

 , Xinping Luan 1,*
, Xinping Luan 1,*1 Cerebral Palsy Center in Neurosurgery, Second Affiliated Hospital of Xinjiang Medical University, 830063 Urumqi, Xinjiang, China
†These authors contributed equally.
Abstract
Cerebral palsy (CP), a common neurological disorder in children, remains a significant research focus. The interleukin (IL) family, pivotal mediators in inflammatory responses, shows increased expression in various neuroinflammatory diseases, markedly influencing their onset and progression. Elevated IL levels in the brains of children with CP, in contrast to healthy peers, reflect similar elevations in neurological conditions linked to CP, indicating a strong association between CP and the IL family. Anti-inflammatory therapies, particularly those targeting ILs, have shown effectiveness in animal models, diverging from traditional CP management methods. This shift suggests IL modulation as a promising therapeutic strategy in pediatric CP. This review consolidates recent findings on the IL family’s role in CP, illuminating their evolving relationship.
Keywords
- pathogenesis of cerebral palsy
- interleukin
- inflammatory factor
- neuroinflammation
Cerebral palsy (CP) is a predominant cause of childhood disability, with numerous risk factors originating in the perinatal period [1]. Diagnosing CP involves integrating clinical presentation, neuroimaging, and the evaluation of CP risk factors. Imaging in high-risk children often reveals white matter injury, periventricular hemorrhagic infarction, deep gray matter structure injury, cerebellar hemorrhage, and developmental brain malformations. Neurological assessments include the Prechtl Qualitative Assessment of General Movements (GMA), the Hammersmith Infant Neurologic Examination (HINE), and the Gross Motor Function Assessment (GMFM) [2]. Recent research has validated the continued necessity and reliability of early standardized assessments for CP diagnosis [3]. Additionally, evolving animal models, such as the 6-arm maze, provide reference standards for CP diagnosis [4]. Pathogenesis typically involves injury to the developing brain during fetal or perinatal stages, resulting in skeletal muscle dysfunction. This manifests as abnormal muscle contractions, postural difficulties, and restricted mobility, often accompanied by neurodevelopmental disorders including sensory and cognitive impairments, intellectual disabilities, epilepsy, and autism spectrum disorders. Clinically, CP is categorized into four primary types: spastic, dyskinetic, ataxic, and mixed [4, 5]. Initial study identified hypoxia as a potential cause of CP, but subsequent research has revealed a multifactorial etiology involving various factors that impair brain function [6]. While the precise mechanisms triggering CP onset remain incompletely understood, emerging evidence highlights the significant role of inflammatory responses in its development. Comparative analyses show elevated levels of pro-inflammatory cytokines, such as interleukin (IL)-2, IL-6, and IL-10, in the blood and cerebrospinal fluid (CSF) of children with CP compared to healthy controls [7]. Biomarkers related to free radical production and antioxidant effects, indicative of cell damage, are also significantly increased among non-inflammatory markers [8]. A comprehensive review on inflammation and CP consistently demonstrates heightened inflammatory markers, particularly those of the IL family, in the CSF of affected children [9, 10]. This review integrates advancements in understanding the IL family’s impact on CP pathogenesis, analyzing their roles in disease development, and proposing novel avenues for inflammation-centric CP research.
The pathogenesis of CP is categorized into four primary stages: fetal, antenatal, perinatal, and post-delivery (Table 1). During the fetal stage, maternal age, lifestyle, intrauterine infections, and genetics are pivotal factors. Intrauterine hypoxia, primarily due to placental insufficiency and abruption in late pregnancy, is a significant prenatal contributor to CP. Prematurity is the predominant risk factor in the perinatal period, attributed to the heightened vulnerabilities of preterm infants due to their lower gestational age and developmental immaturity. Despite advancements in medical care improving survival rates of premature infants, the incidence of CP has remained stable at 0.2% to 0.3% in recent decades. Meconium aspiration and ischemic hypoxic encephalopathy are also critical factors in brain injury during this stage. Additionally, infants and toddlers up to the age of five face a reduced risk of CP following head trauma or infections like meningitis, due to their developing immune systems, classifying such instances as acquired CP [1, 2].
| Main stages of CP | Risk factors for cerebral palsy |
| Foetal | Maternal age older or younger, multiple pregnancies, miscarriages, short or long parity, history of intrauterine death |
| Antenatal | Intrauterine growth restriction, intrauterine infection, vitro fertilization, chorioamnionitis |
| Perinatal | Premature birth, meconium aspiration, hypoxic-ischemic encephalopathy |
| Post-delivery | Neonatal seizures, head trauma, drowning, meningitis |
CP, cerebral palsy.
