the mitochondria-dependent apoptosis induced by the toxicant 6-OHDA. Please, read the text for detailed information.
Mitochondrial dynamics, i.e., fusion (which is the generation of one
mitochondrion by the fusion of two mitochondria) and fission (when one
mitochondrion divides generating two mitochondria), alterations have been seen in
several disorders, including neurodegeneration and cardiovascular diseases [184, 185]. The proteins Drp1 and fission 1 (Fis1) mediates fission, whereas the
proteins mitofusin 1 and 2 (MFN1 and MFN2, respectively) and OPA1 modulate
mitochondrial fusion [186]. OPA1, which is located in the IMM, has been reported
as a target of CA through the stimulation of the parkin/I
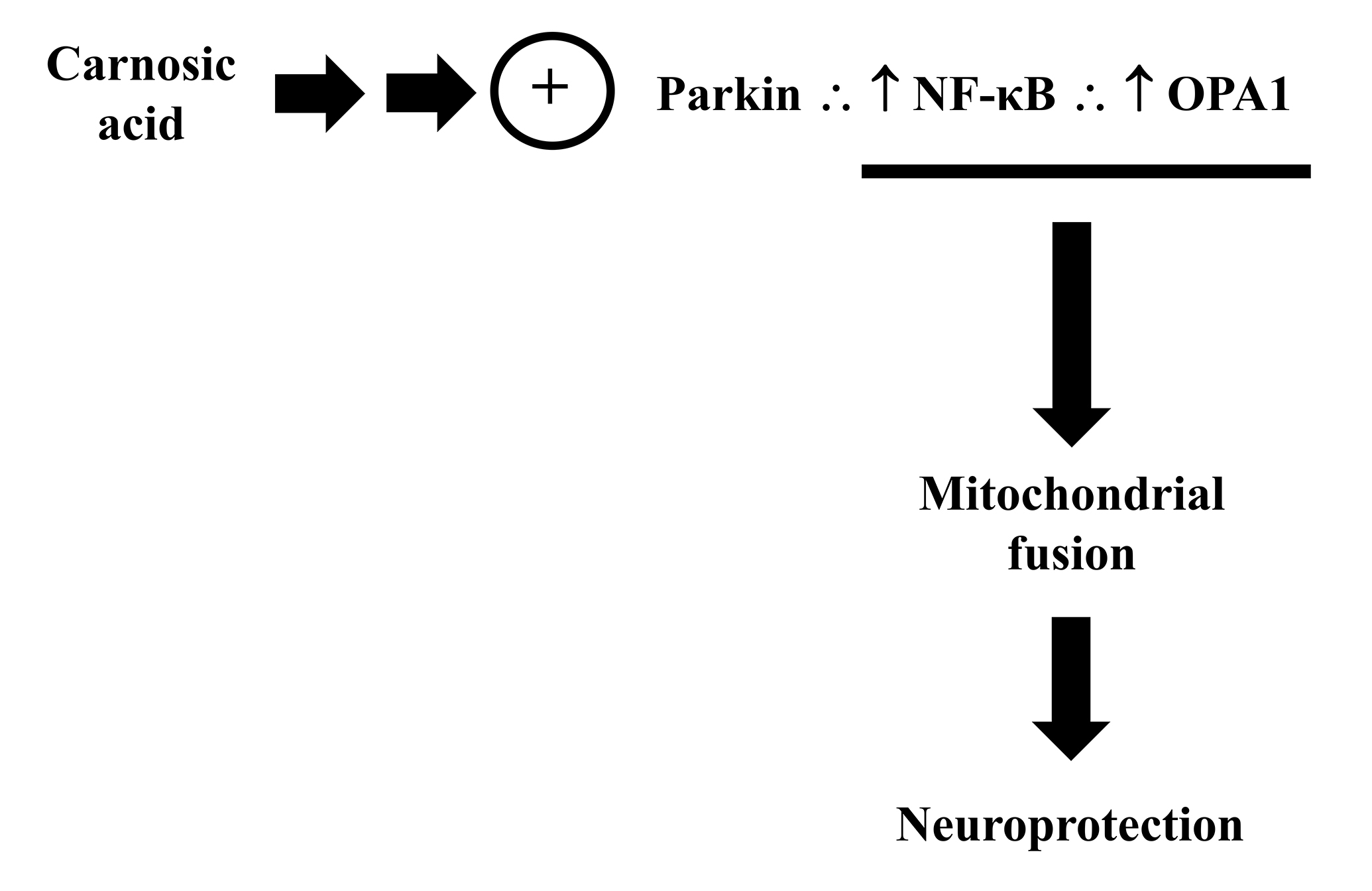 Fig. 12.
