


1 Department of Neurosurgery, University of Florida, Gainesville, FL 32608, USA
Abstract
Craniofacial encephaloceles are rare, yet highly debilitating neuroanatomical abnormalities that result from herniation of neural tissue through a bony defect and can lead to death, cognitive delay, seizures, and issues integrating socially. The etiology of encephaloceles is still being investigated, with evidence pointing towards the Sonic Hedgehog pathway, Wnt signaling, glioma-associated oncogene (GLI) transcription factors, and G protein-coupled receptors within primary cilia as some of the major genetic regulators that can contribute to improper mesenchymal migration and neural tube closure. Consensus on the proper approach to treating craniofacial encephaloceles is confounded by the abundance of surgical techniques and parameters to consider when determining the optimal timing and course of intervention. Minimally invasive approaches to encephalocele and temporal seizure treatment have increasingly shown evidence of successful intervention. Recent evidence suggests that a single, two-stage operation utilizing neurosurgeons to remove the encephalocele and plastic surgeons to reconstruct the surrounding tissue can be successful in many patients. The HULA procedure (H = hard-tissue sealant, U = undermine and excise encephalocele, L = lower supraorbital bar, A = augment nasal dorsum) and endoscopic endonasal surgery using vascularized nasoseptal flaps have surfaced as less invasive and equally successful approaches to surgical correction, compared to traditional craniotomies. Temporal encephaloceles can be a causative factor in drug-resistant temporal seizures and there has been success in curing patients of these seizures by temporal lobectomy