Recent advancements in medical science and technology have highlighted the
pathogenesis of CP as a significant research focus. Despite progress, substantial
knowledge gaps remain, particularly regarding the molecular mechanisms underlying
the disease [11]. Contemporary studies associate CP with neuroinflammation
induced by conditions such as neonatal encephalopathy (hypoxic-ischemic
encephalopathy), fetal inflammatory response syndrome, and white matter
damage/periventricular leukomalacia. Additionally, maternal immune activation is
increasingly recognized as a key factor in CP onset. An animal model of
chorioamnionitis induced by Lipopolysaccharides (LPS) exposure in the cervix of
pregnant rabbits demonstrated that perinatal intrauterine infections could lead
to abnormal motor function in the fetus post-birth and a reduction in the
synthesis of tetrahydrobiopterin in the brains of young rabbits with ischemic
brain injury [12]. The role of tetrahydrobiopterin in preventing ischemia-induced
CP has also been shown [13, 14]. Another animal model of partial and total
cerebral ischemia revealed minimal differences between acute and chronic ischemia
in CP development [15]. However, these models, including most rodent models,
share common limitations, such as the much smaller volume of cerebral white
matter in rodents compared to humans, higher partial pressure of oxygen in human
brain tissue, and differing degrees of cerebral white matter damage due to
ischemia [16, 17]. Following the onset of CP-associated diseases, a notable
commonality is the elevated levels of inflammatory factors in the brain. This
increase is partly due to microglial activation and the subsequent secretion of
inflammatory mediators, with contributions from oligodendrocytes and
oligodendrocyte precursor cells under conditions of oxidative stress and
inflammation. These processes involve distinct mechanisms: (1) Microglia, which
normally serve a neuroprotective function, differentiate into M1 and M2 subtypes
upon activation. M1 microglia release pro-inflammatory mediators (IL-1
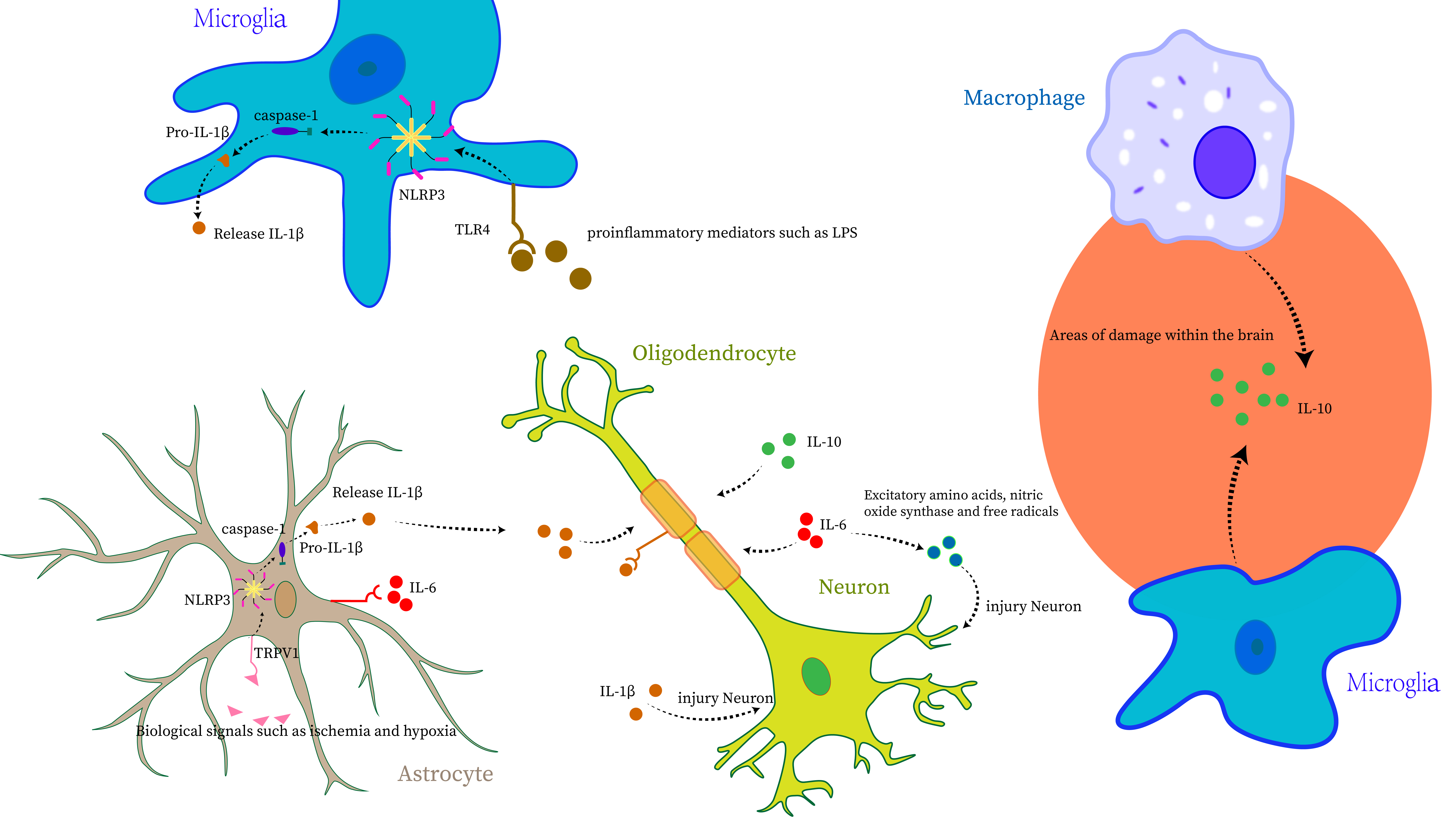 Fig. 1.
Fig. 1.
Interleukin (IL)-1
IL-1
IL-6, secreted by T and B lymphocytes, monocytes, and various non-lymphoid
cells, functions as a critical cytokine with both pro-inflammatory and
anti-inflammatory effects [32]. It is integral to B cell antibody production,
responding to viral and endotoxin infections, as well as cytokines like
TNF-
During mid-pregnancy, maternal IL-6 can traverse the placenta to the fetus.
Research indicates that maternal immune activation (MIA) during this period
enables IL-6 to bind directly to placental receptors, initiating an inflammatory
