







1 Department of Pharmacology, JSS College of Pharmacy, JSS Academy of Higher Education & Research (JSS AHER), 570015 Mysuru, India
2 Centre for Experimental Pharmacology and Toxicology, Central Animal Facility, JSS Academy of Higher Education & Research, 570015 Mysuru, India
3 Section of Molecular Pharmacology and Toxicology, National Institute on Alcohol Abuse and Alcoholism, Bethesda, MD 20892, USA
4 Department of Molecular Nutrition, Council of Scientific and Industrial Research (CSIR)—Central Food Technological Research Institute, 570020 Mysuru, India
5 Council of Scientific and Industrial Research (CSIR), Academy of Scientific and Innovative Research (AcSIR), 201002 Ghaziabad, India
6 Brain, Behavior and Cognitive Neurosciences Research (BBCR), JSS Academy of Higher Education & Research, 570015 Mysuru, India
Abstract
Background: Mitochondrial dysfunction is one of the major hallmarks of
Parkinson’s disease (PD). Recently, angiotensin II type 1 and type 2 receptors
(AT1R, AT2R) were reported to be present on the mitochondrial membrane. Both are
crucial players in the brain renin-angiotensin system (RAS). Current evidence
indicates that blockade of brain AT1R protects dopaminergic neurons in PD.
Methods: Thus, the current study was aimed to explore the effects of
Telmisartan (Tel), a selective AT1R blocker, on mitochondrial function and a
mouse model by exposure to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)
[250 mg/kg body weight (10 divided i.p. injections, each 25 mg/kg body weight at
3.5 days interval) + Probenecid 250 mg/kg]. Gait function was assessed by beam
walk, and mice were euthanized on the 35th day and their brain tissues isolated
for Western blot analysis. Results: Pretreatment with Tel significantly
protected motor functions during the beam walk in MPTP-treated mice. Tel
attenuated the increased levels of AT1R,
Keywords
- Parkinson's disease
- renin-angiotensin system
- mitochondria
- telmisartan
- MPTP
- Adenosine triphosphate (ATP)
Parkinson’s disease (PD) is the second most common neurodegenerative disease and
is characterized by aggregation of alpha-synuclein (
The major causes of PD pathogenesis are mitochondrial dysfunction,
neuroinflammation, and
The brain renin-angiotensin system (RAS) plays a crucial role in the
pathogenesis of Parkinsonism. Angiotensin II (AII), the most important effector
peptide in RAS, exerts its effect via the AII type 1 receptor (AT1R) and the AII
type 2 receptor (AT2R) [14, 15]. In physiological conditions, activation of AT2R
counteracts the function of AT1R [16]. In PD, the upregulation of AT1R expression
contributes to neuroinflammation and apoptosis, leading to dopaminergic cell
death. In our earlier study using a mouse model of PD, we reported that blockade
of AT1R with a selective AT1R antagonist, Telmisartan (Tel), resulted in
neuroprotection [15]. An interesting report by Valenzuela et al. [17]
(2016) revealed the presence of AT1R and AT2R on the mitochondrial membrane of
dopaminergic neurons, thus demonstrating a link between RAS and mitochondrial
function. There is now accumulating evidence that PD is associated with
alterations in mitochondrial dynamics and in the biogenesis of proteins such as
mitofusin protein 1 (MFN1) and Peroxisome proliferator-activated receptor-gamma coactivator-
Telmisartan (Product No. T2861) and Probenecid (Product No. P1975) were
purchased from Tokyo Chemical Industry (TCI) Private Limited (Tamil Nadu, India).
MPTP hydrochloride (Cat. No.: HY-15608) was obtained from MedChemExpress (Middlesex County, NJ,
USA). The specific primary antibodies used in this study were:
anti-
Young male C57BL/6J mice (18–22 g body weight) were procured from Adita
Bioscience (Tumkur, Karnataka, India). The animals were housed in polypropylene
cages in the good laboratory practice standard central animal facility at JSS Academy of Higher Education And Research (JSS
AHER, Mysuru, India). Animals were acclimatized for 7 days in an experimental
room at a controlled temperature of 22
Following acclimatization, mice were trained for the beam walk test (Fig. 1).
The animals were divided into 5 groups (n = 8 per group). Group A (vehicle) was
administered with 0.5% carboxymethylcellulose (CMC) by oral gavage + saline i.p.