and amygdalohippocampectomy, but magnetic resonance-guided laser interstitial thermal therapy has been introduced as a minimally invasive method that has shown success as well. Some of the major concerns postoperatively include infection, cerebrospinal fluid (CSF) leakage, infringement of craniofacial development, elevated intracranial pressure, wound dehiscence, and developmental delay. Depending on the severity of encephalocele prior to surgery, the surgical approach taken, any postoperative complications, and the age of the patient, rehabilitation approaches may vary.
Keywords
- neural tube defects
- Tessier's box osteotomy
- endoscopic endonasal surgery
- vascularized pedicled nasoseptal flap
- HULA procedure
- cerebrospinal fluid leakage
- temporal seizures
- stereo-electroencephalography
- neural crest cells
- sonic hedgehog
- Chiari malformation
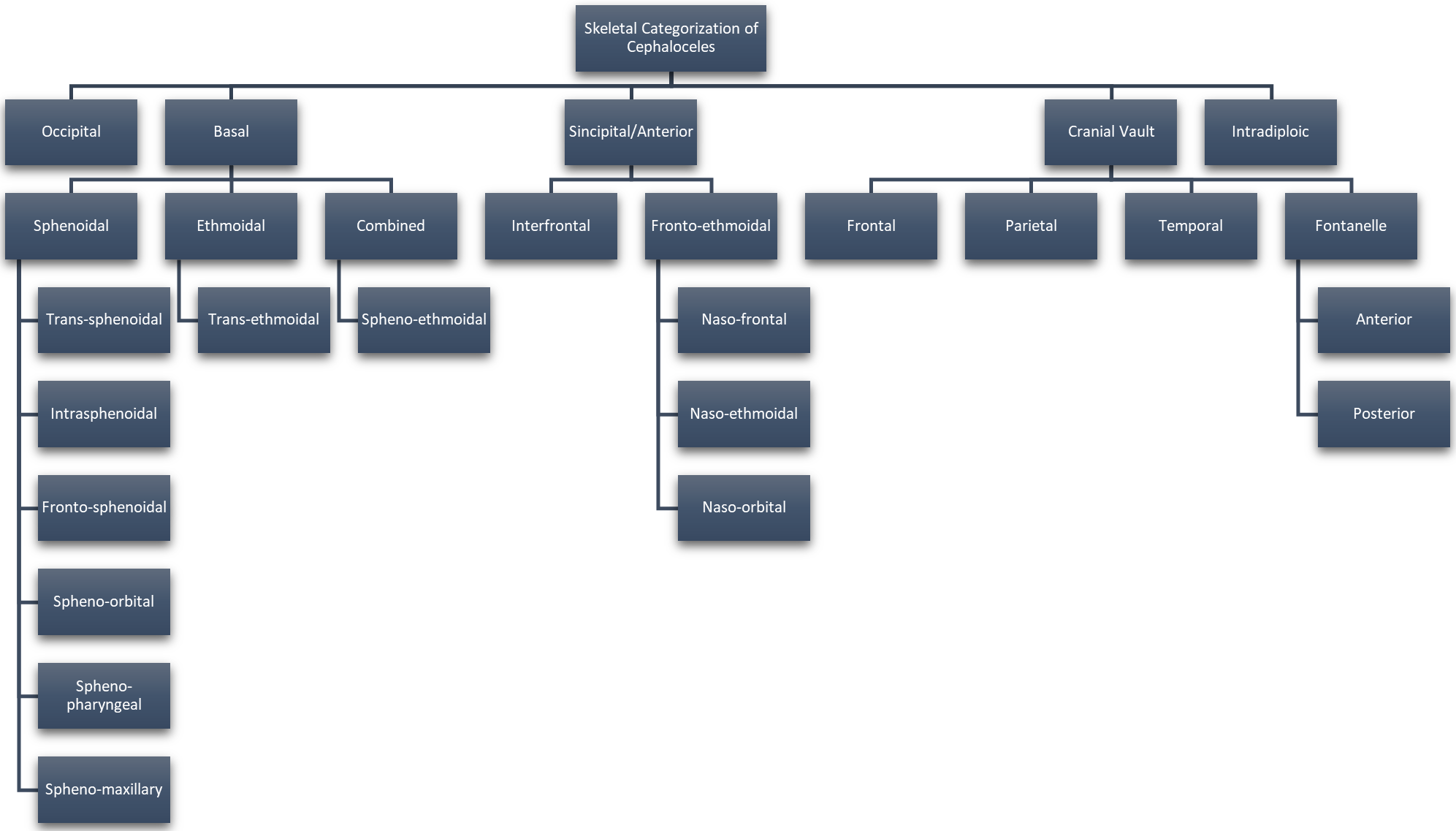
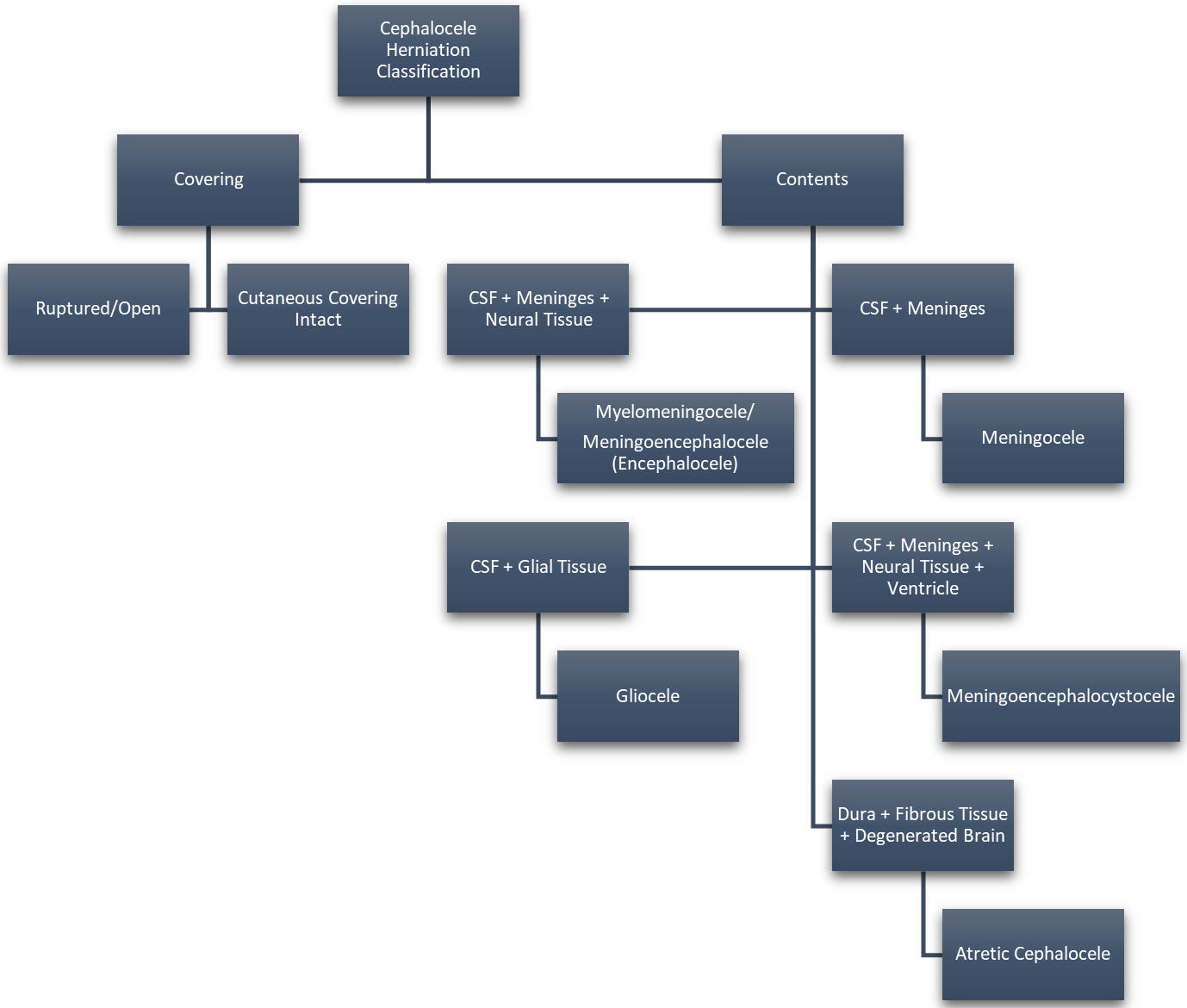
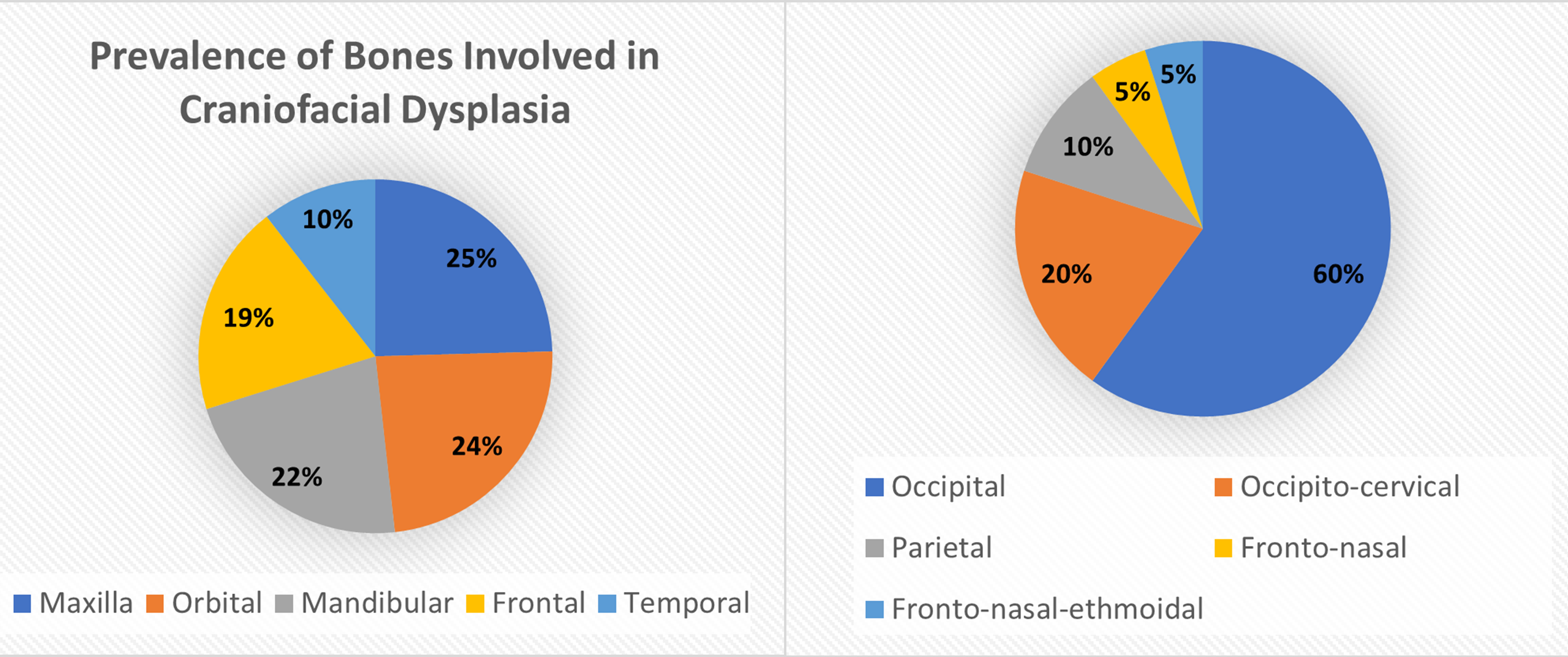
Approximately 1 in every 10,500 infants are born with encephaloceles in the United States [1]. Craniofacial encephaloceles are categorized as primary or secondary, where primary is congenital and secondary is acquired later in life, often due to trauma, neoplasm, surgery, or inflammation. Primary encephaloceles are associated with abnormal neural tube closure and structural weakness within bone junctions, resulting in skeletal cleft formation and sac-like protrusions of nervous tissue, known as encephaloceles. The etiology of craniofacial encephaloceles has not been definitively determined, but recent evidence suggests that failed mesenchymal migration during early development of the fetus results in dysplasia of skeletal features, leading to cleft formation with