cascade within the fetal environment, contributing to neurodevelopmental
disorders at birth [42, 43]. Investigations involving full-term infants who later
developed CP reveal significantly elevated perinatal levels of cytokines,
including IL-1
IL-10, primarily secreted by T helper 2 (Th2) cells, functions as a versatile cytokine. Its expression increases in response to ischemia, hypoxia, and various neurodegenerative conditions through receptor binding. This rise indicates IL-10’s potential in mediating endogenous neuroprotective and neurotrophic activities. IL-10’s critical roles in inflammation modulation include: (1) promoting cluster of differentiation 8-positive (CD8+) T cell proliferation and cytolytic activity, (2) inhibiting antigen presentation and pro-inflammatory cytokine production by antigen-presenting cells (APCs), and (3) reducing tumorigenic effects linked to chronic inflammation [47]. In conditions where IL-10 levels are inadequate and cytotoxic metabolites are elevated, the risk of brain damage increases [48].
Elevated levels of pro-inflammatory cytokines, including IL-6 and
TNF-
The IL family profoundly influences inflammatory responses, particularly in the context of CP and its etiological conditions. Besides the extensively studied ILs with elevated expressions in CP, other ILs—though less frequently encountered or expressed at lower levels—also contribute significantly to the disease’s pathogenesis.
IL-2 emerges as a notable anti-inflammatory factor, primarily secreted by neuronal cells, with lower concentrations in CSF compared to serum. Animal studies highlight its neuroprotective capabilities, especially in promoting regulatory T (Treg) cell expansion and providing neuroprotection [55, 56]. Moreover, IL-2 has been shown to transform activated microglia from the classical disease-associated microglia (DAM) phenotype to an atypical major histocompatibility complex class II (MHCIIhi) phenotype. MHCIIhi microglial cells under-express inflammatory mediators, and this phenotypic switch can decelerate the inflammatory response. This indicates IL-2’s potential as a novel therapeutic approach for neuroinflammatory conditions in future research [57]. Most current studies focus on the beneficial aspects of IL-2 in anti-inflammatory therapy. However, previous research has shown elevated IL-2 levels in patients with white matter lesions, with the increase directly proportional to the severity of white matter damage [58, 59, 60]. Despite these data, the existing studies have limitations, and no consensus exists regarding IL-2’s effects on nerve treatment and injury. Thus, future research should adopt more rigorous and representative designs to elucidate IL-2’s role in nerve loss.