Group B was given 0.5% CMC (vehicle) by oral gavage + MPTP [intraperitoneally at
250 mg/kg b.wt. (in 10 divided injections, each 25 mg/kg b.wt. at 3.5 days
interval) + Probenecid 250 mg/kg i.p.)] MPTP; positive control, and Groups C and
D were given Tel at 3 and 10 mg/kg, by oral gavage, respectively, up to 35 days +
MPTP (Vehicle or Tel was administered 1 hr before the first injection of MPTP and
thereafter daily once for 35 days). Group E (probenecid control) received 250
mg/kg i.p. In the study Probenecid was used here to reduce renal excretion of
MPTP and its metabolites, because its usage is recommended for experimental
protocols of chronic PD [20]. A separate group for Tel alone was not included,
since our earlier study showed that 10 mg/kg Tel did not cause any toxicity [15].
Mice were evaluated for motor functions using a beam walk every 10th day. On the
35th day, mice were euthanized using excess CO
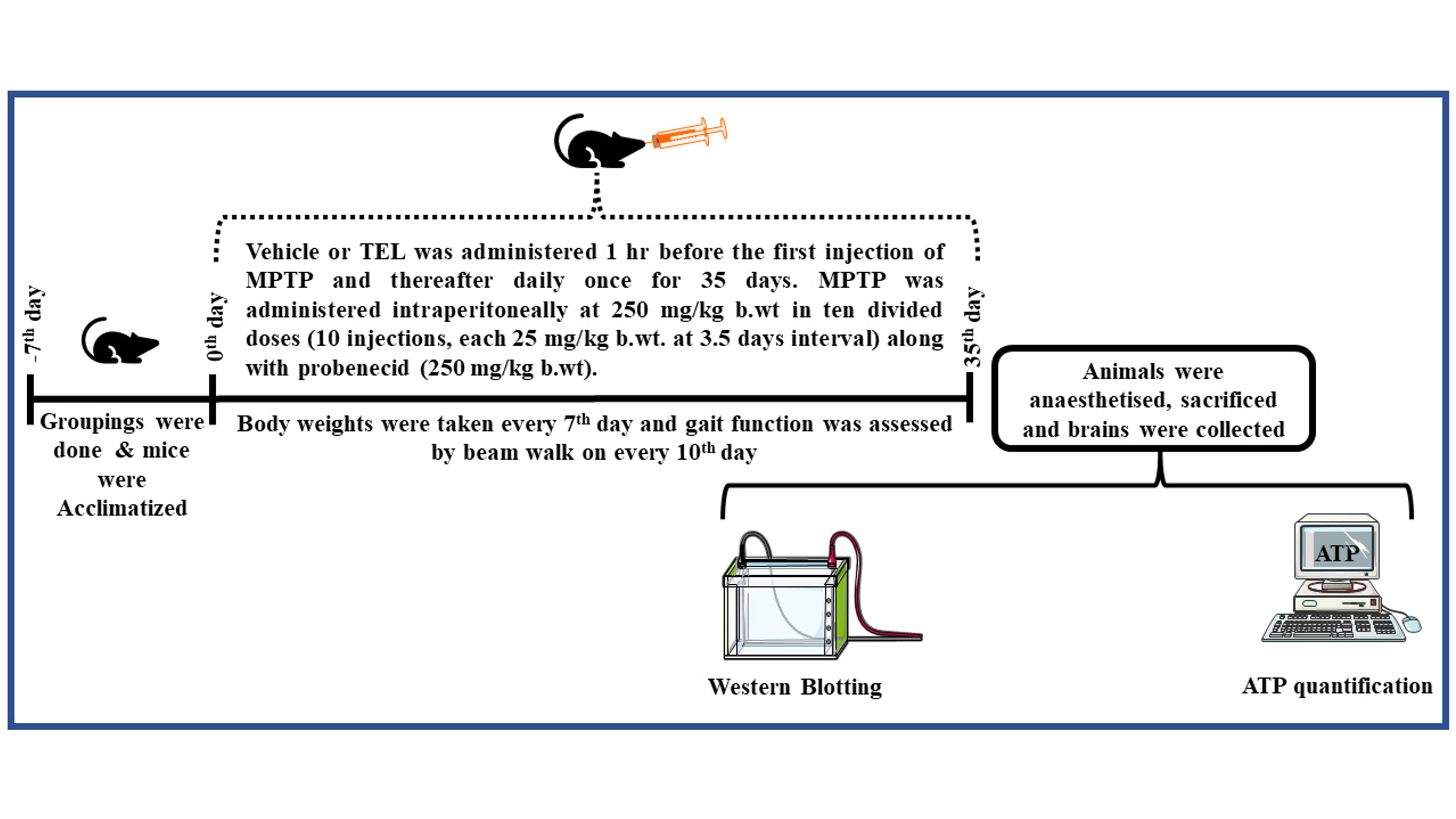 Fig. 1.
