


1 Department of Pathology, Chengdu Women's and Children's Central Hospital, School of Medicine, University of Electronic Science and Technology of China, 611731 Chengdu, Sichuan, China
2 Department of Pathology, The Affiliated Hospital, Southwest Medical University, 646000 Luzhou, Sichuan, China
3 Department of Pathology, Chengdu Anorectal Hospital, 610016 Chengdu, Sichuan, China
4 Department of Pathology, The Yaan People’s Hospital (Yaan Hospital of West China Hospital of Sichuan University), 625000 Yaan, Sichuan, China
†These authors contributed equally.
Abstract
Autism spectrum disorder (ASD) is a neurodevelopmental disorder characterized by impaired social interactions and verbal communication, accompanied by symptoms of restricted and repetitive patterns of behavior or interest. Over the past 30 years, the morbidity of ASD has increased in most areas of the world. Although the pathogenesis of ASD is not fully understood, it has been associated with over 1000 genes or genomic loci, indicating the importance and complexity of the genetic mechanisms involved. This review focuses on the synaptic pathology of ASD and particularly on genetic variants involved in synaptic structure and functions. These include SHANK, NLGN, NRXN, FMR1, and MECP2 as well as other potentially novel genes such as CHD8, CHD2, and SYNGAP1 that could be core elements in ASD pathogenesis. Here, we summarize several pathological pathways supporting the hypothesis that synaptic pathology caused by genetic mutations may be the pathogenic basis for ASD.
Keywords
- autism spectrum disorder (ASD)
- genetic variant
- synaptic pathology
- synapse development
- synaptic transmission
- synaptic plasticity
Autism spectrum disorder (ASD) is a neurodevelopmental condition characterized by a broad spectrum of symptoms, including impaired social interactions and restricted, repetitive patterns of behavior. The term “autism” was first coined by Professor Leo Kanner in 1943 [1] at Johns Hopkins University, USA. He conducted a longitudinal study over nearly three decades on 11 children, aged between 2 and 8 years, who displayed classic ASD behaviors such as repetitive, stereotypical attention to objects [1]. Currently, it is widely recognized that ASD exhibits substantial clinical heterogeneity. Beyond the primary clinical symptoms, a significant number of individuals diagnosed with autism also suffer from various comorbid mental health conditions. These include intellectual disability (ID), epilepsy, anxiety, obsessive-compulsive disorder, sleep disturbances, depression, and physical ailments such as gastrointestinal issues, autoimmune disorders, and obesity [2].
Although certain medications, such as risperidone and aripiprazole, that target these symptoms are clinically approved in the USA, they are not effective for specific autistic characteristics. Presently, early behavioral interventions are widely regarded as the most effective therapeutic approach, as they help individuals acquire specific skills and address problematic behaviors [3].
Globally, the estimated prevalence of ASD is between 1.5% and 2.0%, affecting over 70 million individuals. Males have an approximately four-fold higher incidence than females [4]. Over the past three decades, an increased prevalence of ASD has been observed worldwide. This has been attributed to various factors including environmental changes, shifts in diagnostic criteria, increased awareness of the condition, and enhanced ASD-related services [5].
The etiology of ASD is still unclear, but multiple genetic and environmental factors are thought to be involved. Over the past 50 years, one of the most important achievements in ASD research has been to recognize the role played by genetic/genomic factors [6]. So far, more than 1000 genes or genomic loci have been associated with ASD. Many of these genes play an important role in molecular functions and cellular morphology, including important neurological functions. Genetic abnormalities in ASD include chromatin remodeling disorders [7], DNA mutations [8], mRNA transcription and translation disorders [9], altered protein modifications [10], and altered epigenetic modifications [11]. These can lead to disorders of synaptic structure and functions [12], alterations in ion channels [13, 14], and dysfunctional signaling pathways [15]. Neurons in the brain are connected through synapses, and neural information travels through synaptic transmission. Recent studies have found that genes such as FMR1, NRXN, NLGN, SHANK, MECP2, CDH8, GRIN, and SYNGAP1 are associated with ASD, while also playing major roles in synaptogenesis, synaptic maturation, transmission, and plasticity [6, 15, 16]. Together, the results to date indicate that synaptic disorders may be the most important contributor to the pathogenesis of ASD. Here, we review the genetic mechanisms of synaptic dysfunction in ASD, with the aim of consolidating knowledge in this area.
It has been estimated that chromosomal abnormalities, including deletions, duplications, inversions, and translocations, occur in approximately 2% to 5% of individuals with ASD [17, 18]. Most of the structural aberrations found in chromosomes are relatively rare, and their role in the pathogenesis of ASD remains unclear. Abnormalities in sex chromosomes are observed in individuals with ASD who also have conditions such as Turner syndrome, Klinefelter syndrome, XXX syndrome, and XYY syndrome. These findings support the notion that genes located on the sex chromosomes can significantly influence the structure, function, and development of the brain [19].
Mechanisms such as translocation, regulation of sex-specific gene expression, chromatin regulatory factors, epigenetics, and action potentials can all affect the topological structure of chromatin, which has been associated with ASD and epilepsy. However, due to the lack of mechanistic studies, it has not been conclusively proven that such relationships are causal [11]. It is noteworthy that defects in chromodomain helicase DNA-binding protein 8 (CHD8) can impair synaptic function through their effects on gene expression and chromatin structure [20]. Indeed, copy number variants (CNVs) are a common chromosomal micro-malformation. Duplications and deletions of around 600 kb in the 16p11.2 region are frequently associated with ASD and other neurological disorders, and are the most common CNVs linked to ASD. About 20% of affected individuals carry duplications, and 16% that show ASD-like behaviors carry deletions [21].