IL-4, primarily secreted by microglia and macrophages, with a minor contribution from astrocytes within the brain, functions as an anti-inflammatory factor that promotes neuroprotection. Its receptors are predominantly expressed on these producing cells and oligodendrocytes. Interestingly, IL-4 expression is generally higher in newborn brains compared to adults, yet its overexpression in newborns can inhibit oligodendrocyte differentiation. Moreover, macrophages bearing IL-4 receptors may become activated through overexpression, potentially intensifying inflammatory responses [61]. Similar to IL-10, the inflammatory cytokine IL-8 can recruit macrophages and microglia, exacerbating local damage [54]. Additionally, elevated IL-16 expression in patients with compromised blood-brain barriers indicates its role in mobilizing and activating cells at inflammation sites [62].
IL-17, primarily secreted by T helper 17 (Th17) cells, with additional
contributions from non-lymphoid Paneth cells and neutrophils, comprises six
variants, IL-17A to IL-17F. Among these, IL-17A is a prominent effector, crucial
for neutrophil recruitment and mucosal barrier enhancement. Notably, IL-17
secretion from Th17 cells is tightly regulated by IL-6 levels, with elevated IL-6
correlating with increased IL-17 secretion. IL-17 acts as a potent stimulator for
various inflammatory mediators, including granulocyte colony-stimulating factor,
IL-6, IL-1
IL-35, a novel immunosuppressive cytokine, significantly dampens immune responses in various conditions, including inflammatory and infectious diseases, autoimmune disorders, and cancers. The study indicates that IL-35 also possesses anti-inflammatory and neurotrophic properties, especially in ischemic-hypoxic models [20] (Table 2).
| Anti-inflammatory interleukins | IL-2: ① neuroprotective capabilities. ② converting activated microglia from the DAM phenotype to an atypical MHCIIhi phenotype. |
| IL-4: ① enhancing neuroprotection. ② overexpression in newborns can obstruct oligodendrocyte differentiation. | |
| IL-10: ① supporting neuronal homeostasis and cell activity and accelerating myelin sheath repair. ② elevated levels cause the aggregation of peripheral macrophages and microglia. | |
| IL-35: demonstrating anti-inflammatory and neurotrophic properties. | |
| Pro-inflammatory interleukin | IL-1 |
| IL-6: ① enhancing neuroselective glutamatergic synapses. ② directly affects synapse formation in the fetal brain during pregnancy. ③ infection triggers a reduction of non-heme iron in maternal circulation. ④ direct binding to receptors on the placenta leads to neurodevelopmental disorders in the fetus at birth. | |
| IL-8: high expression results in peripheral macrophage and microglia aggregation. | |
| IL-17A: its secretion is regulated by IL-6, and it promotes the release of pro-inflammatory factors. |
MHCIIhi, major histocompatibility complex class II; DAM, disease-associated microglia; IL, interleukin.
Research consistently identifies neuroinflammation as a primary contributor to
brain injury. The mechanisms by which inflammation leads to early brain injury
include diminished cerebral blood flow, impairing oxygen and glucose uptake;
compromised blood-brain barrier integrity, reducing protection for brain cells;
the generation of free radicals; and the activation of microglia, astrocytes, and
leukocytes, resulting in cerebral edema. These factors collectively lead to
neuronal damage. Additionally, elevated levels of ILs and other inflammatory
markers can adversely affect central nervous system development [48, 63]. Studies
have established a strong link between neuroinflammation in CP and infections.
Infections trigger the release of inflammatory mediators, including microglial
activation and increased levels of IL-1
From a cellular and molecular perspective, the inflammation process, from its
onset to pathogenesis, can be detailed: upon encountering innate immunity
challenges such as bacterial and viral invasions, endotoxin release, or sterile
stimuli, the immune system triggers cellular apoptosis mechanisms. Central to
this process is the NLRP3 inflammasome, a recently identified protein expressed
by myeloid immune cells that functions as a cellular stress sensor. Its
activation, regulated by the nuclear factor kappa-light-chain-enhancer of
activated B cells (NF-
Current therapeutic strategies for CP primarily focus on post-onset
interventions, including rehabilitation training, intrathecal baclofen
injections, and selective posterior rhizotomy (SPR) to reduce limb spasticity.
