







1 Department of Neurology, The Affiliated Nanhua Hospital, Hengyang Medical School, University of South China, 421001 Hengyang, Hunan, China
2 Department of Intensive Care Unit, The Affiliated Nanhua Hospital, Hengyang Medical School, University of South China, 421001 Hengyang, Hunan, China
†These authors contributed equally.
Abstract
Background: Microglia-mediated neuroinflammation is a hallmark of
neurodegeneration. Metabotropic glutamate receptor 8 (GRM8) has been reported to
promote neuronal survival in neurodegenerative diseases, yet the effect of GRM8
on neuroinflammation is still unclear. Calcium overload-induced endoplasmic
reticulum (ER)-mitochondrial miscommunication has been reported to trigger
neuroinflammation in the brain. The aim of this study was to investigate putative
anti-inflammatory effects of GRM8 in microglia, specifically focusing on its role
in calcium overload-induced ER stress and mitochondrial dysfunction.
Methods: BV2 microglial cells were pretreated with GRM8 agonist prior to
lipopolysaccharide administration. Pro-inflammatory cytokine levels and the
microglial polarization state in BV2 cells were then quantified. Cellular
apoptosis and the viability of neuron-like PC12 cells co-cultured with BV2 cells
were examined using flow cytometry and a Cell Counting Kit-8, respectively. The
concentration of cAMP, inositol-1,4,5-triphosphate receptor (IP3R)-dependent
calcium release, ER Ca
Keywords
- metabotropic glutamate receptor 8
- endoplasmic reticulum stress
- mitochondrial function
- neuroinflammation
- M1/M2 polarization
Microglia are immune cells that are abundant in the central nervous system (CNS) and play a pivotal role in immune surveillance, as well as sustaining and nourishing neurons [1]. The interplay between microglia and neurons is critical for synaptic remodeling and pruning in physiological conditions [2]. Microglia are divided into the pro-inflammatory M1 phenotype and the anti-inflammatory M2 phenotype. These cells can transition from a resting state into an activated state in various pathological conditions such as neuronal injury, aging, oxidative stress, and misfolded protein aggregation [3]. Persistent activation of microglia accompanied by excessive pro-inflammatory cytokine release are common events in primary neurodegenerative diseases such as Parkinson’s disease, Alzheimer’s disease, amyotrophic lateral sclerosis, Huntington’s disease, and epilepsy, as well as secondary neurodegeneration such as brain trauma, multiple sclerosis, stroke, and spinal cord injury caused by primary inflammation [4]. Disease-associated microglia and excessive microglial inflammation are thought to trigger irreversible brain damage. Several registered clinical drug trials have focused on attenuating microglial inflammation in neurodegenerative diseases, and some trials have shown significant amelioration of pathological and clinical manifestations [5]. Thus, the control of microglia-mediated neuroinflammation may be a feasible strategy for limiting neurodegeneration.
Almost 30% of the proteome undergoes folding and maturation within the
endoplasmic reticulum (ER). Calcium signaling is crucial in the protein folding
process. As the major organelle for calcium storage, calcium depletion within the
ER may disturb the protein folding process, leading to a condition termed ER
stress [6]. In addition, ER-to-mitochondrial Ca
Metabotropic glutamate receptors (GRMs) are widely distributed in the CNS and
are activated by glutamate. GRMs are classified into three groups according to
their sequence homology and physiological function. Amongst these, group III GRMs
(GRM4, 6, 7, 8) are reported to restrain glutamate release, limit neurons from
excitotoxicity, and promote neuronal survival [11]. GRMs are members of the G
protein-coupled receptor family. Following extracellular ligand binding, GRMs may
facilitate intracellular G-protein (G
In this study, we sought to explore the anti-inflammatory effects of GRM8 in BV2 microglial cells challenged with lipopolysaccharide (LPS). We hypothesize that the anti-inflammatory effects of microglial GRM8 may in part be due to its potential to mitigate calcium overload-induced ER stress and mitochondrial dysfunction.
BV2 microglial cells were purchased from Procell Life Science & Technology
(Wuhan, Hubei, China) and were grown in Dulbecco’s Modified Eagle’s Medium (DMEM,
Gibco, Anaheim, CA, USA) supplemented with 10% Fetal Bovine Serum (FBS) and 1%
streptomycin/penicillin. PC12 cells were grown in dulbecco’s modified eagle medium (DMEM)-high glucose medium
(Gibco) containing 10% FBS, 5% horse serum, and 1% streptomycin/penicillin.
Both BV2 and PC12 cells were incubated at 37 °C in a 5% CO
BV2 cells were pretreated with the GRM8 agonist AZ12216052 (1 µM; 1290628-31-7; MedChemExpress, Monmouth Junction, NJ, USA) or saline vehicle for 2 h. Afterwards, the cells were treated with saline control or LPS (1 µg/mL) for 12 h.