Mouse models with different 16p11.2 genotypes have different phenotypes. Those with 16p11.2 deletions often exhibit significant head malformations, whereas those with duplications typically have smaller head deformities. Moreover, the former have more severe specific autistic features, such as poorer cognitive and behavioral levels, consistent with observations in human subjects [21, 22, 23]. In mouse models and humans with 16p11.2 microdeletions, notable dysfunctional connectivity and poor integration have been observed in prefrontal and parietotemporal regions of the brain [24]. The social and cognitive deficits in 16p11.2dp/+ mice are reportedly caused by downregulation of the gamma-aminobutyric acid (GABA) (inhibitory) synapse regulator Npas4 due to decreased GABAergic synaptic transmission and elevated neuronal excitability in the prefrontal cortex (PFC) [25]. Consistent with studies in the brain of humans with autism, the increased density of dendritic spines and increased number of excitatory synapses, together with the decreased number of inhibitory synapses, causes an imbalance of excitatory/inhibitory (E/I) synapses, thereby resulting in neural dysfunction and disordered network connections in neurons [26, 27]. Furthermore, restoration of Npas4 expression was able to ameliorate the social and cognitive deficits, while reversing GABAergic synaptic impairment and neuronal hyperexcitability [25]. Npas4 is one of the target genes regulated by histone deacetylase 5 (HDAC5). Intervention with LMK235, an HDAC5 inhibitor, can normalize histone acetylation, restore GABAergic signaling in the PFC, and significantly improve social preference in 16p11.2dp/+ mice, thus potentially ameliorating gene, synaptic, and behavioral defects under the conditions of 16p11.2 duplication [28].
Other CNVs linked to ASD are located in the 15q11-q13, 7q11.23, 22q11.2, and 1q21.1 regions [29]. Duplication of the UBE3A gene in the 15q11-q13 region is associated with ASD-like symptoms [30]. This gene encodes a ubiquitin-protease E3A that is involved in protein homeostasis and synaptic plasticity. A mouse model with complete or partial reduction of the Met receptor tyrosine kinase was constructed to study the behavioral impact of altered Met expression. This revealed a complex contribution of Met to the development of circuits mediating social, emotional, and cognitive behavior [31]. Broad disruptions have been observed in the developing synaptic proteome of Met null mice, particularly in proteins related to the ubiquitin-proteasome system essential for synaptic function [32]. Deletions in the 15q13.3 region have been linked to an increased likelihood of autism, ID, schizophrenia, and epilepsy [21]. Additionally, the 22q11.2 locus is crucial for brain development and harbors CNVs that increase the risk of ASD, schizophrenia, and other neurodevelopmental disorders [33]. Deletions in 22q11.2 are associated with abnormal development of functional network connections between the thalamus and cortex, thereby impacting hippocampal interneurons that regulate the balance of excitation and inhibition in the brain [34, 35]. Deletions in 22q11.2 also impair the regulation of interneuron migration through the cytokine C-X-C chemokine receptor type 4 (Cxcr4) [36]. A Df(16)A+/in mouse model with a 1.5 Mb deletion in 22q11.2 shows reduced density of interneurons expressing parvalbumin, decreased feedforward inhibition, and impaired social cognition [33]. The incidence of ASD in carriers of a deletion in 22q11.2 is 11% [21].
Recently, a microdeletion of nearly 2.4 Mb in the 8p23 terminal region was
identified in a Taiwanese boy with ASD. This deletion reduced the expression of
DLGAP2, a postsynaptic scaffold protein, thus potentially contributing to ASD
pathogenesis. DLGAP2 knockout (KO) mice show impaired spatial memory
(indicative of hippocampal dysfunction), abnormal expression of postsynaptic
proteins such as postsynaptic density 95 (PSD95), SH3 and multiple ankyrin repeat domains 3
(SHANK3), Homer scaffold protein 1 (HOMER1), glutamate ionotropic receptor
NMDA type subunit 2A (GluN2A), glutamate receptor 2 (GluR2),
metabotropic glutamate receptor 1 (mGluR1), metabotropic glutamate receptor 5 (mGluR5),
beta calcium/calmodulin-dependent protein kinase II (
Advances in whole genome analysis have highlighted the significant role of CNVs in biodiversity and disease. Sebat et al. (2007) [40] were the first to report that de novo CNVs were associated with ASD. They found that over 10% of children with ASD carry CNVs, compared with less than 1% in a control group. Subsequent research confirmed that CNVs are more common in ASD individuals than in healthy controls [41]. CNVs can be categorized into recurrent and non-recurrent types, with both duplication and deletion mutations affecting gene expression and regulation locally, or more broadly (see Table 1, Ref. [21, 22, 23, 24, 25, 29, 30, 32, 34, 35, 37]). Seventeen CNV loci are listed in the Simons Foundation Autism Research Initiative (SFARI) database, with the most frequent being duplications or deletions in 16p11.2.