possible neural herniation. Cephaloceles are classified based on location of the cleft and content of the sac (Fig. 1, Ref. [2, 3]; Fig. 2, Ref. [2, 4]). The clefts can be monostotic and form in the frontal, parietal, orbital, ethmoidal, maxillary, palatine, or occipital bones, or can be polyostotic and form in a combination of multiple adjacent bones. Encephaloceles can be further subdivided into median, paramedian, orbital, or lateral, depending on the position of the extracranial herniation. The contents of the hernia may include cerebrospinal fluid, meninges, and brain tissue. The herniation may be open to the external environment or closed by a layer of cutaneous tissue [4]. The prevalence of the different subtypes of craniofacial dysplasia are variable based on geographical origin. A 2015 study of 20 patients showed occipital as the most common and a 2013 metanalysis of 18 case studies shows maxillary, orbital, and mandibular as the most prevalent of that sample (Fig. 3, Ref. [5, 6]).
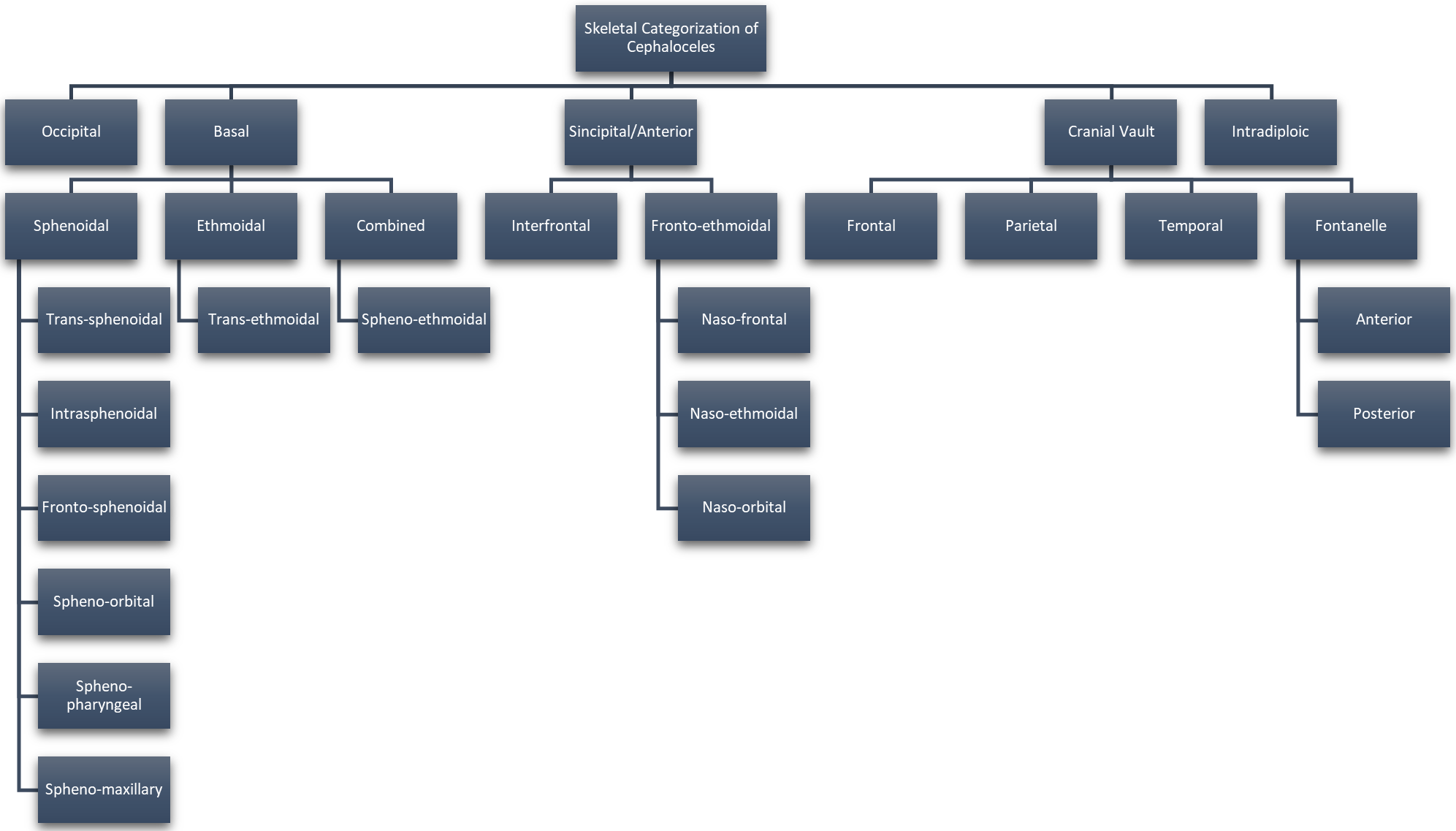 Fig. 1.
Fig. 1.Categorization of Cephaloceles Based on Skeletal Origin. Reproduced with permission from Francis Deng, Cephalocele: Radiology Reference Article, 2021 [2]; and Reproduced with permission from Centers for Disease Control and Prevention, U.S Department of Health and Human Services (CDC), Encephalocele, 2021 [3].
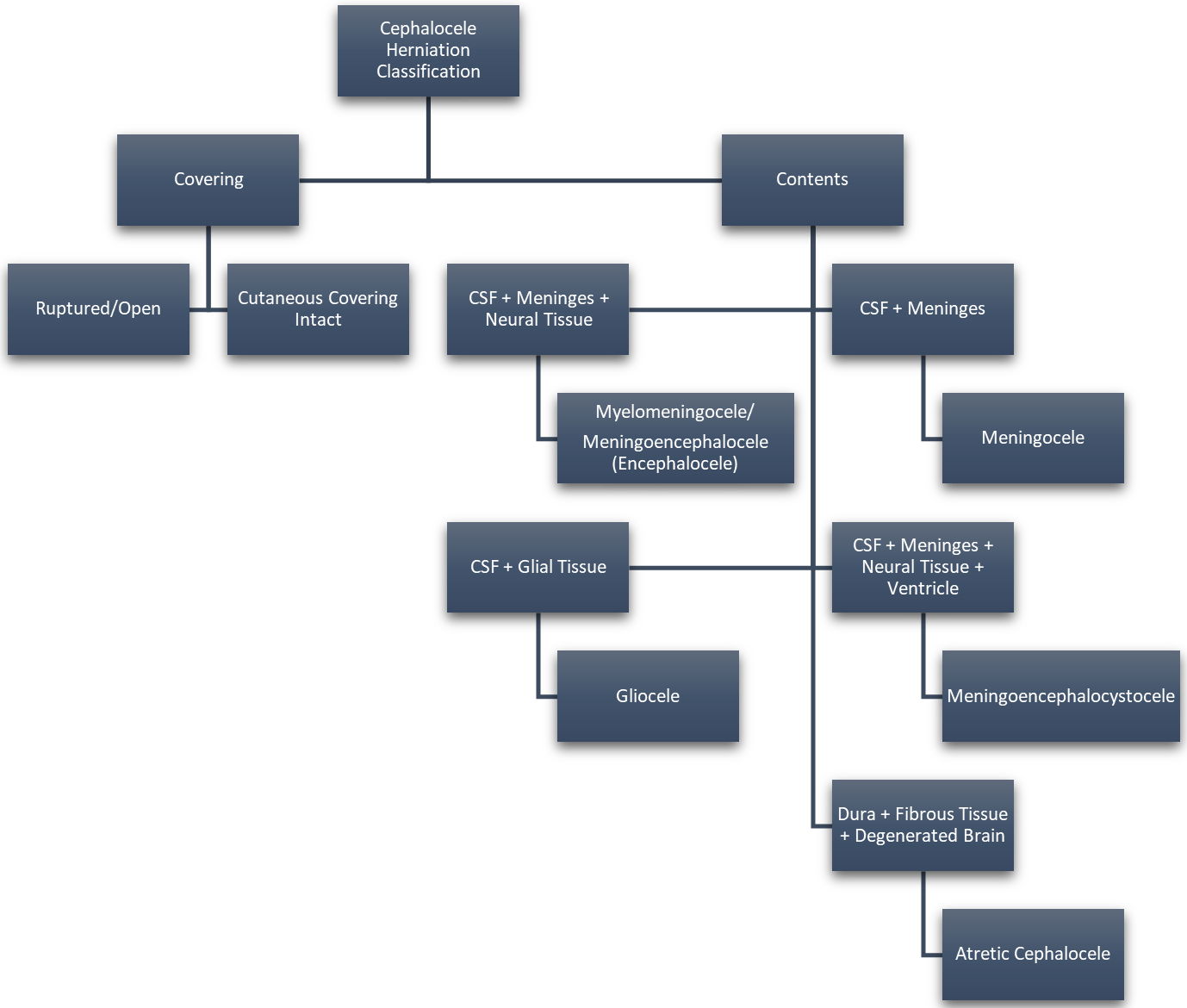 Fig. 2.
Fig. 2.Cephalocele Categorization Based on Cyst Contents. Reproduced with permission from Rüegg EM, Management of median and paramedian craniofacial clefts; published by Elsevier, 2019 [4]; and Reproduced with permission from Francis Deng, Cephalocele: Radiology Reference Article, 2021 [2].
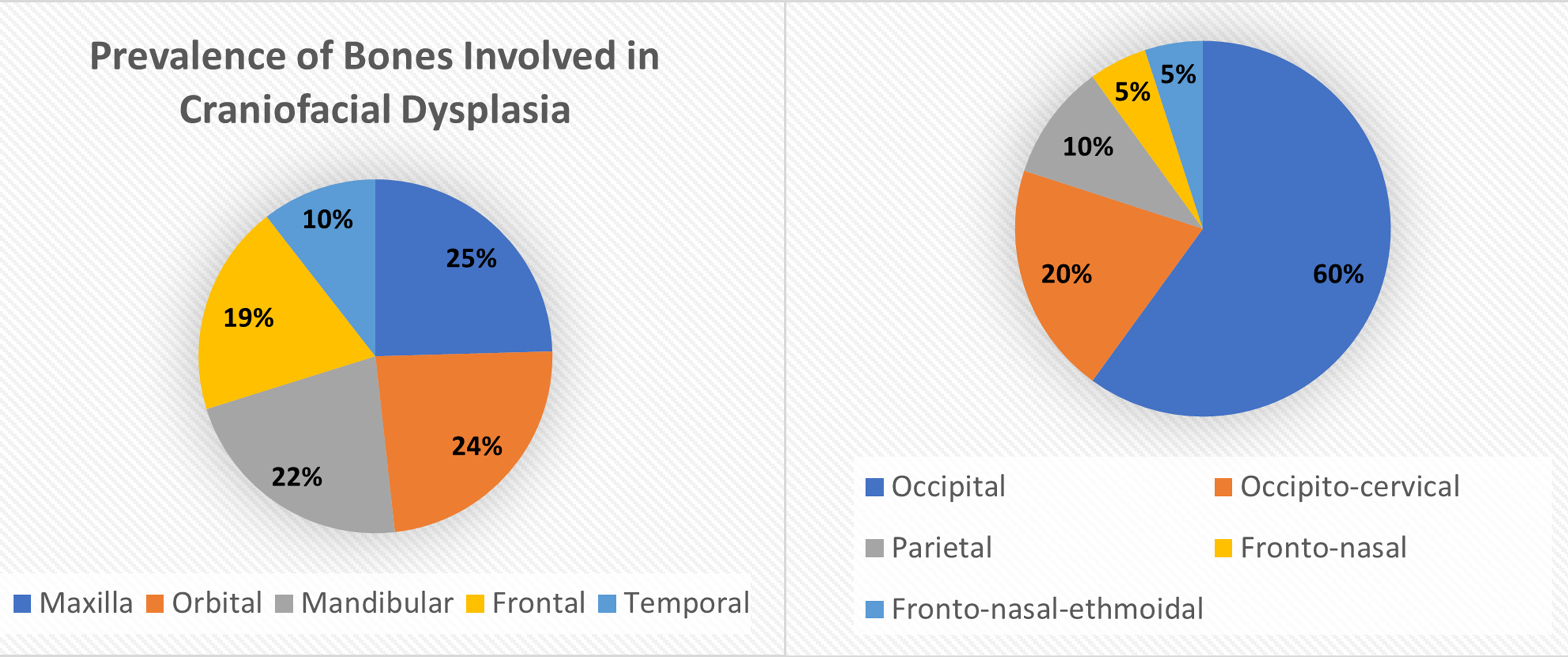 Fig. 3.