Fig. 1.Experimental design. TEL, Telmisartan; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; ATP, Adenosine triphosphate.
Prior to MPTP administration, mice were pre-trained for the beam walk test by allowing them to traverse the narrow 100 cm length runway with a dark escape box at the other end [22]. An aversive stimulus was created by using a bright light (100 lux) placed above the beam to motivate the walk. Mice were allowed a maximum of 60 seconds for the travel. The observer for the experiment was blinded to the identity of the treatment groups.
The SNPc region from either side of the mouse brain was isolated and used for
Western immunoblot analyses. The isolated brain tissues were homogenized using
Radioimmunoprecipitation Assay (RIPA) lysis buffer (Cat #786-490, G-Biosciences, St. Louis, MO, USA) and a
protease and phosphatase inhibitor cocktail (MP Biomedicals, Santa Ana, CA, USA), incubated for 30 min on ice, and
then centrifuged (15,000 g at 4 °C for 10 min) to obtain individual
tissue lysates. Tissue lysate supernatants were collected and used for measuring
the protein concentration with a BCA Protein Assay Kit (Cat #23225,
ThermoFisher, Rockford, IL, USA). Aliquots of 40 µg protein were mixed with sodium
dodecyl sulfate (SDS)-sample loading buffer containing bromophenol blue and
The Adenosine triphosphate (ATP) content of SNPc tissues was measured using an Enzyme-linked Immunosorbent Assay (ELISA) kit as recommended by the manufacturer (Catalogue number: A22066, Invitrogen™, Waltham, MA, USA) and a luminometer (TECAN/SPARK 10M, Männedorf, Switzerland).
Experimental data (mean differences) were analyzed using one-way analysis of
variance (ANOVA) followed by Tukey’s multiple comparison test, with a
p-value
Chronic MPTP + probenecid administration caused motor dysfunction and decreased the body weight of mice in the MPTP control group. However, treatment with Tel protected the normal body weight of MPTP mice (Body weight data is given in Supplementary Materials).