| Copy Number Variant (CNV) | |||
| Locus | Variant | Pathology | References |
| 16p11.2 | Duplication Deletion | Duplication: small head deformities | [21, 22, 23, 25] |
| Deletion: large head malformations | |||
| Microdeletions: remarkable dysfunctional connectivity and remote integration in the prefrontal and parietotemporal region of the brain | [24] | ||
| 15q11-q13 | Duplication | Effects on protein homeostasis and synaptic plasticity | [29, 30, 32] |
| Deletion | |||
| 22q11.2 | Deletion | Effects on hippocampal interneurons and network connections between the thalamus and cortex | [21, 34, 35] |
| 8p23 | Deletion | Hippocampal dysfunction with abnormal expression of PSD (PSD95, SHANK3, HOMER1, GluN2A, GluR2, mGluR1, mGluR5, |
[37] |
CNVs, copy number variants; ASD, autism spectrum disorder; PSD95, postsynaptic
density 95; SHANK3, SH3 and multiple ankyrin repeat domains 3; GluR2, glutamate receptor 2; mGluR1, metabotropic
glutamate receptor 1;
In addition to CNVs, single nucleotide polymorphisms (SNPs) have also received widespread attention due to their potential involvement in the etiology of ASD. Many studies have shown significant relationships between ASD and SNPs in more than 1000 genes or loci [15, 42]. At least 5% of ASD cases have been attributed to SNPs in NLGN3, NLGN4, NRXN1, MECP2, SHANK3, FMR1, TSC1/2, and UBE3A [15], although there are some discordant results. Due to the extensive heterogeneity of clinical symptoms in ASD, many genome-wide association studies (GWAS) have attempted to link specific clinical symptoms with specific genetic variations. Hu et al. [43] identified 18 significant SNPs after separating ASD patients into four sub-types. However, when all patients were analyzed as a single cohort, no clear associations were found between the SNPs and ASD. Although one study reported that SNPs have a minor effect on the etiology of ASD [44], a growing number of more recent studies have found that SNPs are associated with ASD [11, 45]. In addition to the common SNPs mentioned above, Rabaya et al. [46] investigated SNPs in different iron metabolism genes in relation to ASD. They found that the frequencies of SNPs rs11915082 (TFRC gene) and rs1439816 (SLC40A1 gene) were significantly different between the ASD and control groups, and concluded that specific mutations in genes involved with iron metabolism might serve as biomarkers for the early diagnosis of ASD [46]. A meta-analysis also found that the RIT2 rs16976358 SNP was significantly associated with ASD in the Asian population [47]. In a study of 243 ethnic Han Chinese children aged 2–10 years with ASD, Liu et al. [13] found that KCND2 was a genetic risk factor for ASD. Furthermore, these authors reported that three SNPs (rs1990429, rs7800545, and rs7793864) in KCND2 were associated with a reduced risk of developmental ASD, and that rs1990429 and rs7800545 can attenuate the symptoms of restricted repetitive behavior (RRB) in ASD. The KCND2 gene located on 7q31.31 codes for the voltage-gated potassium channel Kv4.2, which has important roles in the function and plasticity of synapses (Table 2, Ref. [13, 46, 47, 48]).
| Gene | Variant | Consequence | Reference |
| TFRC | rs11915082 | Affects iron metabolism and neurodevelopment. | [46] |
| SLC40A1 | rs1439816 | Potential early biomarkers of ASD. | |
| RIT2 | rs16976358 | Involved in many signaling pathways affecting neurogenesis, neuronal differentiation, and neurite growth and branching. | [47] |
| Associated with ASD in the Asian population. | |||
| KCND2 | rs1990429 | Encodes Kv4.2, which is involved in synaptic function and plasticity. | [13] |
| rs7800545 | Reduces the risk of developmental ASD. | ||
| rs7793864 | |||
| CC2D1A | rs747172992 rs1364074600 |
Regulate multiple intracellular signaling pathways affecting neuronal differentiation and neurodevelopment, and activate protein kinase B (PKB) and nuclear factor kappa-B (NF-kB). | [48] |
Associations between SNPs and ASD are continually being confirmed. Moreover, two novel SNP variants in CC2D1A (rs747172992 and rs1364074600) have been reported to be associated with ASD [48]. These findings provide new insights into the pathogenesis and genetic etiology of ASD, while hinting at the complexity of this condition. Meanwhile, the precise contribution of these variants to ASD remains to be conclusively established.
The formation and maturation of synapses is crucial for the development of brain neural circuits, and synaptic dysfunction is a potential pathogenic factor in developmental disorders of the brain. ASD-like symptoms may be related to synaptic dysfunction, and hence the role of genes related to synaptic function has received widespread attention in the pathogenesis of ASD. Mutations in FMR1, NRXN, NLGN, SHANK, MECP2, and potentially other genes, have been linked to damaged synaptic formation, maturation, or plasticity. Therefore, developmental disorders and dysfunction of synapses may play an essential role in the pathogenesis of ASD. A brief overview of the roles of some synaptic function-related genes is shown in Fig. 1.
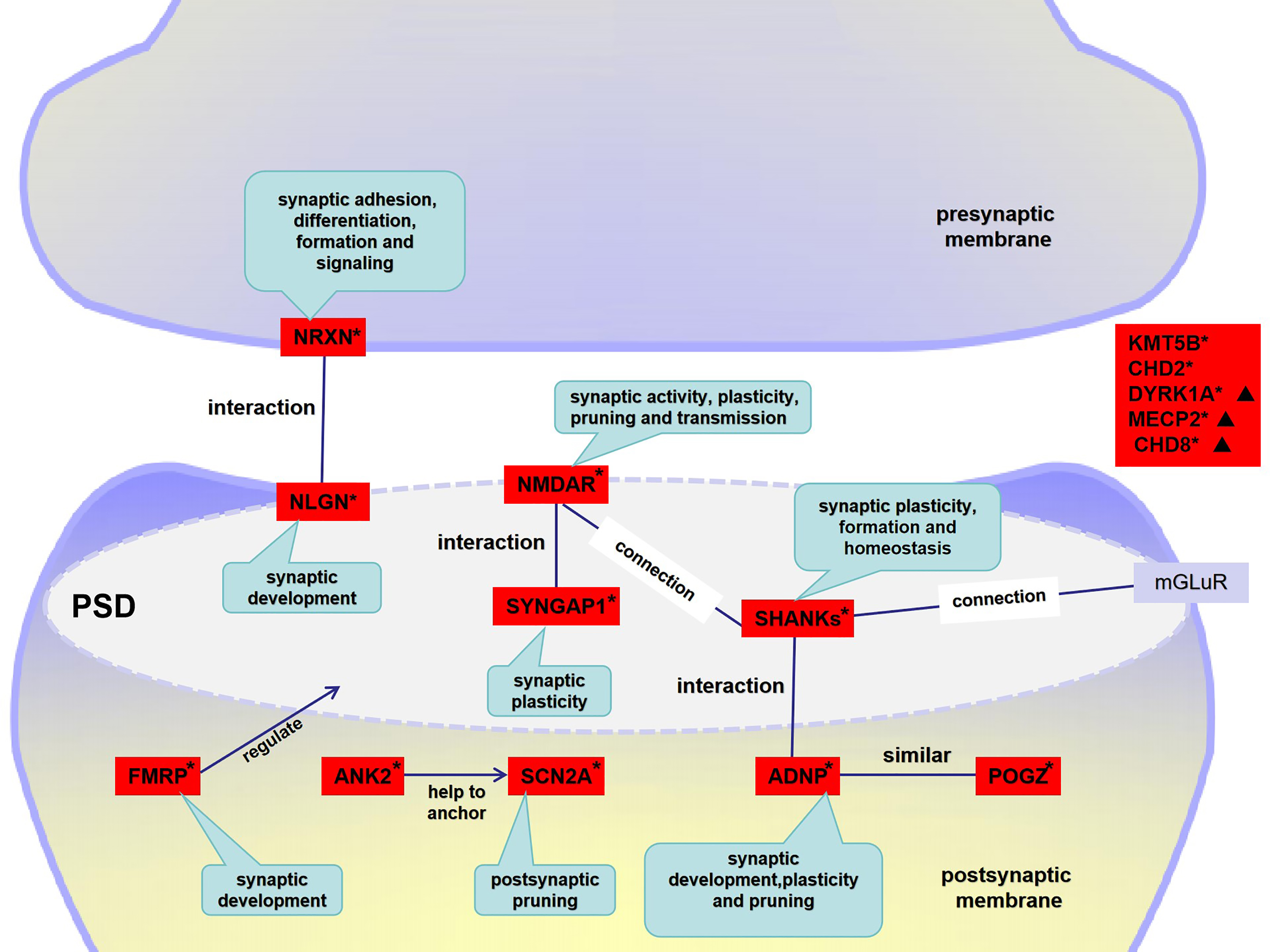 Fig. 1.
