
 , Wenliang Ji 2,*
, Wenliang Ji 2,*1 Department of Physical Education, Henan Normal University, 453007 Xinxiang, Henan, China
2 College of Chemistry, Beijing Normal University, 100875 Beijing, China
Abstract
Alzheimer’s disease (AD) is ranked as the third-most expensive illness and sixth
leading cause of mortality. It is associated with the deposition of extracellular
amyloid-
Graphical Abstract
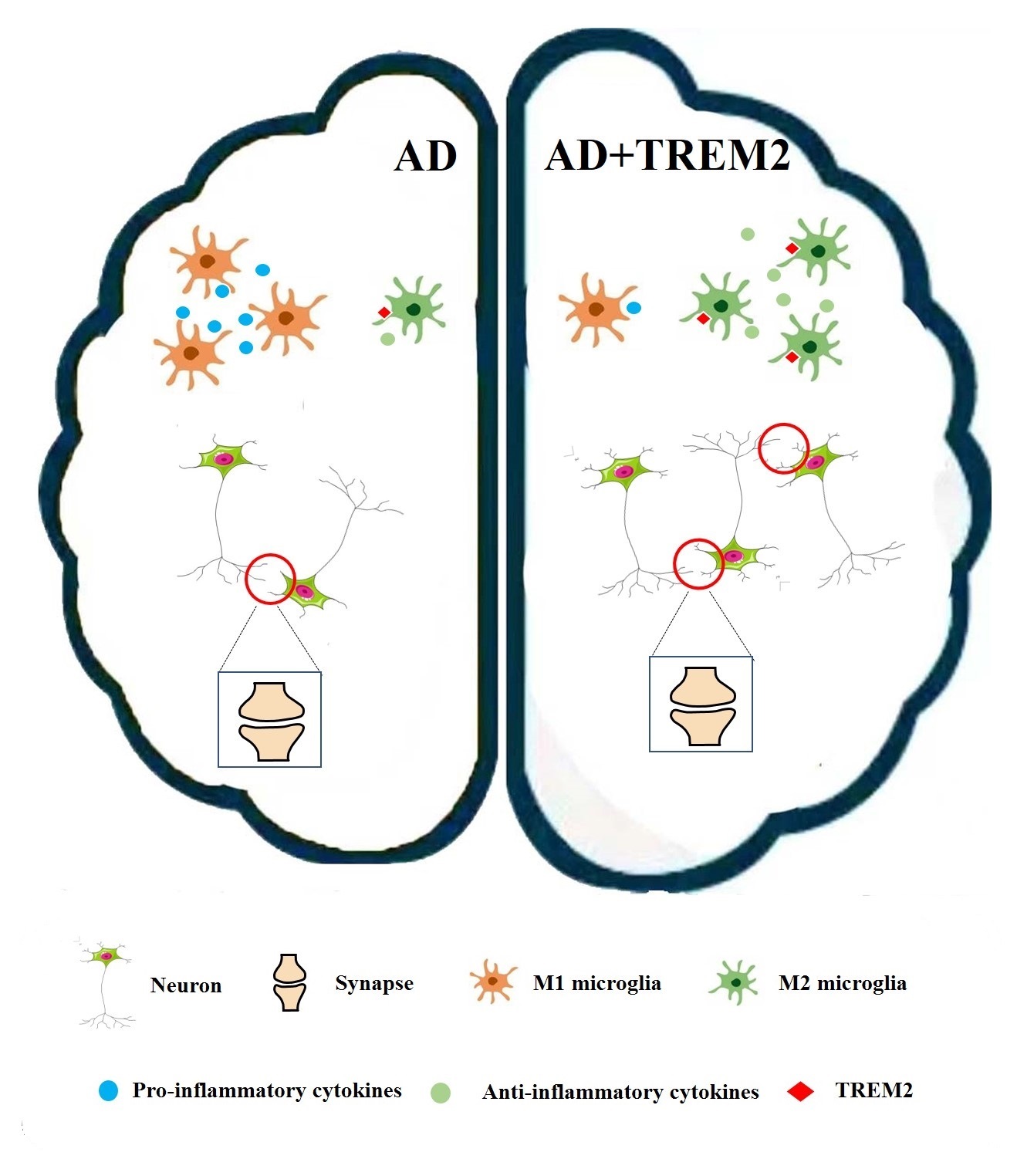
Keywords
- AD
- pathogenesis
- neuroinflammation
- TREM2
Alzheimer’s disease (AD) is an irreversible and progressive neurological
disorder [1, 2]. Various pathological factors are found in the postmortem brains
of AD patients, such as abnormal amyloid-
Triggering receptor expressed on myeloid cells 2 (TREM2) is expressed in myeloid
progenitors. It is an innate immune receptor that is located on the microglial
membrane. Findings from genome-wide association experiments have revealed rare
TREM2 (R47H) mutations that are linked to an increased possibility of developing
AD, as TREM2 is one of the most important receptors because of it’s effects on
neuronal/microglia health [13]. The type I transmembrane glycoprotein TREM2
communicates with an immune adaptor DNAX activation protein of
12 kDa (DAP12) transmembrane area to facilitate signaling via the immune-receptor tyrosine-based activation motif (
ITAM) domain of DAP12 in migration, phagocytosis, cell survival, and inflammatory
cytokine release [14, 15]. TREM2 is also implicated in the etiology of AD as it
regulates microglial function and innate immunity [16]. The function of TREM2 in
neuroinflammation and its relationship with AD have not been fully elucidated.
This review focuses on current understanding of the pathogenesis of AD and the
shift from the initial focus on abnormal A
AD is the most widespread age-related neurodegenerative disorder, with no
effective cure that can help alleviate the financial and emotional burden of the
disease [17, 18]. Studies on the pathogenesis and development of therapies are
vital for preventing or alleviating AD symptoms. A number of hypotheses explain
AD pathophysiology, including the hypotheses of the A
A
A