Cervical perivascular sympathectomy (CPVS) is also employed to address cerebral
ischemia symptoms [77]. While these treatments offer symptomatic relief, they
neither address the underlying causes of CP nor fully reverse neurological
damage. Recent studies exploring the link between CP and inflammation have
demonstrated promising outcomes in treating mild to moderate CP in rabbit models
with anti-inflammatory agents. These treatments not only improved survival rates
but also enhanced motor function recovery, nearly matching that of unaffected
controls [78, 79]. This breakthrough highlights the potential of targeting brain
inflammation in CP treatment, utilizing anti-inflammatory cytokines like IL-4 and
IL-10 to counteract pro-inflammatory cytokine effects and employing IL
antagonists such as the IL-1 receptor antagonist. Anakinra, commercially
available as IL-1Ra, has been used in clinical settings for over two decades to
treat various inflammatory syndromes, including sepsis. Its ability to rapidly
cross the blood-brain barrier [9] has led to its recent use in investigating the
control of neuroinflammation. Study has indicated that Anakinra can
effectively regulate GBS-induced MIA [80]. Addressing IL-6-induced
hypoferritinemia through maternal iron supplementation presents a novel strategy
to prevent fetal developmental issues [34]. Recent study has emphasized the
use of IL-2/IL-2R in the prognostic treatment of brain hypoxia, primarily by
increasing regulatory T cell populations that provide neuroprotection. These
cells can reduce IL-6 levels and promote macrophage polarization to an M2
phenotype, thereby diminishing ischemia-induced inflammation [81]. Additionally,
melatonin, an endogenously produced hormone, has been shown to counteract
inflammation by enhancing mitochondrial autophagy and inhibiting the
TLR4/NF-
This review offers the first systematic analysis and synthesis of ILs’ impacts on CP onset. By integrating insights on neuroinflammation, CP development, and the specific ILs driving neuroinflammation, this study elucidates the significant influence of ILs on CP’s underlying mechanisms. Extensive research consistently indicates that IL levels in children with CP are markedly elevated compared to healthy counterparts and remain elevated over time. This persistent inflammation highlights its continuous role in the development and progression of CP. Although the inflammatory response serves as a protective mechanism against external pathogens, it simultaneously acts as a double-edged sword. By producing cytotoxic effects on normal cells, inflammation can paradoxically exacerbate the condition it initially aims to ameliorate. Experimental studies using animal models of CP have shown that precursor diseases leading to CP trigger inflammatory responses and dysregulate inflammatory factors within the brain. This evidence further solidifies the connection between inflammation and CP onset. At the core of these inflammatory responses is a complex network of signaling among various factors and cells, with the IL family emerging as a significant mediator closely associated with CP.
Current investigations into CP mechanisms face ethical constraints, necessitating reliance on animal models for experimental studies. Consequently, this review predominantly incorporates data from animal research, with human studies remaining largely observational. The absence of experimental human models impedes a detailed understanding of the IL-CP relationship. Addressing this requires further research using primates and other models closely mimicking human physiology, contingent upon increased funding. This review’s limitations include the lack of gender-segregated experimental data, precluding conclusions on potential gender-dependent differences in the findings. In summary, this review addresses methodological considerations and suggests potential therapeutic avenues for future CP research. Future efforts should aim to elucidate the molecular mechanisms of ILs in CP and develop innovative screening methods, essential for translating animal model findings into human health applications.
Although research on inflammatory factors has advanced recently, the exploration of their specific mechanisms and direct links to CP remains limited. Therefore, investigating the role of ILs in CP’s development is a crucial and long-term research priority. The primary aim of ongoing CP research is to deepen understanding of its specific mechanisms, elucidate the role of ILs in its pathology, thoroughly examine the IL family’s impact on the nervous system, and identify the triggers for inflammatory responses through various pathways. Furthermore, monitoring the dynamics of inflammatory responses and IL levels during anti-infection and anti-inflammatory treatments is essential. This comprehensive approach aims to enhance therapeutic strategies for CP, providing more effective interventions.
MBH, JJW and CB were responsible for setting the direction of the study, analyzing the data, and writing the manuscript. HZ was responsible for reviewing the literature and organizing the references. XPL was responsible for developing writing ideas, guiding the writing process, interpretation of data, designing the figures and finalizing the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
During the preparation process, the authors checked all texts for spelling and grammar using translation software tools such as Chatgpt 4. The authors took full responsibility for the content of the publication. We thank Bullet Edits Limited for the language editing assistance.
This research was also funded by the Chinese National Natural Science Foundation’s Regional Project [81660202], Xinjiang Uygur Autonomous Region Science and Technology Support to Xinjiang Project (2016E02057) and the Rehabilitation Program for Disabled Children of the Autonomous Regions Disabled People Federation.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.