Fig. 1.
The roles of genes in synaptic function. This is recognized as a potential pathogenesis of neurodevelopmental disorders. The figure depicts the individual roles of synaptic function-related genes in the pathogenesis of ASD. PSD: the post-synaptic density is a large macromolecular complex consisting of more than 1000 proteins. Several hundred potential fragile X messenger ribonucleoprotein (FMRPs) target mRNAs, including encoded synaptic proteins such as N-methyl-D-aspartate receptor (NMDARs), mGluR5, PSD95, SHANK1-3, and Neurexins. *A fundamental pathological mechanism would be alteration of the NMDAR pathway and disruption of synaptic transmission and excitatory/inhibitory (E/I) balance due to genetic mutation. ▲Genes playing a role in chromatin remodeling could affect synaptic function. ANK2, ankyrin 2; ADNP, activity-dependent neuroprotector homeobox; CHD, chromodomain helicase DNA binding protein; DYRK1A, dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 1A; MECP2, methyl CpG binding protein 2.
Fragile X Syndrome (FXS) is the most prevalent cause of hereditary ID, accounting for approximately 1%–2% of all ID cases. Additionally, FXS is the leading single-gene disorder associated with ASD, contributing to about 60%–74% of such cases. Mutations in the FMR1 gene are the direct cause of FXS. Remarkably, around 2%–5% of individuals with ASD are diagnosed through FMR1 mutation testing [49].
The FMR1 gene codes for fragile X messenger ribonucleoprotein (FMRP), which regulates protein translation by binding to mRNA. FMRP plays a significant role in neurodevelopment, particularly in the pre- and post-synaptic development phases [50]. High-throughput screening methods have recently been used to identify several hundred potential FMRP target mRNAs, many of which are linked to ASD and encode synaptic proteins such as N-methyl-D-aspartate glutamate receptors (NMDARs), mGluR5, PSD95, SHANK1-3, HOMER1, and Neurexins [51]. Dysfunction of FMRP, particularly in relation to its translational activity, is considered to be a fundamental cause of ASD-related synaptic dysfunction and cognitive deficits. FMR1 KO mice exhibit increased local translation of NLGN mRNA, with neuroligin 1 (NLGN1) and NLGN3 specifically targeting the postsynaptic membrane [52].
In mouse models of FXS, the interaction of mGluR5 with the short HOMER1a isoform is enhanced in the hippocampal CA1 area, while its interaction with the long HOMER subtypes is reduced. This alteration leads to a significant increase in the post-synaptic fluidity of mGluR5 [53]. Furthermore, Hurley et al. [54] reported that (R, Z)-3–[6-(dimethyl-amino)-6oxohex-1-en-1-yl)-N-(1-hydroxypropan-2-yl)-benzamide (VSN16R), a cannabinoid-like activator of large-conductance calcium-activated potassium channels, could ameliorate behavioral deficits in a mouse model of FXS. These behavioral changes included less repetitive behavior, improvements in hippocampal-dependent daily living tasks, decreased hyperactivity, and improved memory.
SHANK (also known as ProSAP) is a major scaffold protein that determines the post-synaptic density (PSD) of excitatory synapses in the brain. The SHANK family is composed of three members, namely SHANK1, SHANK2, and SHANK3, encoded by the SHANK1-3 genes, respectively. They all share the following similar domains: an N-terminal ankyrin repeat, a Src-homology 3 (SH3) domain, a PSD95/discs large/ZO-1 (PDZ) domain, an extended proline-rich region, and a sterile motif (SAM) domain. SHANK proteins are related to cell adhesion molecules and are involved in actin remodeling. They also enhance synaptic formation and homeostasis, which are crucial for dendritic spines and the proper formation of excitatory synapses [55].
PSD is a large macromolecular complex consisting of more than 1000 proteins,
including membrane-tethered receptors and channels (e.g., N-methyl-D-aspartate
glutamate receptors (NMDARs), mGluRs, and
The SHANK1 gene is located in the 19q13.33 region. Sato et al.
[59] found SHANK1 deletions in two multiplex ASD families and reported
that these were associated with higher functioning ASD in males. In the maze
test, SHANK1 KO mice showed decreased motor function, impaired fear
conditioned memory, and increased spatial memory [60]. Qin et al. [61]found a specific recurrent missense mutant in SHANK1, c.2621 G
SHANK2 is located in the 11q13.3-q13.4 region [62]. Mutations in
SHANK2 include deletions, missense mutations, truncations, and
alterations in the promoter region. Leblond et al. [63] identified three
ASD sufferers with de novo deletions in SHANK2 (truncations),
one of whom carried a paternally inherited deletion of the synaptic translation
repressor CYFIP1. SHANK2 KO mice show ASD-like behavior. However,
SHANK2 e6–7 KO mice exhibit hypo-function of NMDAR and impaired LTP,
whereas SHANK2 e7 KO mice show hyper-function of NMDAR and enhanced LTP
in the adult hippocampus [57]. Another study reported that restoration of NMDAR
function improved the social behavior of SHANK2-mutant mice [64].