Mice in the MPTP group took significantly longer to reach the goal (dark escape
box at the end of the runway) on all of the locomotor test days [11th day
(p
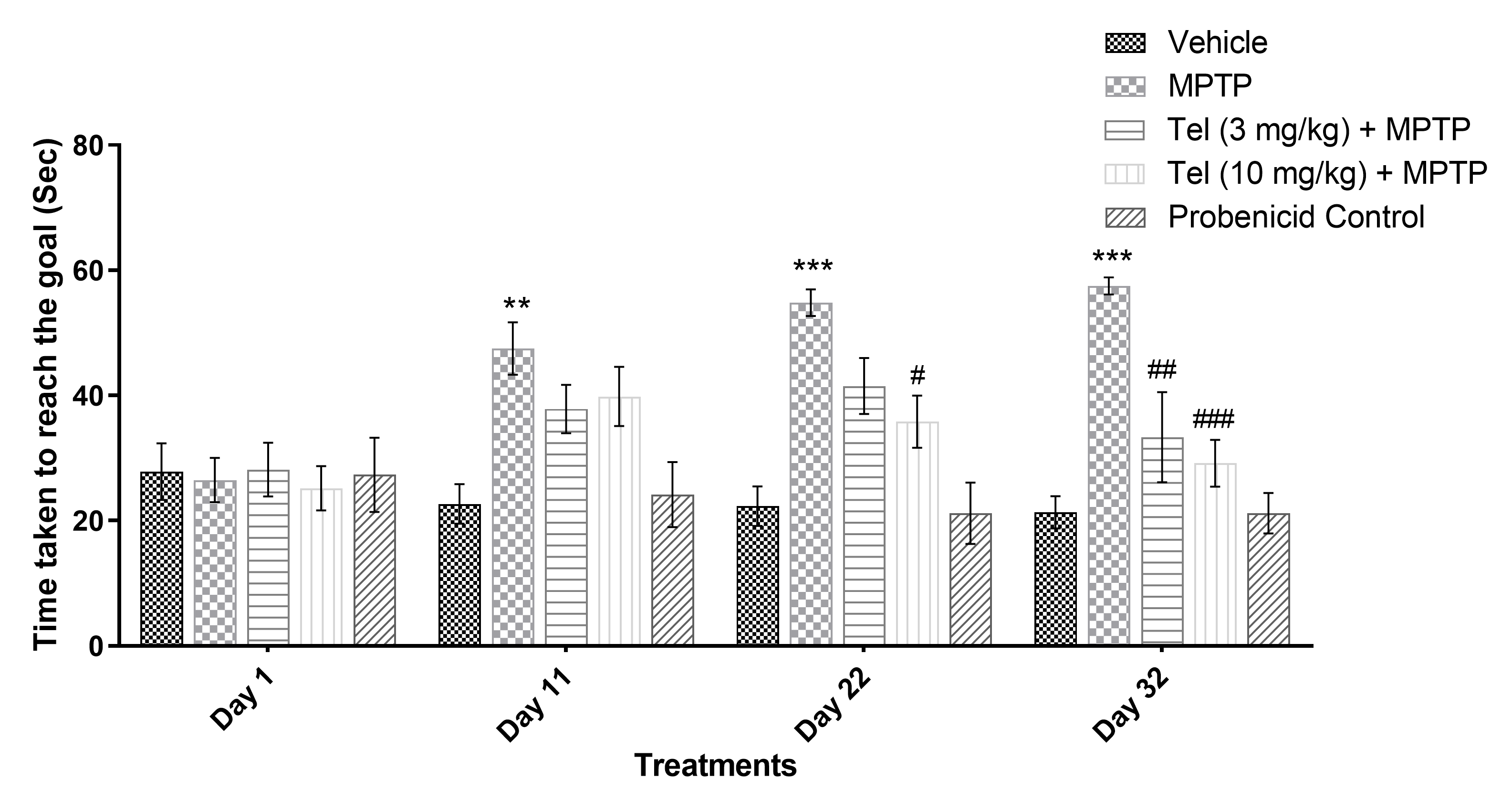 Fig. 2.
Fig. 2.Effect of Tel on the time taken to cross the runway
(sec). ** and *** indicate p
The ATP content in SNPc regions of the mouse brains was determined with an ELISA kit. ATP concentration was significantly lower in the MPTP group (group B) compared to the vehicle (group A) and Probenecid (group E) control groups. However, mice treated with Tel (3 and 10 mg/kg) showed a significantly higher ATP content compared to MPTP-treated mice (Fig. 3).
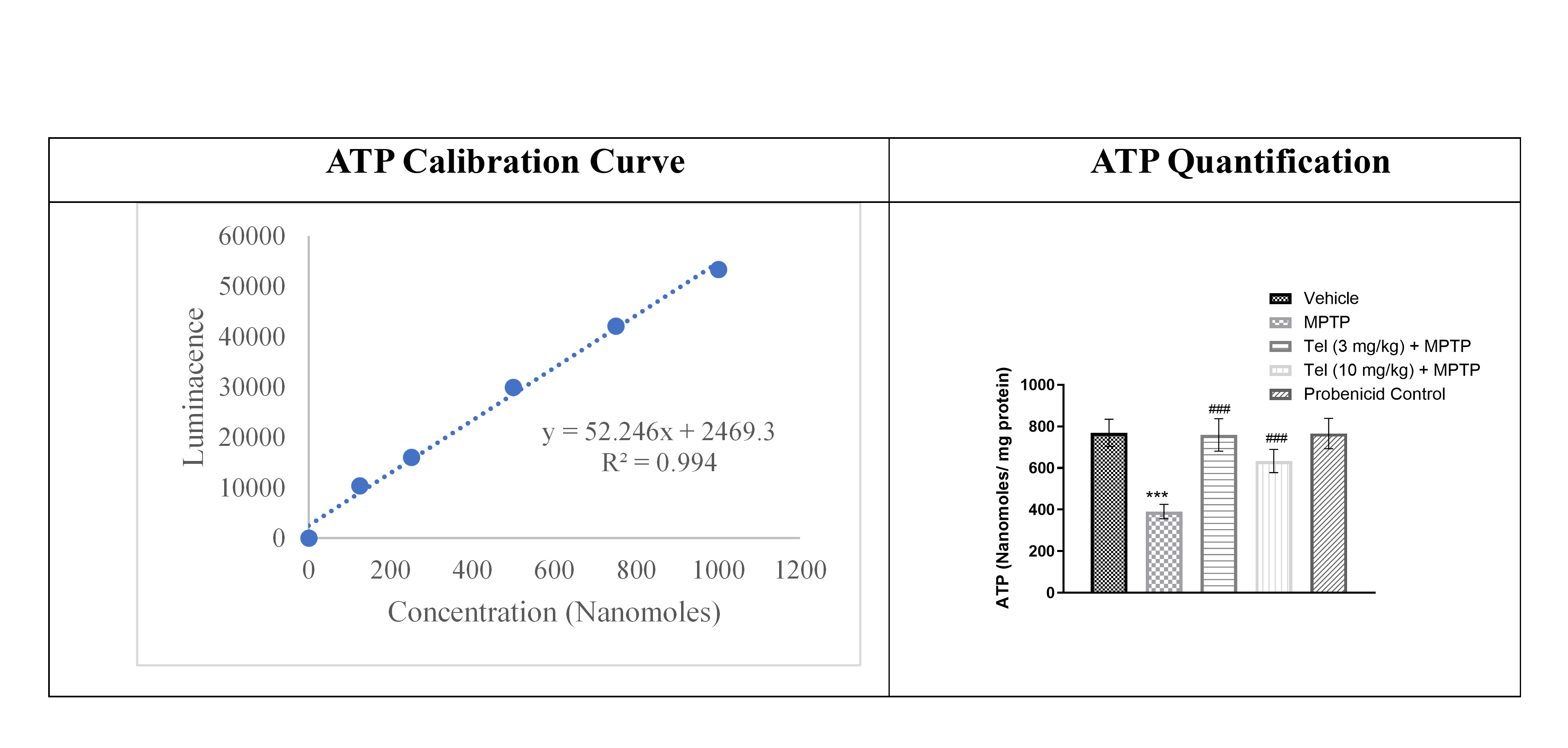 Fig. 3.
Fig. 3.Quantification of ATP content using a commercial ELISA kit.
SNPc region homogenates were diluted 10-fold and processed as