Fig. 12.Carnosic acid modulates the parkin/NF-
In addition to the ability to modulate mitochondrial dynamics, CA also promoted
mitochondrial biogenesis in in vitro experimental models. Lin et
al. [166] observed that a pretreatment with CA attenuated the downregulation
induced by 6-OHDA on the proteins NRF1 and TFAM, that modulates mitochondrial
biogenesis [43, 191]. CA also upregulated PGC-1
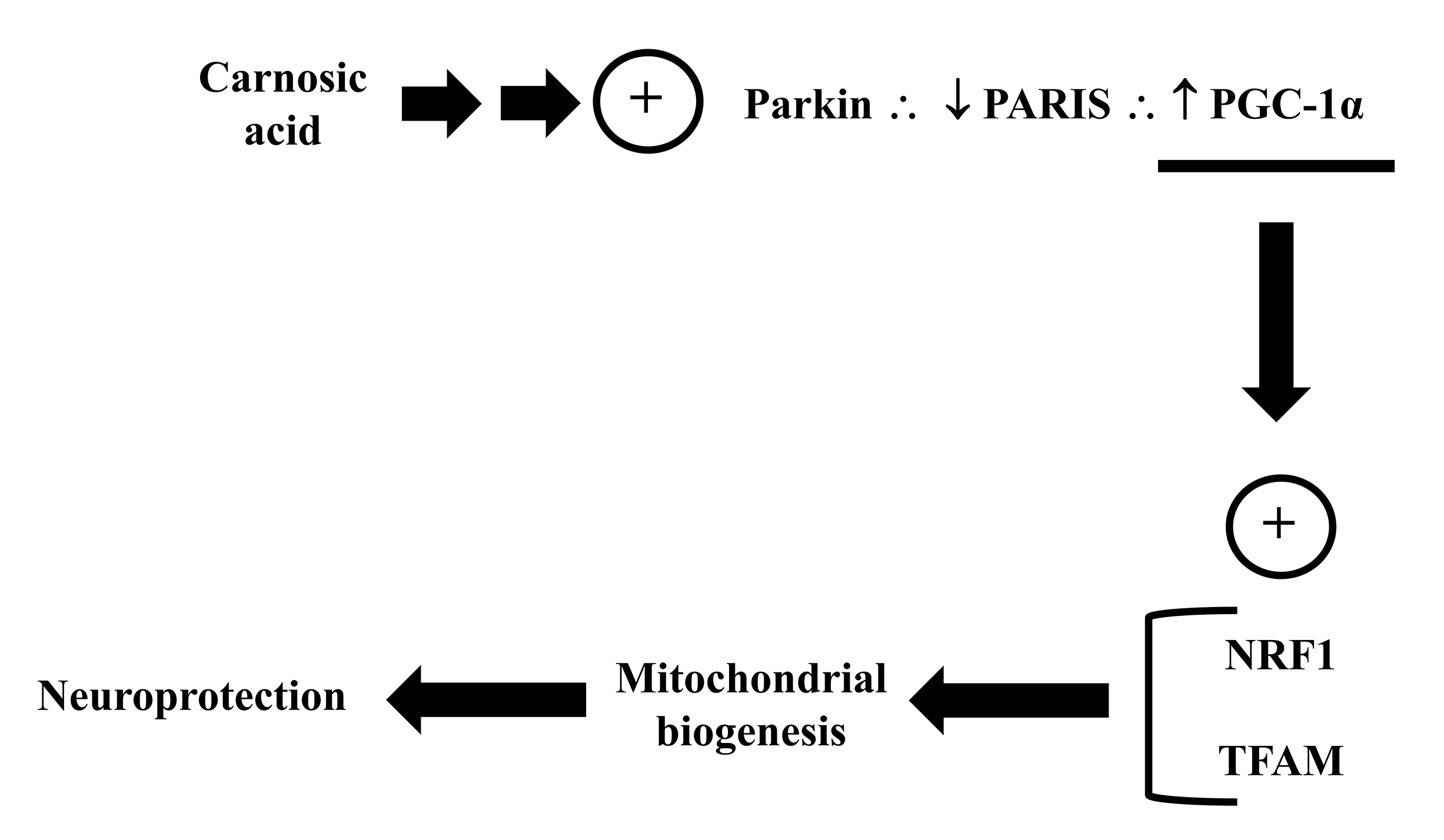 Fig. 13.