Recently, Wu et al. [62] identified two novel mutations in
SHANK2, one of which was a heterozygous splicing substitution
(NM_012309.5:c.2198-1G
SHANK3 is a master scaffolding protein encoded by SHANK3 located at 22q13.3. It is mainly responsible for synapse construction, and for maintaining synapse structure and function through glutamate receptors and interactions with the cytoskeleton [55]. 22q13.3 deletion syndrome, also known as Phelan McDermid syndrome (PMS), is characterized by developmental delay with disrupted SHANK3. More than half of PMS individuals show ASD-like behaviors [65]. Durand et al. [66] first reported an ASD sufferer carrying a de novo deletion mutation of SHANK3 in 2007. Subsequently, Gauthier et al. [67] reported a novel SHANK3 de novo deletion mutation in ASD. Since then, many mutations in SHANK3 have been reported in ASD sufferers. SHANK3 mutations lead to decreased PSD proteins in neurons, including guanylate kinase-associated protein (GKAP), HOMER1b/C, the AMPA-type glutamate receptor subunit-1 (GluA1) subunit of AMPAR, and the NR2A subunit of NMDAR [58, 68]. SHANK3a/b homozygous mice exhibit more severe reductions in synaptic transmission and LTP in the hippocampal CA1 compared with heterozygous mice [69]. SHANK3a/b KO mice have lower parvalbumin signals in the thalamic nuclei and are more prone to epilepsy after treatment with kainic acid compared with wildtype mice [70]. These results suggest that SHANK3 isoforms are essential for normal hippocampal function.
Various SNPs in SHANK3, in particular rs9616915, have been studied for
their association with ASD, with mixed results across different populations.
Study in Northern Chinese Han, Bangladeshi, Japanese, and Caucasian populations
found no association, whereas associations with ASD were observed in Iranian and
Wenzhou populations [55]. SHANK3 mutant mouse models have been
invaluable in understanding ASD neurobiology. A novel model that combined
SHANK3
NLGNs are a group of cell adhesion factors located in the neuronal post-synaptic membrane. They are single transmembrane proteins composed mainly of a large ectodomain (including an acetylcholinesterase homology domain, EF-hand type motif, and O-glycosylation domain), a single pathway transmembrane domain, and intracellular domains. The five NLGN proteins in humans are NLGN1, NLGN2, NLGN3, NLGN4, and NLGN4y. NLGN1 is mostly expressed at excitatory post-synaptic membranes [72], and NLGN2 at inhibitory postsynaptic membranes [73]. NLGN3 and NLGN4 are expressed at both excitatory and inhibitory postsynaptic membranes [74]. Recently, it was reported that human NLGN4 is highly expressed in the cerebral cortex and especially at excitatory synapses. NLGN4 is located almost exclusively in neurons, with a wide cytoplasmic distribution pattern [75]. NLGNs have a significant effect on the development of functional synapses. They bind to axonal proteins (neurexins) of the pre-synaptic membrane to regulate the balance between excitatory and inhibitory synapses. Jamain et al. [76] were the first to report an association between mutations in Nlgns and ASD. These authors found an r45lc missense mutation in Nlgn3 and a c.1186insT frameshift mutation in Nlgn4 on the X chromosome in two ASD families from Sweden. Xu et al. [77] identified four rare missense mutations through direct sequencing of 318 Chinese Han with ASD: p.g426s (Nlgn3), p.g84r (Nlgn4), p.q162k (Nlgn4) and p.a283t (Nlgn4). Each of these mutations was located in the ectodomain of Nlgns, namely the acetylcholinesterase homology domain. This is an essential domain that triggers synapse activity by binding to axonal proteins [77]. Several mouse models have been used to explore the contribution of the Nlgns gene to ASD pathogenesis. Almost all these models show disorders of neurotransmitter transmission in excitatory or inhibitory synapses, with the study finding changes in hippocampal synaptic density and morphology [78]. Nlgns mutant mice exhibit some behaviors that resemble ASD symptoms. For example, Blundell et al. [78] reported that Nlgn1 KO mice show poor social behavior, impaired spatial learning and memory, and repetitive stereotypical grooming behavior. Moreover, the hippocampus in these KO mice shows significant increases in synaptic number, length, and connections, and increased dendritic spine density in excitatory synapses. Consistent with this, electrophysiological study showed a significant increase in the ratio of excitatory post-synaptic potential (EPSP) and inhibitory post-synaptic potential (IPSP) (E/I), resulting in impaired LTP [79]. Similar phenotypes were also found in other mouse models of Nlgn2, Nlgn3, and Nlgn4. The expression of NLGN2 and NLGN3 were both elevated in a maternal diabetes-related, streptozotocin-induced ASD mouse model. Using cryogenic electron microscopy (cryo-EM), the NLGN structures were found to be highly conserved in both NLGN2 and NLGN3. The small differences between them possibly cause large differences in function. NLGN2 can inhibit synaptic transmission in neurons, and NLGN3 can control AMPAR-mediated basal excitatory transmission [80]. The Nlgn3 R451C mutant mouse model shows impaired social communication, a core autism-like phenotype, but increased ability for spatial learning. One pathogenic feature was increased synaptic turnover, which was also observed in the 15q11-13 duplication model [81]. Another mechanism could be imbalance of inhibition/excitation (I/E) inputs to Purkinje cells, leading to synaptic development disorder in the cerebellum [82]. Interestingly, two brothers with the Nlgn3 R451C mutation showed gastrointestinal tract dysfunction, possibly caused by an imbalance in the activity of neurons in the gastrointestinal tract [83]. Furthermore, Nlgn3 mRNA was found in most submucosal and myenteric neurons, and Nlgn3 was also strongly expressed in enteric glia, indicating that neuroligin-3 plays a role in mediating enteric neuron–glia interactions [84]. Increased paired-pulse inhibition of dentate granule cell population spikes due to altered network activity can be observed in the hippocampal dentate gyrus of adult Nlgn4 KO mice [85]. NLGN4 is expressed in different types of neurons and is highly expressed in oxytocin (OXT)/vasopressin (AVP) producing cells in the hypothalamus. Most OXT/AVP-producing neurons express NLGN4, which is involved in intelligence, social abilities, sleep, and arousal. Deficiencies in these are common in autism [75].