per the kit instructions. Luminescence was read at ~560 nm using
a luminometer. One-way ANOVA and Tukey’s multiple comparison tests were applied.
*** denotes a p-value
Protein-protein interactions (PPI) were analyzed using the
Search Tool for the Retrieval of Interacting Genes/Proteins
(STRING) database (https://string-db.org/). Ten nodes of predicted protein
interactions were built based on genomic context analysis. The PPI enrichment
value (p-value: 2.81
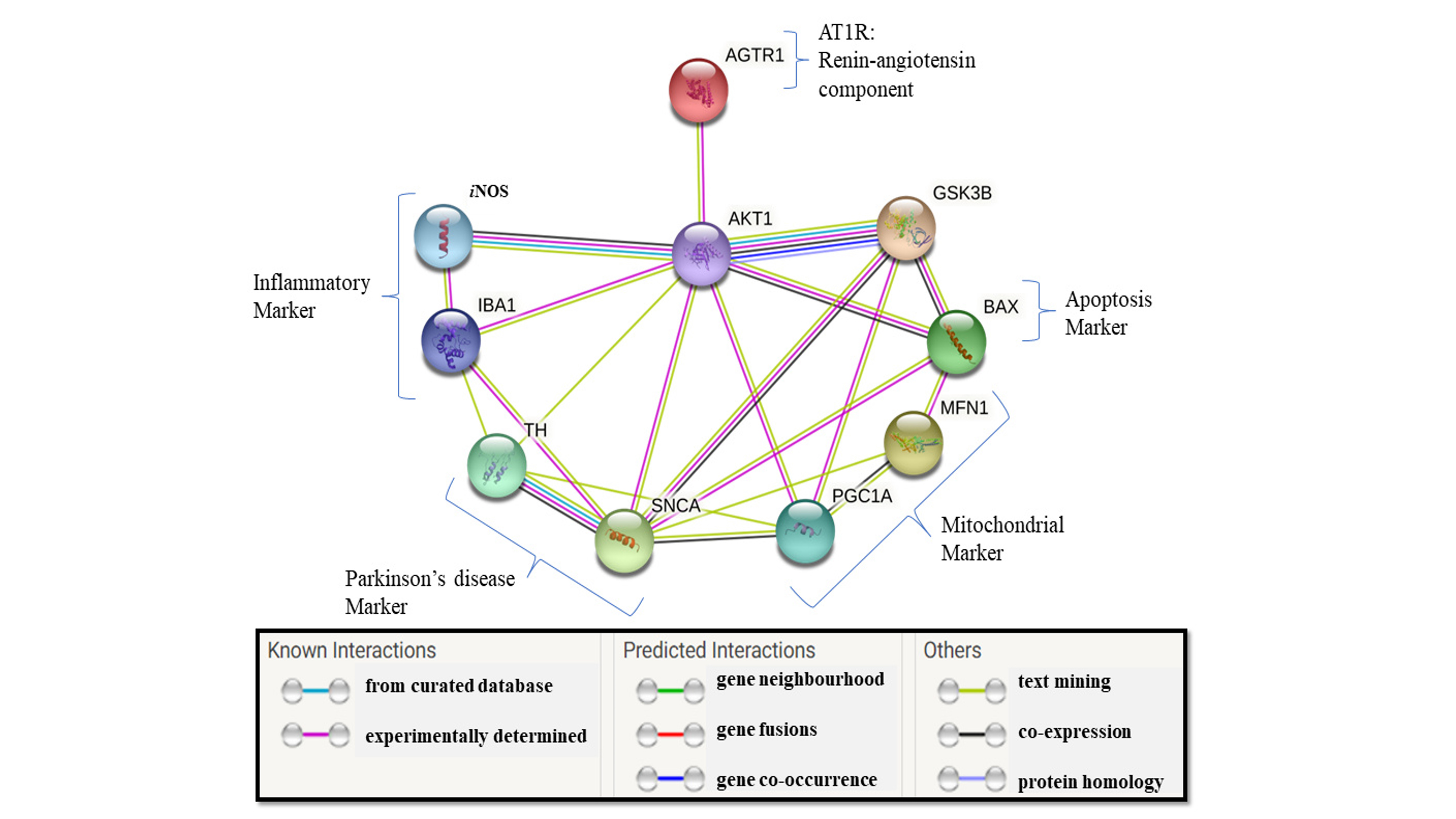 Fig. 4.
Fig. 4.Protein-protein interaction networks (STRING Database). The lines between the protein connections represent the types of interactions with other proteins with different color. These are based on experimentally proven or predicted results from other available databases. STRING, Search Tool for the Retrieval of Interacting Genes/Proteins; AGTR1 (AT1R), Angiotensin II Receptor Type 1; AKT1, AKT Serine/Threonine