Tau pathology is a hallmark of AD progression [32]. Tau present in the medial temporal lobe binds and stabilizes the microtubule architecture, where it is normally concentrated. However, hyperphosphorylated tau protein produced in AD dissociates from microtubules and accumulates inside neurons [33]. Once it spreads from the medial temporal lobe to the surrounding neocortex, hyperphosphorylated tau protein can cause cognitive impairment.
Findings from animal models have shown that A
Cholinergic neurons have been demonstrated to modulate neuronal circuits in the hippocampus and cortical regions and play an important role in hippocampus-dependent memory [40, 41]. Normally, endogenous nerve growth factor (NGF) is produced by hippocampal and post-synaptic cortical neurons and is expressed through related receptors present on presynaptic cholinergic terminals [42, 43]. Stimulated NGF is then transported from the target regions (hippocampus and cortex) to cholinergic nuclei, consequently initiating cholinergic signaling in these brain areas [44].
Findings from various animal models and human studies have shown that cholinergic neuron degeneration in the basal forebrain is linked to cognitive impairments in AD [45, 46]. The cholinergic hypothesis primarily focuses on the gradual damage of limbic and neocortical cholinergic innervations, which is linked to neurofibrillary degeneration and inefficient axonal transport and signaling [47]. The crosstalk between postsynaptic cortical/hippocampal neurons and presynaptic cholinergic terminals is altered during AD. These variations are related to the accessibility of mature NGF (mNGF) to basal forebrain cholinergic neurons, which subsequently results in cholinergic degeneration in hippocampal and cortical areas [48, 49].
The cholinergic hypothesis has transformed different areas of AD research, from the field of neuropathology to the modern concept of synaptic neurotransmission [50]. It was developed based on three parameters: (1) diminished presynaptic cholinergic indicators in the cerebral cortex [51, 52], (2) the nucleus basalis of Meynert (NBM) in the basal forebrain is a source of cortical cholinergic innervation that undergoes intense neurodegeneration in AD [53, 54] (3) cholinergic antagonists impair memory whereas agonists have a contrasting effect on memory [55].