The NRXN genes encode
The MECP2 gene located on Xq28 encodes for methyl CpG binding protein 2 (MECP2). MECP2 is a calcium-dependent transcription repressor critical for heterochromatin formation and for maintaining chromosomal structure and transcriptional regulation. MECP2 is located in the Xq28 region in humans [93]. It is predominantly expressed in neurons, with its expression increasing after birth in parallel with neuronal maturation and synaptic formation during developmental stages [93, 94]. MECP2 protein facilitates various brain functions, including neuronal growth, development of dendritic spines, synaptogenesis, enhancement of neural circuits, and synaptic plasticity [94].
Mutations in MECP2 lead to neurodevelopmental disorders such as Rett syndrome and ASD, although it is not a prevalent cause of the latter. In murine models, MECP2 deletions are associated with reduced brain weight, neocortical thinning, and delayed neuronal and synaptic development, resulting in lost synaptic plasticity and disrupted excitatory synaptic transmission [95]. These disruptions manifest as broad defects in synaptic maturation that affect learning and memory, thereby explaining the ASD-like features observed in affected individuals [95]. Conversely, MECP2 overexpression can also impair synaptic integrity and present with an autistic phenotype. MECP2 duplication syndrome is a subtype of ASD that is associated with various behavioral phenotypes [96]. It is characterized by significant disruption to neurological development, altered gray matter volumes in the hippocampus and thalamus, and dysfunctional development in a core network comprising the dorsal medial prefrontal cortex (dmPFC) and retrosplenial cortex (RSP). Furthermore, transgenic rats that overexpress human MECP2 show increased anxiety and social behavioral disorders but not movement disorders [97].
MECP2 duplications in males often result in ID, language disorders,
anxiety, and ASD behaviors [98]. MECP2 transcriptionally regulates the
expression of other genes that impact brain development. For example, BDNF is a
critical protein for brain development, maintenance, and function, and its
expression level is altered in MECP2 KO and overexpression models [94].
BDNF interacts with tropomyosin-related kinase B (TrkB) to activate downstream
signaling pathways such as the mitogen-activated protein kinase (MAPK), the
phosphoinositide-3 kinase (PI3K), and the phospholipase C
MECP2 deficiency also reduces the expression of ubiquitin protein ligase E3A (UBE3A) and GABRB3. Significant downregulation of MECP2 is observed in MECP2-deficient models and in human brains affected by Rett syndrome, Angelman syndrome, and autism, suggesting a shared mechanism of gene dysfunction in the 15q11–q13 region [100]. MECP2 also regulates fibroblast growth factor 13 (FGF13), a critical accessory protein for voltage-gated Na+ channels that are essential for maintaining synaptic E/I balance [101]. Interactions have been reported between MECP2 and the PTEN gene, which is frequently mutated in ASD, with phosphatase and tensin homolog (PTEN) involved in neural development processes such as synaptic formation [102]. Additionally, transcription factor 20 (TCF20) modulates downstream pathways of MECP2, potentially ameliorating MECP2-induced synaptic and behavioral dysfunction. Mutations in this complex can potentially lead to neurological deficits [103]. These insights highlight the multifaceted role of MECP2 in brain development, which is mediated through the regulation of, and interaction between, multiple genetic pathways.
Chromodomain helicase DNA binding protein (CHD) regulates gene expression by
remodeling chromatin structure and influencing histone acetylation. CHD8 is a
transcription regulator encoded by the CHD8 gene located in the 14q11.2
region. It acts as a regulator in many biological pathways [104], including Wnt
signaling, by binding to
CHD8 mutations are much more commonly found in male ASD sufferers, with a notable characteristic being the large head malformations found in 85% of cases [104]. Almost 4% of people diagnosed with autism have CHD8 mutations [20]. CHD8 deficiency impairs neuronal axon development and migration [107], and impairs synapse function by altering gene expression and chromatin remodeling [20]. Individuals with CHD8 mutations exhibit autism behaviors and other comorbidities, including social deficits and communication disorders, repetitive behaviors, anxiety, and cognitive delays [107]. In vitro and animal study has shown that CHD8 deficiency is causally related to neural cell proliferation, synaptic function, myelin formation, and ASD-like behaviors [107]. In mice, homozygous CHD8 KO is embryonically lethal [108], while CHD8+/- mouse models exhibit neuronal delay, increased brain capacity, anxiety, ASD-like behaviors, and gastrointestinal abnormalities [104, 107]. Abnormalities in the gastrointestinal tract can affect the symptomology and behavioral phenotypes of autism [109]. In Drosophila, CHD8 can interact with RIMS1 and participate in the plasticity of pre-synaptic homeostasis and the stability of synaptic transmission [104].
People with haploinsufficiency of CHD2, another member of the CHD family, show recurrent clinical symptoms including developmental delay, ID, epilepsy, behavioral problems, and autism-like features. This may be due to altered synaptic transmission, with an imbalance of E/I synaptic interactions [110].
N-methyl-D-aspartate receptor (NMDAR) is a glutamate receptor and also a ligand-gated ion channel [111, 112]. It is composed of two obligatory glycine-binding GluN1 subunits and two glutamate-binding GluN2 or GluN3 subunits, encoded by the GRIN1, GRIN2A–D, and GRIN3A–B receptor genes, respectively. NMDAR plays an important role in regulating synaptic activity [113]. Each NMDAR subtype has a different temporal and spatial expression pattern in various cells, resulting in different functions. These play vital roles in excitatory neuron transmission, brain development, and synaptic plasticity as well as learning, memory, and advanced cognition [114]. The GluN2B sub-type plays an essential role in synaptic plasticity, channel function and dendritic spine formation [112]. GluN2B mutations impair dendrite growth and development by interfering with NMDAR signal transmission [111] and by promoting retraction and pruning [114].