Kinase 1; iNOS, inducible nitric oxide synthase; BAX, BCL2 Associated X Protein; GSK3B, Glycogen Synthase Kinase 3 Beta; IBA1, Ionized calcium binding adaptor molecule 1; TH, Tyrosine Hydroxylase; SNCA, Synuclein Alpha; PGC1A, PPARG Coactivator 1 Alpha; MFN1, Mitofusin 1.
To further validate the PPI data, the expression levels of specific proteins
were analyzed by Western blot analysis. Chronic MPTP injections significantly
upregulated the expression of AT1R (p
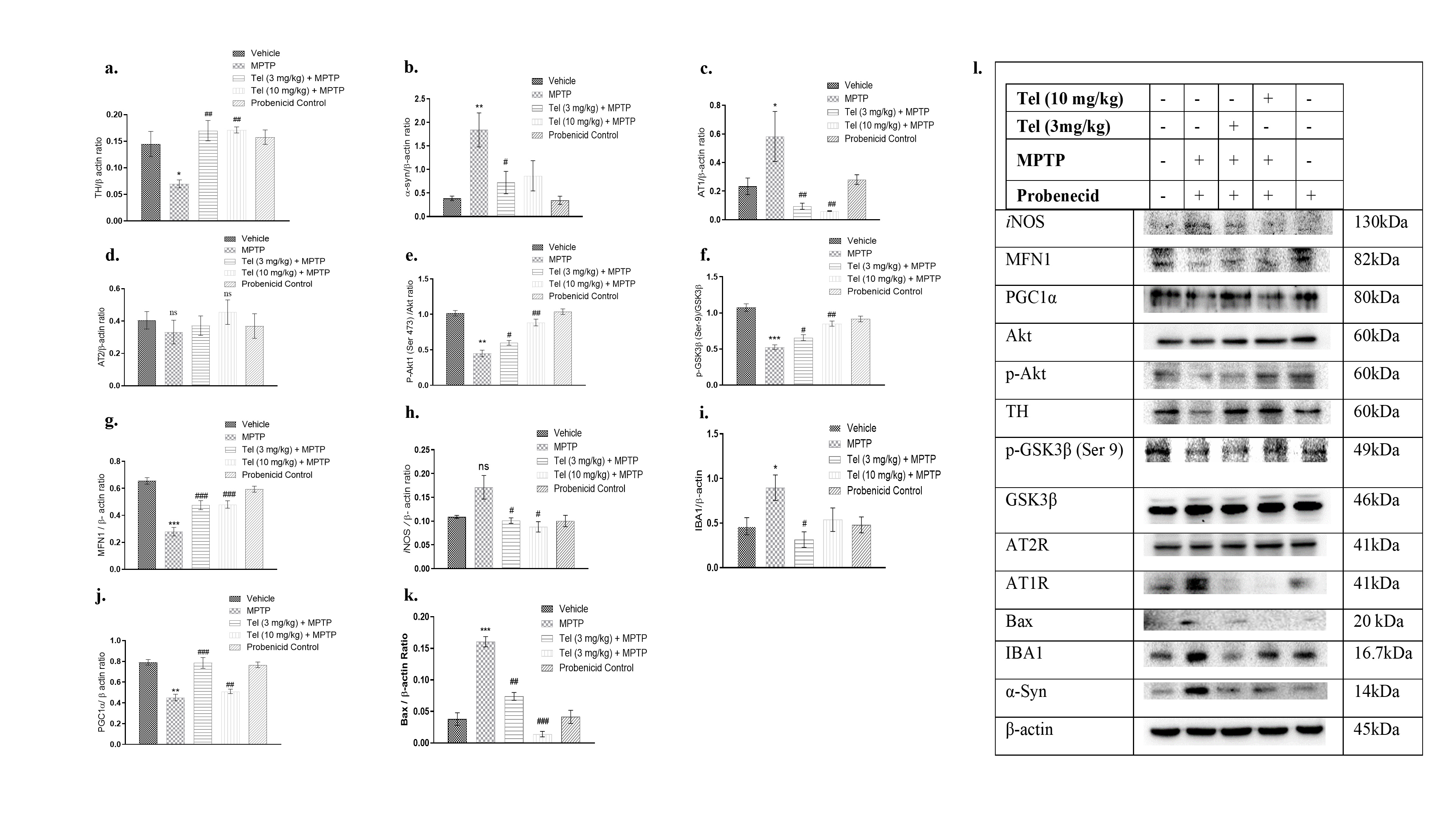 Fig. 5.
Fig. 5.Effects of Tel treatment on the protein levels. (a) TH, (b)
Tel significantly restored the levels of AT1R,
The findings of this study suggest the mechanism by which Tel, a selective AT1R
blocker, can protect mitochondrial function, gait function, and neuronal
apoptosis in a mouse model of PD. Moreover, the current study validated the
STRING database PPI associated with the renin-angiotensin system in PD (Fig. 4).