Fig. 3.Distribution of Bones Typically Dysplastic in Craniofacial Abnormalities. Data derived from [5, 6]. Reproduced with permission from Liya Yang, Prevalence of Different Forms and Involved Bones of Craniofacial Fibrous Dysplasia; published by LIPPINCOTT WILLIAMS & WILKINS, 2017 [5]; and Reproduced with permission from Shashank Ravindra Ramdurg, Pediatric encephaloceles: A series of 20 cases over a period of 3 years; published by Wolters Kluwer Health, 2015 [6].
Radiographic diagnosis of craniofacial encephaloceles is possible during the second trimester of pregnancy and proper planning can be investigated at that time, though the clinical manifestations are variable and cannot be determined until birth. Craniofacial clefts can be observed with prenatal ultrasound as a cystic structure with or without echogenicity, depending on the content of the hernia. Computed tomography (CT) scans and magnetic resonance imaging (MRI) are the preferred imaging modalities for definitively categorizing the contents of the herniated tissue and specific skeletal dysmorphia. For patients with severe bone deformities and herniations containing brain tissue, early surgical intervention is critical to address the compromised neural tissue and reconstruct the surrounding skeletal and cutaneous tissue [2].
When determining management strategies and surgical intervention the age, degree of skeletal deformation, volume and contents of the herniated sac, possible presence of cerebrospinal fluid (CSF) leakage, ulceration, airway obstruction, visual impairment, hemorrhage, and other associated defects that could imminently affect patient wellbeing should be considered. Thorough preoperative imaging using CT/MRI is essential for adequate planning by confirming the location and contents of the encephalocele. magnetic resonance (MR) angiography is also useful for avoiding, resecting, or sparing any major vessels involved with the encephalocele [7]. If a CSF leak is present, the herniation is open to the external environment, or hemorrhaging is occurring, surgery should be performed immediately. If the herniation is covered and the patient is presenting with typical neurological responsiveness, surgical intervention can be delayed until the patient is stable enough to withstand anesthesia and surgical blood loss. Prolonged delay may put the patient at risk for interrupted craniofacial skeletal development due to the infringement of the herniated mass on a newborn’s immature cranium. Definitive surgery is typically carried out anywhere from 3 to 10 months of age, though many anterior encephaloceles are treated as early as 4 days after birth [8]. The use of a multidisciplinary team consisting of both neurosurgeons and plastic surgeons is critical to the typical two stage approach to correcting encephaloceles. The first stage consists of removal of the herniation and closure of the neural tissue, the second stage involves correcting any persisting facial deformities. Maxillofacial surgeons and otolaryngologists may be called on to assisted in surgeries in which the herniation infringes on specific, critical structures or in the case that endoscopic surgery is pursued.