For the establishment of PC12 and BV2 cell co-culture conditions, PC12 was first incubated in a 24-well plate for 72 h. Following this, BV2 cells were transferred to a 0.4 µm pore-sized Transwell insert. PC12 and BV2 cells (BV2:PC12 = 1:2) were subsequently co-cultured for 48 h.
Cells were seeded on a coverslip and subsequently treated with 0.5% Triton
X-100 for 20 min. After washing, cells were incubated overnight at 4 °C with
primary antibody. The primary antibodies used were rabbit anti-GRM8 (1:100;
PA5-33830; Invitrogen, Waltham, MA, USA) and rat anti-CD11b (5 µg/mL;
ab8878; Abcam, Cambridge, UK). Cells were subsequently incubated with secondary
antibodies in a dark room. The secondary antibodies used in this experiment were
fluoresceine isothiocyanate (FITC)-conjugated goat anti-rabbit immunoglobulin G (IgG) H&L (1:800; ab6717;
Abcam) and Cy5-conjugated goat anti-rat IgG Heavy&Light (H&L) (1:800; ab6565; Abcam). Nuclei
were stained with 4
PC12 apoptosis in the co-culture system was detected using flow cytometry by staining PC12 cells with Annexin and propidium iodide (PI) (AP101; Multi Sciences, Hangzhou, Zhejiang, China) according to the manufacturer’s instructions. Preparations were analyzed on a CytoFLEX flow cytometer (Beckman, Pasadena, CA, USA).
Cell viability was assessed using a Cell Counting Kit-8 (CCK-8; C0039; Beyotime,
Shanghai, China). Briefly, PC12 cells were inoculated into a 96-well plate and 10
µL of CCK-8 stock solution was added to each well and incubated for 4 h.
Absorbance at 450 nm was measured using a Varioskan LUX microplate reader
(Varioskan LUX, Thermo Fisher, Moorhead, MN, USA). Cell viability was calculated
using the equation: [ODexperiemnt group – ODblank]/[ODControl group – ODblank]
TRIZOL reagent (Sigma-Aldrich, St. Louis, MO, USA) was used to extract total RNA
from BV2 cells according to the manufacturer’s instructions. RNA was subsequently
reverse transcribed into cDNA using a PrimeScript
| Target gene | Forward primer sequence (5 |
Reverse primer sequence (5 |
| iNOS | GGTGAAGGGACTGAGCTGTT | ACGTTCGTTCTCTTGCA |
| TNF- |
CAGGCGGTGCCTATGTCTC | CCATTTGGGAACTTCTCATCCCTT |
| Arg-1 | CACCTGAGCTTTGATGTCG | TGAAAGGAGCCCTGTCTTG |
| CD206 | AAGGAAGGTTGGCATTTGT | CTTTCAGTCCTTTGCAAGC |
| GAPDH | GCCAAGGCTGTGGGCAAGGT | TCTCCAGGCGGCACGCAGA |
RT-PCR, reverse transcription-polymerase chain reaction; iNOS, nitric oxide synthase;
TNF-
Commercially available enzyme-linked immunosorbent assay (ELISA) kits were used
to measure cAMP level (Abcam) and pro-inflammatory cytokine levels for
interleukin (IL)-1
Inositol-1,4,5-triphosphate receptor (IP3R) is expressed at the surface of the ER and when bound by IP3, calcium is released from the ER into the cytoplasm. Mag-Fluo-AM (GMS10189; GENMED, Shanghai, China) is a fluorescent probe with low affinity for calcium that can be specifically captured by the ER. This property is exploited for the detection of alterations in the concentration of ionized calcium within the ER. In this way, Mag-Fluo-AM can be used as a surrogate to examine the transfer activity of IP3R. Data were obtained 48 h after LPS treatment.
An IP3R functional fluorescence detection kit (GMS10189; GENMED, Shanghai,
China) was used to evaluate IP3R activity. All procedures strictly followed the
manufacturer’s instructions. In brief, Relative Fluorescence Units (RFUs) were
detected using a fluorescence microplate reader (Varioskan LUX, Thermo Fisher,
Moorhead, MN, USA). The induced calcium release rate was calculated using the
following equation: [(RFU
The ER Ca
A commercially available kit (MAK159; Sigma-Aldrich) was used to measure mitochondrial membrane potential (MMP) according to the manufacturer’s instructions. Cells were seeded into a 96-well plate and 25 µL of buffer A containing JC-10 was then added to each well and incubated for 45 min. The cells were then incubated with 25 µL of buffer B. Fluorescence measurements were made at an excitation wavelength of 540 nm and an emission wavelength of 490 nm in a microplate reader (Varioskan LUX, Thermo Fisher). The 540 nm/490 nm fluorescence intensity ratio indicates the MMP.