Fig. 13.Carnosic acid regulates the parkin/PARIS/PGC-1
The ability of CA in modulating mitochondria-related parameters in mammalian brain was tested in different experimental models. Prior studies have proved that CA affects the function and dynamics of mitochondria and, therefore, is a potential agent to be utilized as a mitochondria-related protectant in brain disorders.
CA exhibits a potent antioxidant action by modulating both non-enzymatic and enzymatic antioxidant defenses in mammalian cells [89, 156]. In a study conducted in aged rats, it was found that supplementation with rosemary extract containing different concentrations of CA improved antioxidant defenses in the rat brain. Rats exposed to rosemary extract showed decreased levels of ROS, lipid peroxidation (LPO), catalase (CAT), and nitric oxide synthase (NOS), indicating a better redox balance in comparison with the control [194]. In another study, the authors reported that an extract of rosemary presenting 60% CA decreased the levels of 4-hydroxynonenal (4-HNE, a LPO indicator) in the cerebral cortex of the senescence accelerated mice (SAMP8) [195]. The authors also demonstrated that rosemary extract combined with 10% CA significantly attenuated the levels of protein carbonylation in the mice hippocampus.
CA presents a catechol loop that can be oxidized to form an electrophilic quinone [146]. This quinone can interact with particular cysteine bonds on target proteins through thiol S-alkylation. The primary response against certain reactive agents, such as ROS and electrophiles, is the Kelch-like ECH-associated protein 1 (Keap1)/Nrf2 pathway [196]. Satoh et al. [146] provided the first description of the role of the Nrf2 signaling in mediating the neuroprotective action of CA against oxidative damage and excitotoxicity in an experimental model of middle cerebral artery occlusion (MCAO). The authors demonstrated that the crucial cysteine thiol of the Keap1 protein was alkylated by the electrophilic quinone-type of CA, which triggered the activation of the transcription element Nrf2, and upregulated several cytoprotective enzymes, as commented before.
Numerous studies showed that CA stimulates Nrf2, thus promoting cytoprotective benefits. It was observed that an in vivo treatment with CA at 1 mg/kg attenuated the mitochondrial impairment caused by the in vitro exposure of rat cerebral cortex to 4-HNE in an ex vivo experimental model [197]. The authors also reported that administering CA in vivo, reduced the levels of 4-HNE associated with mitochondrial proteins probably by a mechanism related to the stimulation of the Nrf2/ARE/HO-1 pathway. It was also shown that a combination of CA and sulforaphane considerably reduced the induced-4-HNE decline in the respiration rates of mitochondria. CA also attenuated mitochondrial dysfunction in an experimental model of controlled cortical impact (CCI) in mice. Post-administration of 3 mg/kg CA preserved the respiratory action of mitochondria and alleviated oxidative damage by lowering the protein nitration levels, LPO, and cytoskeletal disintegration in the CCI mice [198].
The effects of CA therapy in a recent study included the stimulation of the
HO-1, thioredoxin-1, Nrf2, and brain-derived neurotrophic factor (BDNF)
expressions, as well as an increase in the brain serotonin levels in
ovariectomized mice [199]. In the same study, CA attenuated oxidative stress, as
well as caused a decline in the expression of tumor necrosis factor-
Additionally, CA reduced inflammation in mice subjected to the pesticide
chlorpyrifos (CPF) [201]. A pretreatment with CA reduced the activity of IL-6,
TNF-
The most widely used drug for the development of PD model is 6-OHDA, a neurotoxic organic compound that specifically kills dopaminergic and noradrenergic neurons [204]. It was found that CA increased the GSH levels with decrease in lipid peroxidation levels along with enhanced protein expression of GCLC, GCLM, SOD, and glutathione reductase (GR) in rats exposed to 6-OHDA in PD experimental model. Simultaneously, CA deactivated c-Jun NH2-terminal kinase and p38, upregulated the Bcl-2/Bax ratio, downregulated the cleaved caspase 3/caspase 3 and cleaved PARP/PARP ratios, and upregulated tyrosine hydroxylase (TH) protein in the brain of 6-OHDA lesioned rats [152].
The mitochondria-related anti-apoptotic CA-induced effect has also been demonstrated in an animal model of neurotoxicity induced by acrylamide. Treatment with 40 mg/kg CA for 11 days enhanced the GSH content and lowered the levels of MDA, the Bax/Bcl-2 ratio, and the activation of caspase-3 in the brain of the rats exposed to acrylamide [205]. Another study showed that CA, in combination with Trolox and human chorionic gonadotropin (HCG), reduced the activity of caspase-3 and Bax in the hippocampus of rats administered to an experimental model of ischemia-reperfusion induced by occlusion of the carotid artery [206]. It was also reported that CA upregulated the levels of SIRT1, Mn-SOD, and Bcl-2, while downregulated the expression of p66shc and Bax and the activation of caspase-3 in an experimental model of subarachnoid haemorrhage (SAH) in rats [207].