MPTP treatment in the present study was observed to upregulate
The current work found that Tel prevented the increase in expression of AT1R,
while at least partially preventing AT2R in MPTP-exposed mice. In a rat model of
insulin resistance, inhibition of AT1R was shown to cause neuroprotection via the
Akt-mediated pathway in dorsal root ganglion (DRG) neurons [30]. MPTP treatment
decreased the active form of Akt (phosphorylated at Ser-473) and downregulated
p-GSK3
Cytosolic GSK3
The Akt and GSK3
In conclusion, the present study showed that Tel upregulates p-GSK3
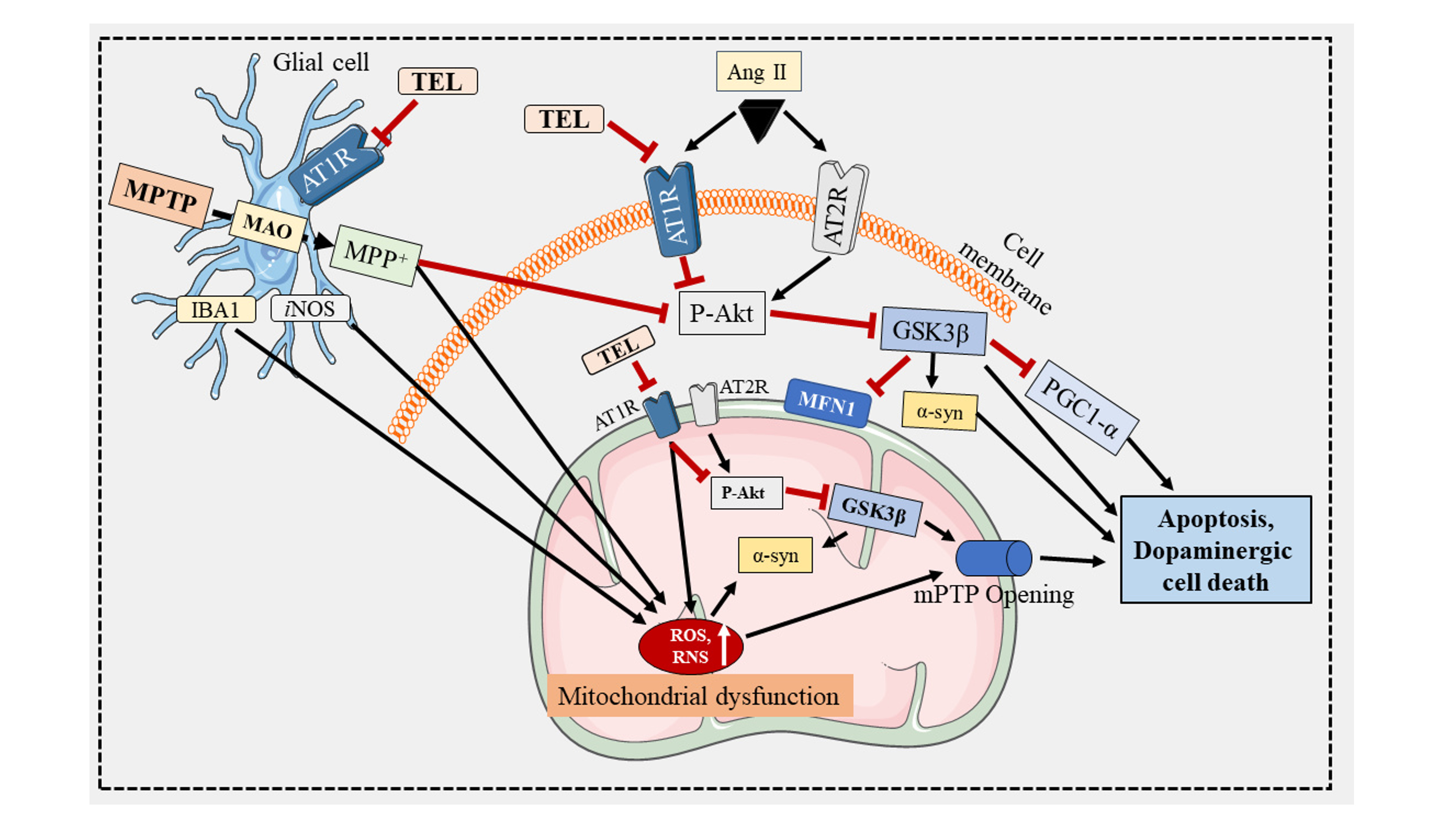 Fig. 6.
Fig. 6.Tel protects mitochondrial function in the brain of a mouse
model of PD. MPTP is converted to MPP+ in glial cells by MAO (monoamine
oxidase). AT1R and AT2R are present on the mitochondrial membrane surface. AT1R
performs the opposite function to AT2R, and its blockade is neuroprotective. The
expression of AT1R in PD is elevated compared to AT2R. AT1R activation inhibits
p-Akt, triggers the aggregation of
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
BR: conceptualized and designed the study, Performed, Data acquisition, analysis, and Manuscript writing; ST, PGN and AS: Data acquisition, analysis, and Manuscript Writing; AMM, PP and BJS: Data analysis and manuscript editing; SBC: conceptualized and designed the study, Manuscript Editing, and supervision. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The current study was approved by the Institutional Animal Ethics Committee (IAEC) (JSS AHER, Mysuru, India) (Approval number: JSSAHER/CPT/IAEC/016/2020).
The authors are thankful to National Institute on Alcohol Abuse and Alcoholism (NIAAA) and other respective institutions for providing the facilities.
This study was partially supported by the Indian Council of Medical Research (ICMR) (No-45/3/2019-PHA/BMS/OL).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.