A luminescent ATP detection assay kit (ab113849; Abcam) was used to measure the ATP levels in BV2 cells. Briefly, detergent solution was added to cell cultures for cell lysis and ATP stabilization. Substrate solution was then added to each well and preparations were analyzed on a luminescence plate reader (Varioskan LUX, Thermo Fisher,) after incubating for 10 min in the dark.
Cells were seeded into 96-well plates and stained with
2
Cells were lysed using radioimmunoprecipitation assay (RIPA) buffer supplemented with Phenylmethanesulfonyl
fluoride (PMSF). The extracted proteins were separated by 10% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and
subsequently electrotransferred onto polyvinylidene fluoride (PVDF) membranes.
These were blocked for 1 h and then incubated separately with primary antibody
against ATF6 (1:1000; #DF6009; Affinity,
Cincinnati, OH, USA), IRE1
All values are expressed as the mean
Immunofluorescence microscopy was used to examine the co-localization of GRM8 and CD11b, a marker for microglia. As shown in Fig. 1, GRM8 co-localized with CD11b, indicating that GRM8 is expressed in microglia.
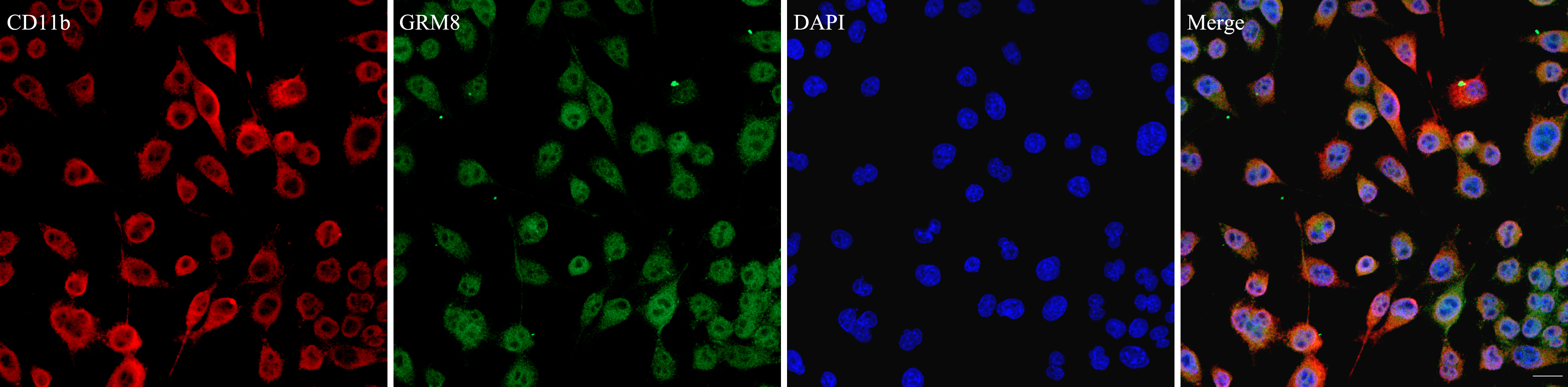 Fig. 1.
Fig. 1.Metabotropic glutamate receptor 8 (GRM8) and CD11b co-localize
in BV2 cells. Red fluorescence indicates CD11b-positive cells, while green
fluorescence indicates GRM8-positive cells. Nuclei were counter stained with
4
Quantitative RT-PCR was used to measure relative mRNA levels of microglial M1/M2
markers. Compared with the controls, LPS markedly upregulated the expression of
pro-inflammatory M1 phenotypic markers, including inducible nitric oxide synthase (iNOS) and
TNF-
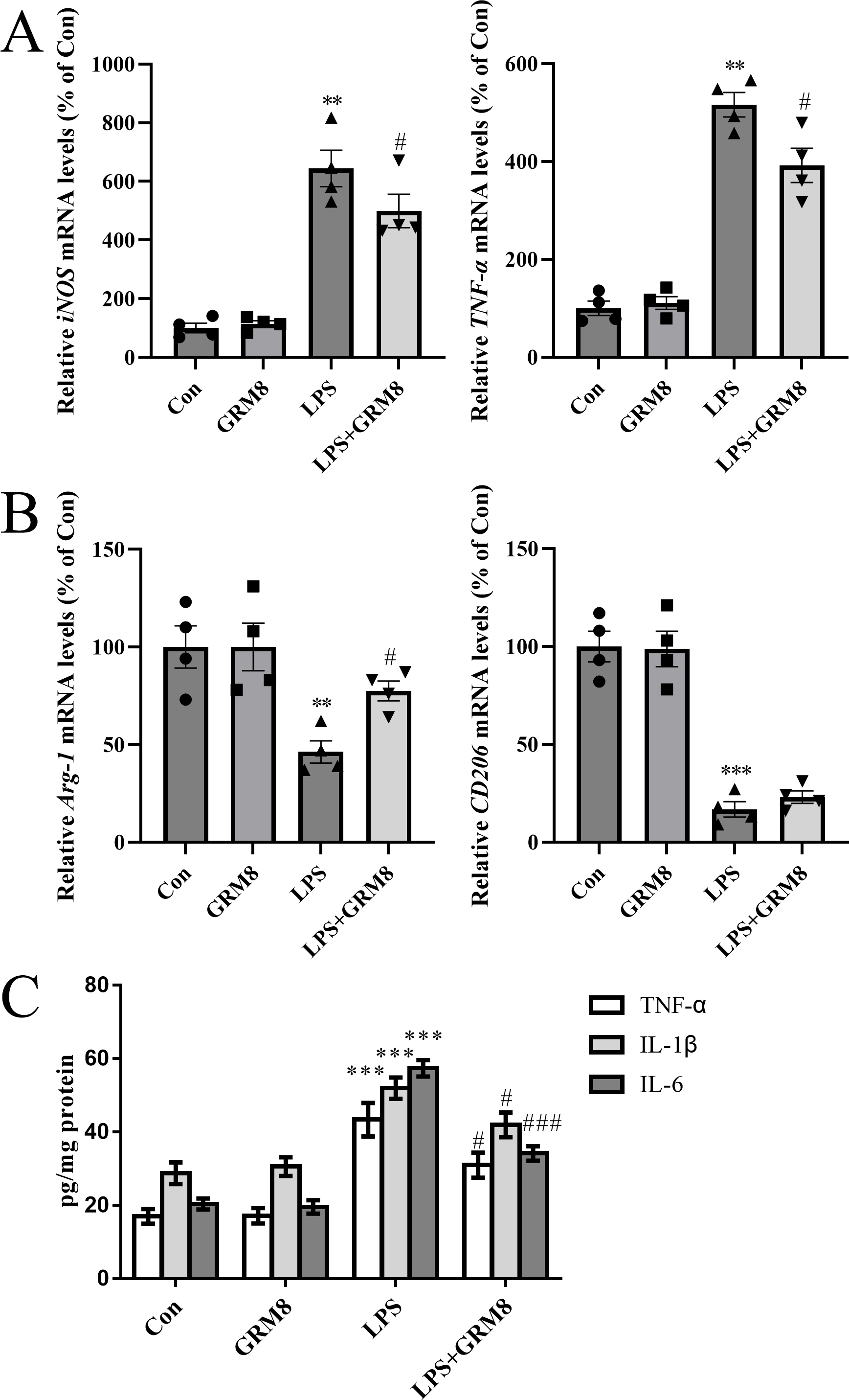 Fig. 2.
Fig. 2.GRM8 activation shifts M1/M2 polarization and inhibits the
release of pro-inflammatory cytokines. (A) Relative mRNA expression of the
pro-inflammatory M1 phenotype markers iNOS and TNF-
We used ELISA to further evaluate pro-inflammatory cytokine levels in BV2 cells.
GRM8 activation significantly inhibited the release of LPS-induced
pro-inflammatory cytokines (TNF-
After incubating with LPS, GRM8, or LPS combined with a GRM8 agonist, BV2 cells
were transferred to a co-culture system with PC12 cells and grown for 48 h (Fig. 3A). PC12 cells were then analyzed using flow cytometry (Fig. 3B) and CCK-8 (Fig. 3D) assays. The flow cytometry results showed that BV2 cells challenged with LPS