GABAergic activity is important for brain development and
plasticity. Under normal conditions, GABA is produced in the presynaptic membrane
and helps synthesize glutamic acid decarboxylase (GAD) from glutamate. GABA can
bind to GABA receptors present on the postsynaptic membrane through the synaptic
cleft, which inhibits the post-synaptic neuron [56, 57]. However, in AD,
imbalances in GABAergic neurons is associated with several disorders. In the AD
brain, A
These outcomes have shown that GABAergic dysfunction is related to the E (excitation)/I (inhibition) imbalance in AD [59]. As mentioned Govindpani et al. [60], because of the narrow scope of existing treatments for disease modification, better insights into GABAergic remodeling in AD may help introduce innovative and unique therapeutic options.
AD pathology involves the clustering of A
In recent decades, AD investigations have prioritized the mechanisms related to
extracellular neural plaques (NPs) and intracellular neurofibrillary tangles (NFTs). Data from genetic, preclinical, and
clinical findings have focused on the neuroinflammatory mediation of the innate
immune system [66]. Many neurodegenerative diseases, particularly AD, have been
linked to inflammation [67, 68]. Increasing evidence suggests that systemic
inflammation plays a key role in AD [69]. Tau-protein tangles and A
Microglia are the primary macrophages and provide basic immunological protection in disorders and injuries of the central nervous system [75, 76]. Plastic cells have dual functions in neuronal damage and healing and can adopt multiple phenotypes [77, 78]. Microglia have a ramified phenotype under normal physiological conditions, characterized by a small cell body and several processes, such as in “resting microglia”. Even in the resting state, microglial processes are dynamic and constantly scan to preserve central nervous system (CNS) cell types with neurons [79]. Evidently, they scan the brain environment continuously and contact synapses by using their fine branches to detect infections and damage in their environment [80].
Microglia, which represent the immune cells of the brain, engage in diverse
functional programming, known as polarization, for responding to external
stimulating factors [81]. In the activated state, microglial phenotypes are
categorized by two major states: classical activation (pro-inflammatory
microglia) and alternative activation (anti-inflammatory microglia) [82, 83].
Pro-inflammatory microglia primarily release proinflammatory substances, whereas
anti-inflammatory microglia release anti-inflammatory cytokines. When triggered
by lipopolysaccharide (LPS), the pro-inflammatory phenotype is acquired by
microglia, which leads to neurotoxicity via secreted pro-inflammatory factors
such as tumor necrosis factor-
In neurodegenerative conditions, especially in AD models, persistently activated
microglia may restrict CD3
Microglia show different phenotypic states in AD modesl, especially under
chronic inflammatory conditions [89]. Some receptors (receptor for advanced glycation endproducts [RAGE] and
NLRP3) of the proinflammatory microglial phenotype secrete proinflammatory
cytokines, such as TNF-
Receptors located on microglial membranes are composed of soluble and membrane
proteins that can receive various stimuli and trigger a series of responses to
maintain microglial homeostasis [93]. The receptor TREM2 is primarily expressed
on the microglial surface. Substantial evidence has shown that TREM2 has
bioactive potential and the ability to connect with ligands, stimulate microglia,
and regulate the immune system in AD progression, including proliferation,
survival, and phagocytosis. Furthermore, a lack of TREM2 expression has been
shown to increase the accumulation of A
TREM2 is an innate immunological receptor found only on the surface of myeloid cells in the brain, such as microglia, macrophages, and monocytes [95, 96]. Mature TREM2 protein weights approximately 40 kDa. In situ hybridization and immunohistochemical labeling have been used to detect the protein [97]. TREM2 has a long ectodomain that interacts with the extracellular setting to control microglial function [98].