Overall, it has been demonstrated both CA can modulate mitochondrial physiology by both indirect and direct mechanisms in the brain cells also in vivo (Fig. 14; Table 3, Ref. [146, 152, 160, 194, 195, 197, 198, 199, 201, 203, 205, 206, 207]). Further studies are necessary to confirm this ability in other contexts involving mitochondrial disturbances.
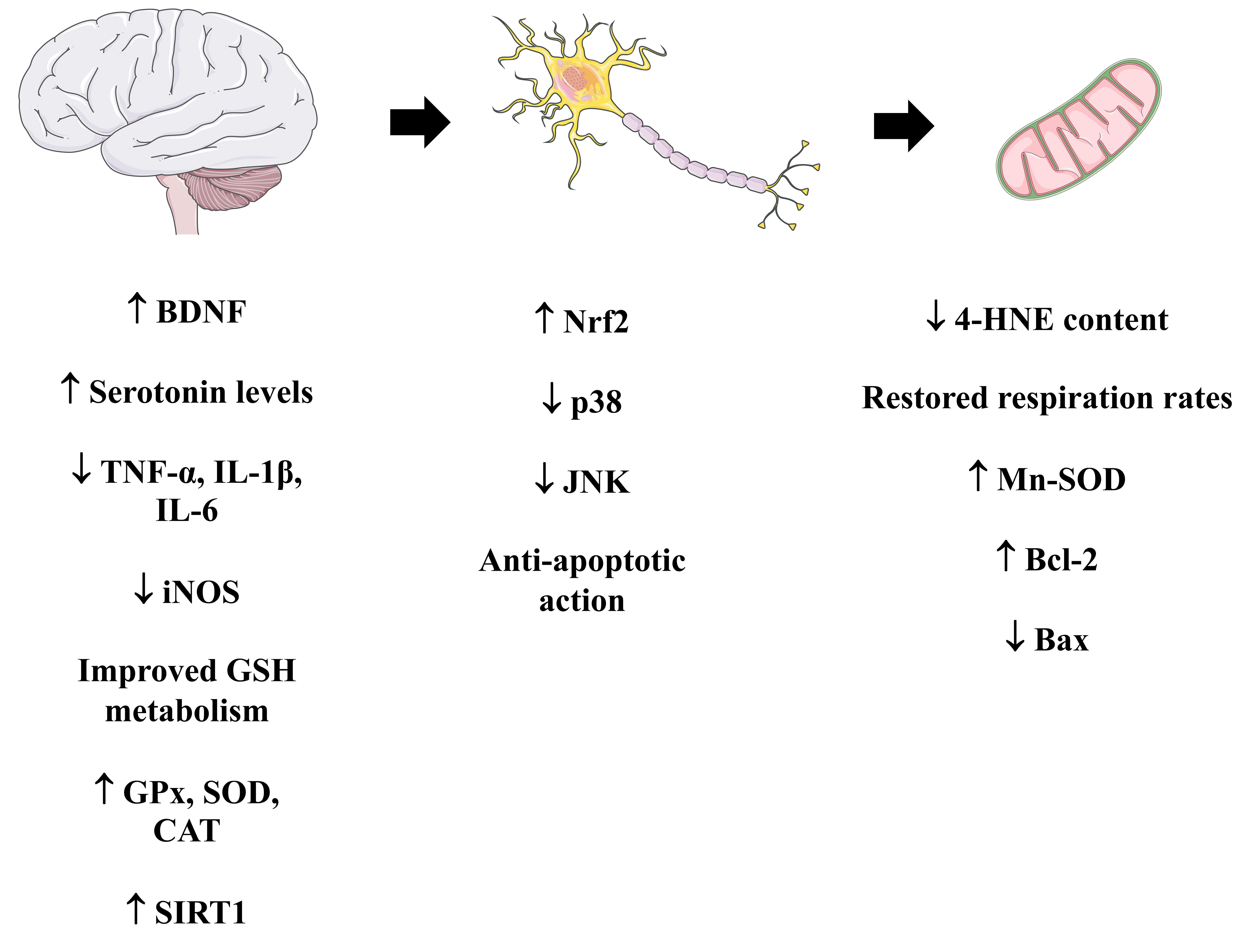 Fig. 14.
Fig. 14.A summary of the mitochondria-related effects induced by
carnosic acid in vivo. Please, read the text for detailed information.