significantly increased the apoptosis of PC12 cells in the co-culture model
system relative to controls (p
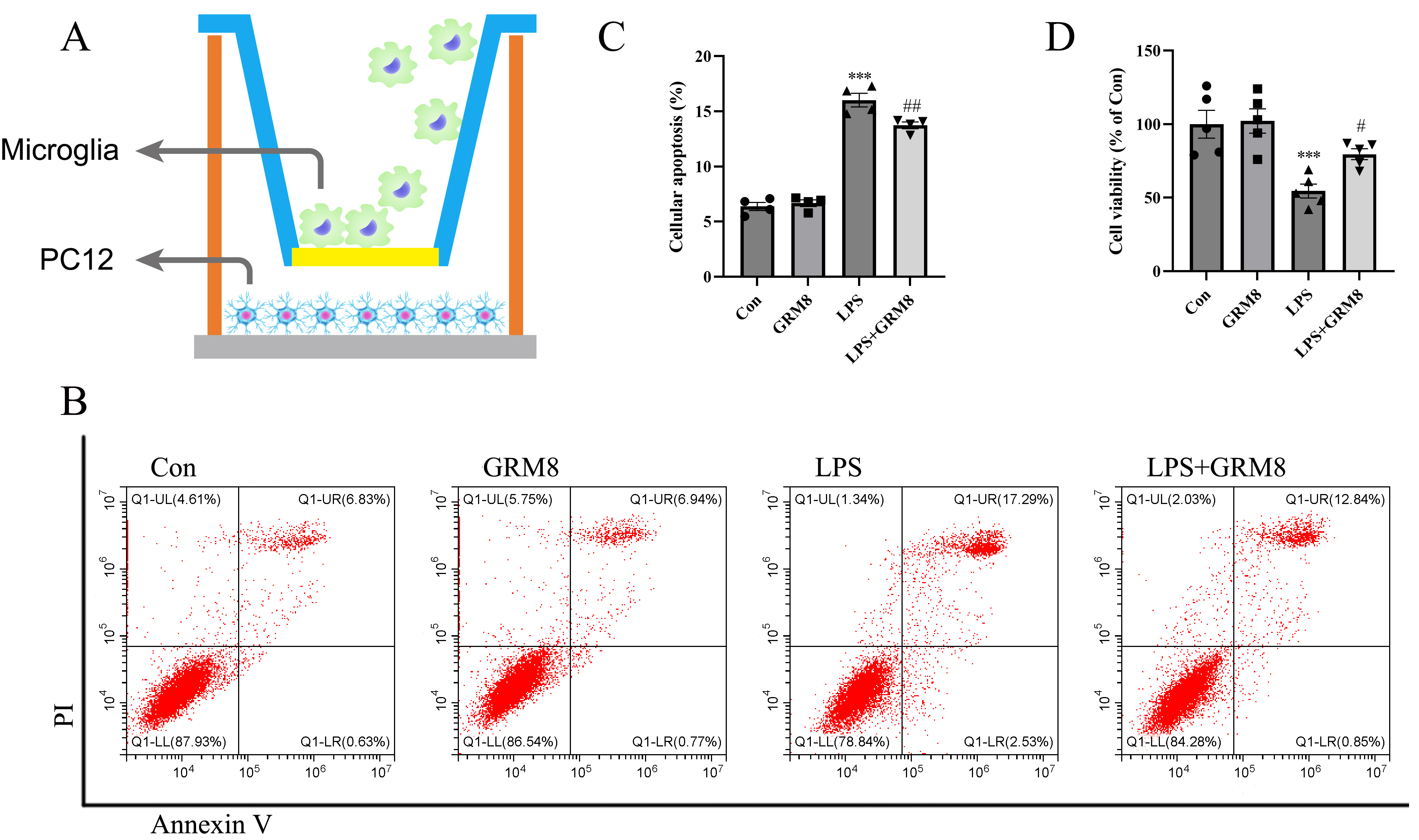 Fig. 3.
Fig. 3.Microglial GRM8 activation promotes neuronal survival in a
co-culture model system. (A) Simplified schematic of the co-culture model. (B)
Representative flow cytometry analyses showing apoptotic cell populations. (C)
Bar graphs indicating mean numbers of apoptotic cells measured in the indicated
experimental groups. n = 4 per group; dots show the individual assay values. (D)
Cell viability examined by CCK-8 assays. n = 5 per group. ***p
In CCK-8 viability assays, LPS treatment markedly reduced the viability of PC12
cells relative to untreated controls (p
To explore the mechanism controlling the anti-inflammatory response to GRM8 in
microglia, the effects of GRM8 on IP3R-dependent calcium release were assessed.
As shown in Fig. 4A, LPS treatment significantly increased calcium release from
the ER to the cytoplasm relative to controls (p
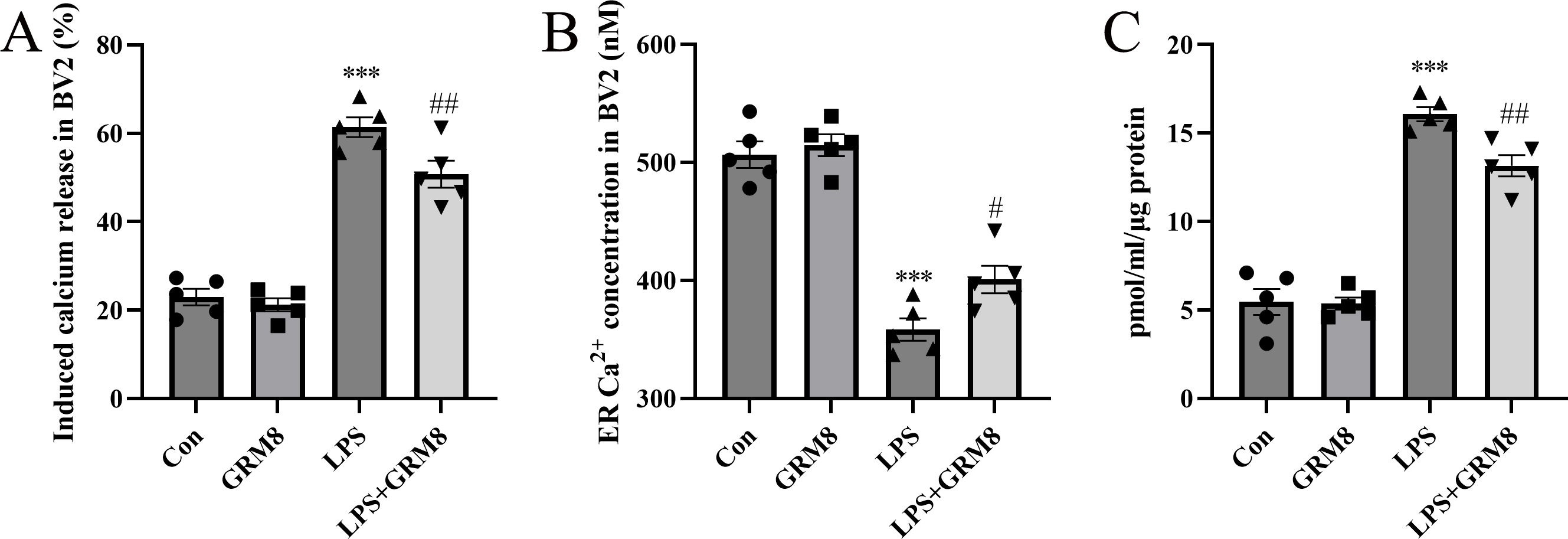 Fig. 4.