Current research indicates that minor changes in soluble TREM2 (sTREM2) levels (approximately 7%–10%), are sufficient for modulating AD risk [99]. According to Piccio et al. [100, 101], AD and other inflammatory central nervous system diseases have considerably higher CSF sTREM2 levels than normal controls. Deming et al. [102] discovered for the first time that, as a vital contributor in TREM2’s biological processes, the membrane-spanning 4-domains subfamily A (MS4A) gene cluster plays a significant role in regulating soluble TREM2 expression as well as AD pathology.
TREM2 is crucial for cell maturation, proliferation, and survival during development under homeostatic conditions [103, 104]. Multiple TREM2 functions have been identified in the last decade, including the regulation of phagocytosis and modulation of inflammatory signaling [105, 106].
The CNS, with its greater phagocytic potential, has myeloid cell subsets expressing TREM2 [107, 108]. Knockout animal studies have shown reduced phagocytosis in apoptotic neurons [109]. The overexpression or activation of TREM2 has been shown to increase substrate uptake [110, 111]. According to Yao et al. [112], phagocytosis depending on TREM2 requires the activation of the SYK/PI3K/AKT/PLC pathways. A novel TREM2 agonistic antibody treatment accelerated the clearing of myelin debris from the CNS demyelination model [113].
TREM2 is traditionally classified as an anti-inflammatory receptor
[114, 115, 116]. Its silencing stimulates an early pro-inflammatory response through
the PI3K/NF-
Genetic variations in TREM2 have been linked to several neurodegenerative diseases [121, 122]. Recent research has shown that microglial TREM2 expression is associated with AD pathology. The most common AD-associated TREM2 variant is rs75932628, a single-nucleotide polymorphism encoding an arginine-to-histidine missense substitution at amino acid 47 (R47H), which significantly increased sporadic AD incidence and impaired phospholipid ligand binding [123, 124]. Recent studies have proposed that R62H and R47H are partial loss-of-function variants and predominantly lead to minimized affinity for TREM2 ligands [125]. In contrast, upregulated TREM2 signaling can lead to several possibilities for immune-linked AD treatments [126, 127]. The regulation of TREM2 in AD represent a novel therapeutic approach that is currently under investigation in clinical trials [128, 129]. Therefore, microglial TREM2 is a potential therapeutic target for ameliorating neurodegeneration disease. However, caution should be exercised when targeting TREM2 as a therapeutic entry point for AD until its involvement in tau aggregation and propagation is better understood [130].
The accumulation of neurotoxic forms of tau and A
Recent evidence demonstrates that TREM2 insufficiency has varying effects on AD progression and initially suppresses amyloid pathology. However, the pathology is exacerbated in later disease stages [138]. Furthermore, deficiency of microglial TREM2 leads to heightened tau pathology coupled with widespread increases in activated neuronal stress kinases [139]. Tau is a neuronal intracellular protein. TREM2 insufficiency enhances the hyperphosphorylation of tau, even in the preliminary stages of AD and other neurodegenerative diseases [140]. Jiang T et al. [141] described that suppressing brain TREM2 expression increased tau pathology, a phenomenon that can be linked to neuroinflammation-induced tau kinase hyperactivation. The authors also pointed out that TREM2 suppresses kinase activity through tau by limiting neuroinflammation and therefore protects against tau pathology.
An increase in inflammation and the classical
activation of microglia are associated with AD [142, 143]. Human and in
vivo studies have revealed that the NLRP3 inflammasome is triggered in AD and
ASC specks are indicated in the amyloid plaque core [144]. Some studies have
shown that TREM2 has anti-inflammatory functions. In cell lines, TREM2
insufficiency enhanced levels of pro-inflammatory mediators, such as
TNF-
Furthermore, a critical function of TREM2 is to modulate microglial function in
the CNS [147, 148]. A decrease in microglia surrounding plaque coverage was shown
in TREM2-deficient mice, especially in plaques with higher volume [149]. In an AD
model, TREM2-mediated early microglial responses reduced amyloid plaque transport
and toxicity [150]. A genomic transgene-guided enhancement in TREM2 expression
was shown to reprogram the responsivity of microglia and improve behavioral and
neuropathological symptoms in AD rodent experients [151]. Recent reports suggest
that TREM2 is an A
Dendritic spines are important for forming neuronal connection and signal transmission in the nervous system and can alter the motility, density, and morphology of neurons in relatively shorter periods [156]. The characteristics of dendritic spines have attracted attention in investigations on neurobiological behavioral platforms. For instance, the structural aspects of dendritic spines are linked to variations in learning and memory, synaptic efficiency, and other cognitive functions [157]. Reportedly, microglia reshape synapses through presynaptic nourishment and spinal head filamentous foot induction [158].