The Figure was partly generated using Servier Medical Art, provided by Servier,
licensed under a Creative Commons Attribution 3.0 unported license. BDNF,
brain-derived neurotrophic factor; iNOS, inducible nitric oxide synthase; GPx,
glutathione peroxidase; SOD, superoxide dismutase; CAT, catalase; SIRT1,
NAD
| Experimental Model | Dose and Duration | Effects | References |
| C57BL/6 mice | 1 and 3 h CA at 3 mg in 0.3 mL olive oil prior to MCAO | [146] | |
| Wistar rats | 20 mg/kg CA treatment for 3 weeks prior to 6-OHDA exposure | [152] | |
| Wistar rats | CA at 20 mg/kg 3 times/3 weeks prior to the administration of 6-OHDA on day 22 | [160] | |
| Wistar rats | Rosemary extract 0.2%, 0.02% for 12 weeks | [194] | |
| SAMP8 mice | 60% and 10% CA with 32, 16, 1.6 mg/kg for 16 weeks | [195] | |
| CF-1 mice | 1.0 mg/kg CA given intraperitoneally 48 h prior to the exposure of 4-HNE | [197] | |
| CF-1 mice | CA at 0.3, 1.0, and 3.0 mg/kg I.P. 15 min post TBI | [198] | |
| Balb/c mice | 20 mg/kg/day CA for 3 weeks in bilateral ovariectomy | [199] | |
| Swiss albino mice | 30, 60 mg/kg/day once/day for 14 days before exposure to chlorpyrifos for last 7 days | [201] | |
| C57BL/6 mice | CA at 1.0 mg/kg given intraperitoneally 30 min after injury | [203] | |
| Wistar rats | CA at 5, 10, 20, and 40 mg/kg injected 30 min prior exposure to acrylamide for 11 days | [205] | |
| Syrian mice | CA at 10 mg/kg was injected immediately after ischemia-reperfusion and then CA at 3 mg/kg for 10 days | [206] | |
| Sprague Dawley rats | 3 mg/kg CA given 24 h prior to SAH | [207] |
MCAO, middle cerebral artery occlusion; LPO, lipid peroxidation; GR, glutathione reductase; TH, tyrosine hydroxylase; ROS, reactive oxygen species; NOS, nitric oxide synthase; MDA, malondialdehyde; AChE, acetylcholinesterase; TBI, traumatic brain injury; SAH, subarachnoid haemorrhage; OS, oxidative stress; PCC, protein carbonylation; 3-NT, 3-nitrotyrosine; GFAP, glial fibrillary acidic protein; Iba1, ionized calcium-binding adapter molecule 1; CAT, catalase; 4-HNE, 4-hydroxynonenal; BDNF, brain-derived neurotrophic factor; iNOS, inducible nitric oxide synthase; GPx, glutathione peroxidase; SIRT1, NAD+-dependent deacetylase sirtuin 1; i.p., intraperitoneal.
The constellation of effects induced by CA on the mitochondria clearly demonstrates that this diterpene is a potential candidate to be utilized as a drug in mitochondrial therapy. Even though some of the findings were confirmed in in vivo experimental models, further investigations are necessary to better understand CA bioavailability and to examine whether CA would exert toxic effects in brain cells (as well as in other organs), for example. Moreover, more research needs to be done on this diterpene’s potential to alter some signaling pathways connected to mitochondria in vivo. Overall, in various brain cells, CA has been shown to be a powerful agent that protects the mitochondria.
4-HNE, 4-hydroxynonenal; 6-OHDA, 6-hydroxydopamine; AChE, acetylcholinesterase;
ACLY, ATP-citrate lyase; AD, Alzheimer’s disease; AIF, apoptosis-inducing factor;
ALS, amyotrophic lateral sclerosis; AMP, adenosine monophosphate; AMPK,
AMP-activated protein kinase; Apaf-1, apoptotic protease activating factor; ARE,
antioxidant-responsive element; ARTS, apoptosis-related protein in the
TGF-
Conceptualization: MRdeO, VI, IP, AS, ST, GS; writing—original draft preparation: VI, IP, AS, ST, GS, MRdeO; writing—review and editing: VI, IP, AS, ST, GS, MRdeO. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
MRdeO receives a “Bolsa de Produtividade em Pesquisa (Research Productivity Grant) 2—PQ2” fellow from the CNPq (protocol number 301273/2018-9).
The authors declare no conflict of interest. Marcos Roberto de Oliveira is serving as one of the Guest editors of this journal. We declare that Marcos Roberto de Oliveira had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Gernot Riedel.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.