Fig. 4.Microglial GRM8 restores calcium homeostasis in the endoplasmic
reticulum (ER) and reduces cAMP levels in BV2 cells. (A) Induced calcium release
from the ER to the cytoplasm in BV2 cells. n = 5 per group. (B) ER Ca
cAMP is a common second messenger that may facilitate calcium release by
sensitizing IP3R. Therefore, we next measured cellular cAMP levels using ELISA.
cAMP levels increased significantly following LPS stimulation (p
Since calcium overload can trigger mitochondrial dysfunction and promote
excessive ROS production, ROS could also be responsible for increasing ER stress.
We observed a marked decrease in MMP in BV2 cells treated with LPS relative to
controls (p
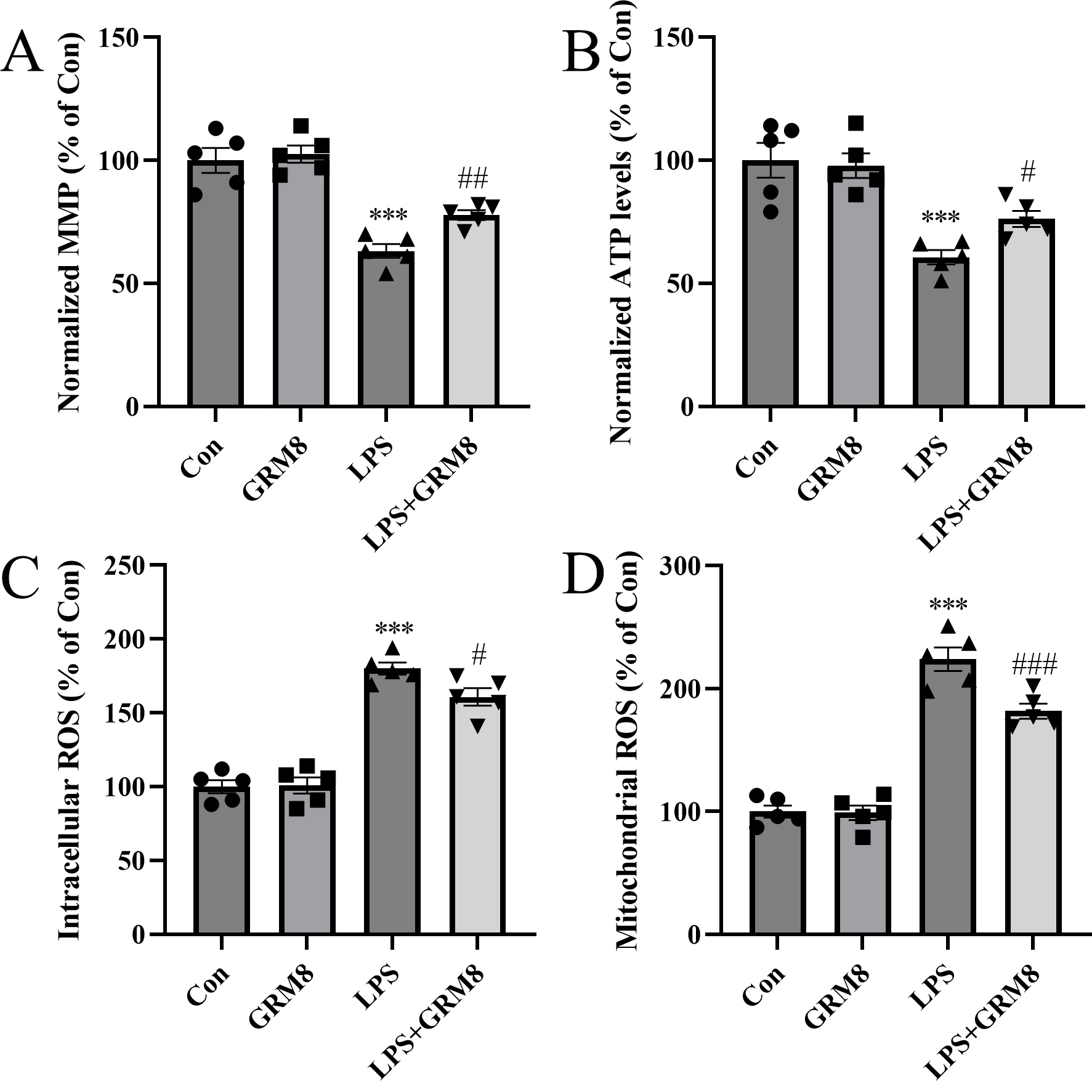 Fig. 5.