Many animal models of ASD show NMDAR dysfunction, suggesting that they share molecular mechanisms with changing NMDARs functions. Mice with neuroligin-1 deletion show reduced NMDAR function in the hippocampus and striatum [115]. Functional reduction of NMDAR in the hippocampus was also observed in a mouse model with deletion of exons 6 and 7 in SHANK2 [57]. In contrast, enhanced NMDAR function is observed in mice that lack only exon 7 of SHANK2 [57], as was also observed in IRSp53-/- mice [116]. IRSp53, also called BAIAP2, is a scaffolding protein that regulates excitatory synaptic dendritic spines [116]. The above findings suggest that both enhanced and reduced NMDAR functions in the brain can induce an ASD-like phenotype. As part of the glutamate receptor, SHANK3 connects NMDAR with mGlu5 through interaction with the complex scaffolding protein PSD95-GKAP-SHANK3-HOMER [117]. In addition, KO of the Dock4 gene (located at 7q31.1) [118] or of the Pcdh10 gene [119] can impair NMDAR function. Zinc has also been shown to regulate NMDAR function and to enter the post-synaptic membrane via NMDARs [120]. Moskal et al. [121] reported that GLYX-13, an NMDAR-modulating drug, may have potential for ASD intervention.
The SYNGAP1 gene is another risk factor for ASD. It encodes the SYNGAP1 protein, which is mainly expressed in the excitatory neuronal post-synaptic density (PSD) [122]. SYNGAP1 protein interacts with NMDAR to negatively regulate the Ras and Rap GTPase pathways. By inserting AMPA receptors into the post-synaptic membrane, Ras signaling can induce and maintain LTP. The Rap pathway regulates long-term depression (LTD) via p38MAPK [123]. SYNGAP1 plays a vital role in synaptic function and plasticity [122, 123, 124]. The characteristics of SYNGAP1-Related Intellectual Disability (SYNGAP1-ID) are developmental delay, epilepsy, ID, sleeping disorders, and behavioral problems, with an ASD prevalence of 50%–73% [124]. Mice with SYNGAP1 mutations have severe cognitive disorders, including social and memory defects [123]. Heterozygous mice with SYNGAP1 deletions show impaired synaptic plasticity, dendritic spine morphogenesis, and learning [125].
Neuronal activity has been shown to control the expression of KLHL17 protein through NMDAR signaling and protein synthesis. KLHL17 regulates the distribution of synaptopodin in the synapse, both of which are actin-binding proteins related to ASD. Mouse genetic models further demonstrate that KLHL17 haploinsufficiency and KO lead to decreased dendritic spines containing endoplasmic reticulum (ER) clusters, activity-dependent dendritic spine enlargement, and impaired neuronal activation. These models found that KLHL17 plays a role in controlling synaptic plasticity by regulating synaptopodin and synaptic ER clustering, and suggest that dysfunction of synaptic plasticity contributes to the pathogenesis of KLHL17-related diseases [126].
Interestingly, ASD individuals with ANK2 mutations have a normal IQ. ANK2 is a rare but high-confidence autism-associated gene. It encodes for giant ankyrin-B (ankB) protein, which is expressed only in the nervous system. A mouse model (giant ankB mutant mice) mimics the human ANK2 frameshift mutation in exon 37, with similar findings of normal brain morphology and cognitive function. However, the number of axonal branches is increased and ectopic connections are also observed [127]. It was reported that ANK2 regulates neural differentiation and neuronal migration, and that mutations of ANK2 cause neurodevelopmental disorders associated with an increased risk of ASD [128]. ANK2 is not directly involved in synaptic function. Instead, its function is to ensure that other membrane proteins anchor in the correct domain. For example, ankB helps Nav1.2 to localize in the dendrites, where the two proteins interact to regulate dendritic excitability in neocortical pyramidal cells [129].
Nav1.2 is encoded by SCN2A, one of the common autism-related genes. This protein has an important regulatory role in axonal excitability during development. In addition, Nav1.2 also has an important role in maintaining dendritic excitability and the synaptic function of pyramidal cells in the mature PFC [130]. A recent study found that SCN2A deficiency can lead to excessive pruning of post-synapses by microglia, which are the brain’s immune cells [131]. Neuronal dysfunction could activate partial microglia-like traumatic brain injuries, causing synapses to be over-pruned. SCN2A-deficient mice show reduced synaptic transmission and lower spine density, resulting in impaired learning and memory [131].
Conrow-Graham et al. [132] reported an increased number and function of
microglia in mice with POGZ or ADNP deficiency. This may lead
to synaptic over-pruning and the dysfunction of glutamatergic transmission. A
2016 study indicated that mutations in the POGZ gene are associated with
ID and ASD [133]. Moreover, the authors postulated that the resulting changes
could increase protein complexes that modify chromatin structure and gene
regulation. This view has been confirmed in subsequent studies. Matsumura
et al. [134] created a mouse model that was heterozygous for POGZ
mutation. These authors found that POGZ was able to regulate
transcriptional networks to control neuronal differentiation through chromatin
remodeling, and then complete the regulation of cortical excitatory neuron
development. Recent studies have shown that POGZ forms a complex with activity-dependent neuroprotector homeobox (ADNP) to
co-regulate transcription, suggesting that they have a similar or shared
mechanism, such as activation of microglia in the PFC or regulation of the
AMPAR/NMDAR-mediated synaptic signal pathway [132, 135]. Furthermore, POGZ and
ADNP have connections with other autism-related genes. For example, POGZ
and CH8 share common downstream targets [135] and ADNP is
connected to SHANK3 through SH3- and actin-binding domains [136]. A
recent study found that hyperphosphorylation of CaMKI
The dual-specificity tyrosine-(Y)-phosphorylation-regulated kinase 1 A (DYRK1A) gene is located on chromosome 21. This regulatory gene plays an important role in neural development and has been implicated as a risk factor for autism. Dysregulation of DYRK1A dosage can impair dendritic growth, dendritic spine density, cortical migration, synaptic transmission, and plasticity [138, 139]. DYRK1A is involved in many regulatory signals, including the REST/NRSF-SWI/SNF chromatin remodeling complex, AMPARs/NMDARs, tropomyosin receptor kinase B (TrkB)-BDNF (also seen in MECP2), ERK/MAPK, and mammalian target of rapamycin (mTOR) signaling [139, 140].