TREM2 is crucial for microglia-assisted synaptic refinement during the
preliminary stages of brain development. A lack of TREM2 expression causes
ineffective synapse pruning, increases excitatory neurotransmission, and
minimizes long-range functional connectivity [159]. Maturation studies have shown
that variations in the structure and activity of synapses and dendrites are
preliminary and critical occurrences in the pathogenesis of AD neurodegenerative
procedures [160]. A
There is plenty of literature demonstrating that regular physical exercise may
slow disease progression or ameliorate symptoms in AD by increasing cerebrospinal
biomarkers [163] and cardiovascular fitness [164], as well as decreasing
AD-related biomarkers [165]. In addition, various animal models have demonstrated
that exercise exerts neuroprotective effects on cognition in AD, such as object
recognition memory [166], spatial learning and memory [167], and anxiety behavior
[168]. Dao AT et al. [169] demonstrated that AD-impaired basal
synaptic transmission and suppression of early-phase long-term potentiation in
the dentate gyrus was prevented by prior moderate treadmill exercise. Using a
transgenic model, Mu L et al. [170] demonstrated that strengthening
structural synaptic plasticity may represent a potential mechanism by which
treadmill exercise prevents decline in spatial learning and memory and synapse
loss in 3
The interaction between microglia and neurons, mediated by TREM2, has been shown to contribute to AD pathogenesis. Therefore, the regulation of TREM2 to alleviate AD has garnered major interest in AD research. Recently, findings from an animal study showed that upregulation of TREM2 ameliorated inflammatory responses, neuronal injury, and cognitive deficits [174, 175]. More importantly, exercise can be viewed as a safe and economic option for improving cognitive performance in both normal and diseased states, including AD. However, few studies have revealed the critical role of TREM2 in exercise and AD.
Currently, the mechanism underlying the exercise-induced neuroprotective effect
on cognitive function associated with TREM2 in an AD model can be illustrated
from three major findings. First, exercise has been shown to regulate microglial
function through TREM2 regulation in the brain. Improvement in recognition memory
in an AD rat model is linked to the upregulation of the hippocampal TREM2/DAP12
pathway, which can inhibit excessive microglial activation as well as
neuroinflammation [176]. Recent research using the APP/PS1 mouse model
demonstrated that long-term running inhibits TREM2 shedding to maintain the
levels of TREM2 protein as well as microglial metabolic activity [177]. Second, exercise can increase dendritic spine density by
modulating TREM2 expression. TREM2 deficiency was shown to exacerbate dendritic
spine loss in an AD mouse model [162]. In contrast, Mu et al. [170]
showed that exercise protects memory function by enhancing dendritic spine
density in the brain in a 3
The pathology of AD is well-studied. The demand for AD prevention or treatment methods has increased owing to more than 15 years of failure in clinical studies. Fortunately, research on AD is currently underway. Emerging evidence implicating TREM2 can provide new insights into AD diagnostics and treatments because early TREM2-participated microglial inflammation may be present decades before the onset of AD-related cognitive impairments. TREM2 deficiency is closely associated with AD and other neurodegenerative disorders. In contrast, TREM2 upregulation in response to exercise can ameliorate AD-related neuropathological changes. Further studies are required to elucidate the protective mechanism of TREM2 in AD.
GTL, LLZ and WLJ designed the research study. YZF conducted a literature review and revised the manuscript. YZF and WLJ supervised the study. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.