Fig. 5.Effects of GRM8 on mitochondrial function in BV2 cells
challenged with LPS. (A) Relative mitochondrial membrane potential (MMP). (B)
Relative ATP levels. (C) Intracellular reactive oxygen species (ROS) levels. (D)
Mitochondrial ROS levels. n = 5 per group. ***p
Increased ER stress can activate inflammatory signaling pathways such as the
canonical nuclear factor kappa B (NF-
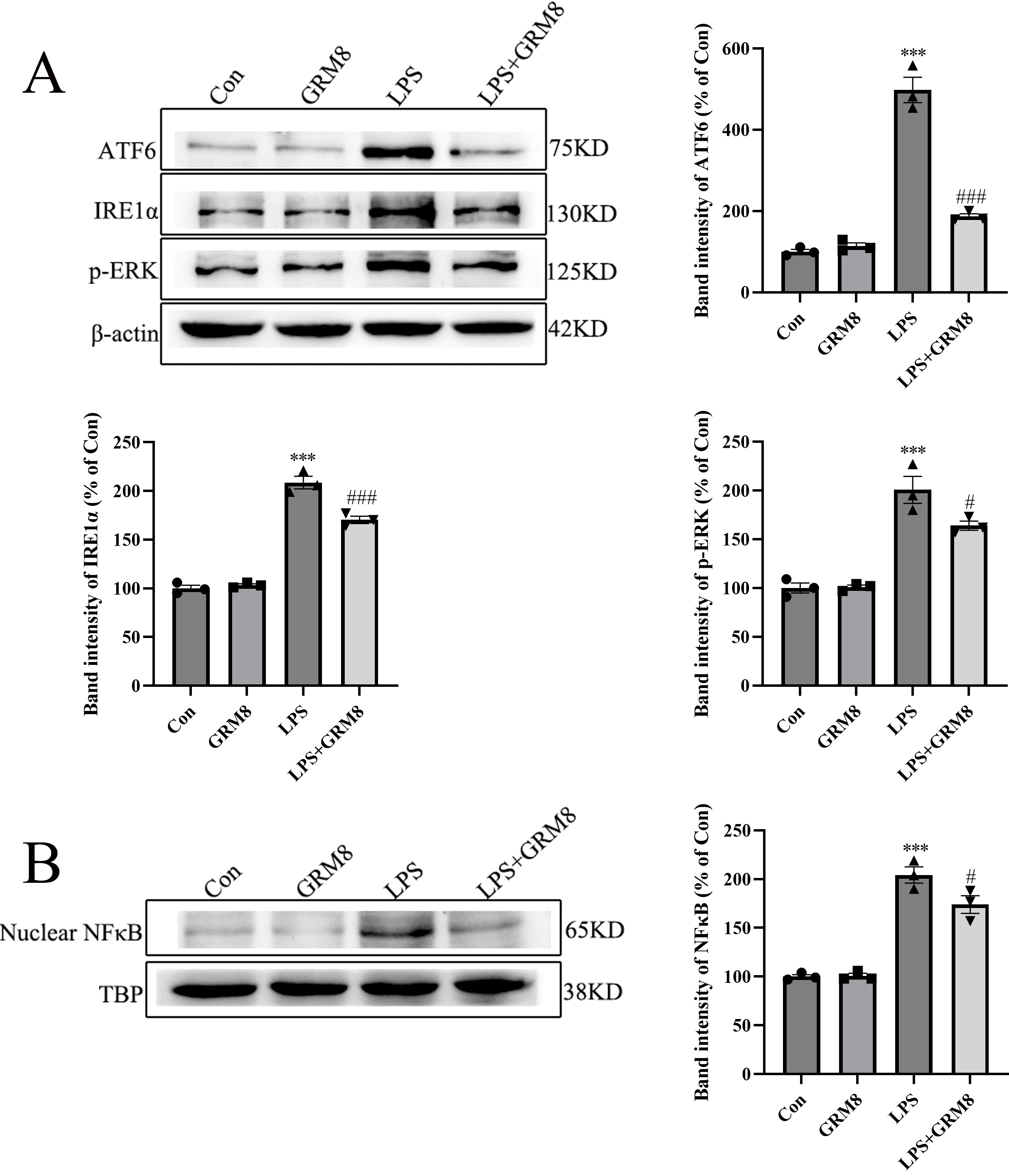 Fig. 6.