In addition, the decreased glutamatergic transmission observed in KMT5B-deficient mice has been attributed to decreased expression of the AMPAR and NMDAR subunits, and to increased expression of DNA-damage-inducible transcript 4 (DDIT4), a downstream target of p53 [141]. The dysfunction of synaptic transmission has also been observed in other models [25, 92, 129, 132]. KMT5B encodes a lysine methyltransferase consisting of a histone H4 K20 di- and tri-methyltransferase with an important role in neuronal development in the PFC. KMT5B is also a high risk autism-related gene [142]. Pathogenic mutations in KMT5B cause an autism-like phenotype, with social deficits and/or macrocephaly [141, 142]. This occurs through the disruption of normal epigenetic regulation during the neurodevelopmental process [142], thus inducing synaptic dysfunction and transcriptional aberration [141].
Synaptic pruning is crucial for resolving disorders of synaptogenesis, synaptic neurotransmission, and synaptic plasticity as well as for the proper functioning of several ASD risk genes, including MEF2C, FMR1, DLG4, and Pcdh10. This process plays a vital role in maintaining the proper balance and functionality of neural circuits, which are fundamental for normal cognitive development and behavior.
Synapse formation occurs predominantly during prenatal brain development. This process leads to the creation of more synapses than are physiologically required, which can result in pathological conditions if not adequately pruned. In later developmental stages, the number of synapses is reduced through the process of pruning, ensuring that only functional synapses are retained. This selective elimination is essential for the accurate formation and maintenance of mature neuronal circuits, upon which cognitive function and expected behavioral outcomes are heavily dependent.
Research has shown that the absence of MEF2C in mice leads to a significant increase in the number of excitatory synapses, whereas hyper-activation of MEF2C results in a decrease in the number of excitatory post-synaptic sites. MEF2C works in conjunction with FMRP to regulate the expression of Protocadherin-10 (Pcdh10). Activation of MEF2C leads to ubiquitination of PSD95 by the ubiquitin E3 ligase Murine Double Minute-2 (Mdm2), thereby targeting it for proteasomal degradation mediated by Pcdh10 [143, 144, 145]. Ube3a E3 ligase is crucial for the control of synaptic number and activity. It can inhibit pre-synaptic bone morphogenetic protein (BMP) signaling to promote pre-synaptic elimination, thus effectively managing the synaptic landscape. Conversely, increased pre-synaptic Ube3a activity can precipitate early synapse elimination and disrupt synaptic transmission [146].
The study on Pcdh10 KO mice has highlighted the significant role of Pcdh10 in the development of excitatory synapses in the dorsal basolateral nucleus of the amygdala, with no observable defects in axon development [147]. These findings link Pcdh10 to the regulation of behaviors associated with anxiety, fear, and stress [147]. Furthermore, study of the medial PFC suggests that microglia influence the maturation of both excitatory and inhibitory synapses through various complement-dependent, phagocytic, and non-phagocytic signaling pathways [148].
Together, these findings indicate that disruptions in synaptic pruning may underpin the cellular and neuronal pathogenesis of ASD, highlighting its importance in neurodevelopmental disorders.
Animal models have contributed greatly to the study of ASD. In this review, results from models including 16p11.2dp/+ mice show that the number and transmission of excitatory synapses is increased, with loss of E/I balance, similar to observations in the human brain. Similarly, mouse models with CNTNAP2 or SCN2A deficiency show an E/I imbalance, but with reduced transmission of excitatory synapse. In addition, studies have shown that mutations in FMR1, SHANKs, NRXNs, NLGNs, SYNGAP1, CHD2, SCN2A, POGZ, ADNP, and DYRK1A share pathways with altered NMDAR (glutamate receptors). These findings suggest that dysfunctional synaptic transmission and E/I imbalance represent a baseline pathological mechanism for ASD. Different genetic variants share a common molecular pathway. For example, changes in oxidative phosphorylation and Rho family small GTPase signaling at the synapse in several mouse models including FMR1 KO mice; TrKB-BDNF alterations caused by MECP2 or DYRK1A mutation can influence neuronal differentiation, growth, and plasticity; genetic mutations can enhance microglia activity, resulting in excessive pruning of synapses and reduced transmission of excitatory synapses. These all suggest that the E/I imbalance caused by alteration of synapse formation, remodeling, and transmission function is an important pathological mechanism in ASD and may be achieved through several common molecular pathways.
With advances in gene sequencing technologies, the role of hereditary factors in the pathogenesis of ASD has garnered increasing attention. The discovery of numerous risk genes for ASD highlights their critical role in various mechanisms that are characterized by complex and overlapping signaling pathways. This also underscores the intricate and multifaceted nature of ASD inheritance. Despite this progress, the specific contributions of individual genetic components to ASD remains challenging to fully delineate because of the diversity of genetic abnormalities and the number of genes involved.
Focusing on the genes associated with synaptic function should eventually lead to an increased understanding of the genetic and neuronal mechanisms underlying ASD. This approach should deepen our comprehension of ASD and also facilitate the development of tailored interventions that could significantly improve the quality of life of those affected by this condition.
YZ and RT identified appropriate references, drafted and the manuscript and prepared the tables and figures. ZMH and XHW identified appropriate references, drafted the manuscript. XG and TW drafted the manuscript, they also analyzed and interpreted the data. MXT designed the literature retrieval strategy, reviewed the manuscript, and obtained funding. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We express our gratitude to all those, excluding the authors, who helped during the writing of this manuscript.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.