Fig. 6.GRM8 activation deactivates ER stress and NF-
We studied the anti-inflammatory effects of GRM8 activation in BV2 cells
challenged with LPS. GRM8 activation was found to ameliorate the pro-inflammatory
response observed in LPS-treated BV2 cells, and to attenuate neuronal injury in a
BV2/PC12 cell co-culture model. Mechanistically, microglial GRM8 activation
inhibited LPS-induced excessive cAMP production, resulting in desensitization of
IP3R and reduction of IP3R-dependent calcium release from the ER to the
mitochondria. Calcium homeostasis is thought to inhibit ER stress by restoring
the process of protein folding. In addition, the return of calcium homeostasis
restores mitochondrial function and inhibits ROS generation. ROS is widely
thought to play an essential role in mitochondrial-ER crosstalk. In accordance
with this, reduced ROS levels also deactivated ER stress signaling. GRM8
activation was found to disrupt the canonical NF-
GRM8 is expressed extensively in the CNS, particularly in the substantia nigra,
basal ganglia, thalamus, dorsal striatum, globus pallidus, subthalamic nucleus,
and nucleus accumbens [16]. The neuroprotective effect of GRM8 has been confirmed
in several animal models of neurological diseases, but a detailed mechanism of
action has yet to be reported. GRM8 was initially thought to be expressed in the
presynaptic membrane and to modulate excitatory neurotransmission [17]. However,
GRM8 is not restricted to the control of synaptic transmission. By using
immunofluorescent staining, we found that GRM8 was also expressed in microglia.
Microglial inflammation can be a double-edged sword in neurodegeneration. Under
physiological conditions, microglia function to clear debris and toxins from the
CNS. Under pathological conditions, excessive microglial activation contributes
to neuronal degeneration. To investigate a putative anti-inflammatory effect for
GRM8, we established an LPS-induced, pro-inflammatory microglial cell model.
Using this model, GRM8 activation was found to significantly reduce the release
of pro-inflammatory cytokines (IL-6, TNF-
Calcium is one of the most widely distributed biological second messengers, and
hence calcium balance is crucial for several biological functions. The ER is a
major site for calcium storage. Upon binding with IP3, the inositol triphosphate (IP3R) calcium channel
located within the ER is opened and facilitates calcium release [19]. Ca
Our work suggests a vital role for GRM8 in modulating ER calcium homeostasis,
based on its potential to inhibit cAMP production. Calcium sequestered in the ER
is crucial for ER-mitochondrion interaction, as calcium released through IP3R is
transferred to the intermembrane of the mitochondrion. Such calcium overload is
widely believed to cause mitochondrial dysfunction. We evaluated mitochondrial
function by quantifying MMP, the ATP level, and ROS production and found that
GRM8 activation restored mitochondrial function in microglia following LPS
challenge. The interplay between ROS and ER stress is complex in nature.
Nevertheless, ROS, and particularly mitochondrial ROS, is thought to induce ER
stress. Excessive ROS production may result in injury to ER Ca
Based on evidence obtained from the use of knock-out (KO) mice, a definitive role for GRM8 in neurological disease remains to be confirmed. GRM8 deletion has protective and detrimental effects in neurological disorders, as well as contradictory behavioral effects in KO mice [28, 29, 30]. It is worth noting that GRM4 may compensate for GRM8 deficiency, as a neuroprotective effect of GRM4 has been comprehensively reported [16]. Future studies should explore how GRM4 might influence GRM8-mediated anti-inflammatory potential. GRMs have also been shown to exhibit sex-related differences in brain function, stemming from circulatory estrogen levels. Estrogen receptors are coupled to GRMs, and upon binding with estrogen, intracellular G-protein signaling is engaged and ultimately affects cellular function [31]. However, whether estrogen receptors impact GRM8 function awaits further clarification.
The results of this study suggest that GRM8 strongly protects microglia from potentially harmful LPS-induced inflammatory responses in BV2 cells. It does this by reducing ER stress and improving mitochondrial function through mechanisms linked to IP3R-mediated calcium homeostasis. These findings indicate that targeting GRM8 may be a feasible therapeutic strategy for limiting neuroinflammation.
The data used and analyzed during the current study are available on reasonable request.
YX, LC, JC, YL and YC designed the research study. YX, ZPeng, HZ and LC performed the research. YX, ZPan and JC conducted experiments. YC analyzed the data. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We thank EJEAR’s English editing service for assisting in the preparation of this manuscript.
This research was funded by key projects of Hunan Provincial Health and Health Commission (grant number: 20201910).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.