Historically, craniotomies have been the management strategy of choice for direct surgical intervention, though this puts the patient at risk of infection, blood loss, and injury to the neurovascular tissue surrounding the area of surgical exposure. The general trend of encephalocele surgical intervention involves delineation of the defect, excision of the herniated neural tissue, closure of dural defects, cranial bone reconstruction, and closure of cutaneous tissue. The cosmetic reconstruction of craniofacial abnormalities typically involves special attention to the supraorbital bar, nose, and possible presence of hypertelorism or telecanthus. If craniotomies are performed, prophylactic intravenous antibiotic usage has shown to benefit patients to avoid postoperative infection. A major concern with skeletal operations in newborns is the fragility of their bones and the success of bone graft remodeling, considering the thinness of their craniofacial structures [8]. Despite that, a review of 418 cranioplasties shows that remolding the skull of infants and utilizing the patient’s own skull in a bone graft was successful in every case with minimal complications [9]. In the following sections, cases in which different surgical approaches took place to treat various types of encephaloceles will be reviewed, and recommendations on how to move forward with specific craniofacial encephaloceles will be suggested.
Occipital encephaloceles are the most common type seen in North America, but anterior encephaloceles are some of the most common forms seen in Southeast Asia, Russia, and Central Africa. For either case, early surgical intervention has shown to significantly improve prognosis [10]. A traditional surgical algorithm is used to determine the appropriate age for operating on most encephaloceles, but the urgency with which intervention is sought also depends on the encephalocele categorization, especially when considering the contents of the hernia and subsequent chance of infection. Open herniations pose the greatest threat for an infection like meningitis and encephalitis and therefore should be treated with urgency. It is recommended that newborns with herniated sacs containing defective cutaneous envelopes have interventional surgery within the first days of life [8].
A retrospective review of cases from 1971–2002 analyzed the clinical data, surgical procedures, and postoperative outcomes of 103 anterior encephaloceles operated on at the Department of Neurosurgery, All India Institute of Medical Science [11]. Of the 103 children with ages ranging from 1 day to 17 years, 80% had frontoethmoidal encephaloceles, 8% had orbital, 8% had transethmoidal, 3% had transsellar, and 1% had interfrontal type. CSF leaks, meningitis, neurofibromatosis, hypertelorism, and facial clefts were present in several of the patients prior to surgical intervention. 13 of the patients had hydrocephalus, which required a ventriculoperitoneal shunt to be placed before corrective surgery could be pursued. Surgical intervention consisted of Tessier’s procedure and hypertelorism correction using medial advancement of the orbits. These procedures utilize bone grafts and cranial vault expansion, which have shown cosmetic success when treating craniosynostoses [12]. In terms of postoperative complications, 22 of the patients suffered CSF leaks, 4 children had recurrent seizures, and one patient suffered osteomyelitis 1 year postoperatively. There was an attempt to manage the CSF leaks with lumbar drainage, but 5 of the patients required placement of percutaneous lumboperitoneal shunts in addition to the drainage technique [11].
Median craniofacial clefts can lead to anterior encephaloceles in addition to hypertelorism, nasal and maxillary deformities, cleft lip and palate, and eyelid coloboma. One study examined 30 patients operated on for median and paramedian craniofacial abnormalities in Switzerland between 1986 and 2017. The ages of the patients ranged from 4 months to 18 years, with a mean age of 6.4 years for patients treated with cranioplasty. Most of the patients received multiple operations as part of a long-term reconstructive plan. 23 of those patients had anterior encephaloceles and all underwent fronto-orbital remodeling to excise and repair the herniations. The surgeries involved neurosurgeons and plastic surgeons who contributed to the removal of interorbital obstacles and reconstruction of the cranium and soft tissue. The anterior cranial vault was exposed via a midline excision using a coronal approach to remove the encephalocele and a box osteotomy was used to address the hypertelorbitism in most patients. Skeletal abnormalities were addressed with a split calvarial bone graft and median transosseous canthopexy with a frontal bone flap approach. Pericranium flaps and fibrin glue were used to fortify the primary dural closure. In the patients with more aggressive cranial bone defects, total bone reconstruction was done using additional split calvarial bone grafts from adjacent bones to close the cleft. Several patients underwent internal orbitotomy and bilateral medialization of orbital walls in combination with the encephalocele surgical intervention to address their telecanthus. Maxillary clefts were treated with facial bipartition in two patients. Patients who were treated with craniotomies recovered in the intensive care unit for the first 24 hours and were given prophylactic intravenous antibiotics for at least 2 days postoperatively. Patients in this study were followed for 1 to 30 years postoperatively. Frontal infections occurred in three instances, which were treated with wound debridement, one of which manifested 2 years postoperatively due to trauma and led to the development of osteomyelitis. Some reoperations occurred, one which was due to bone resorption and required surgical re-correction of hypertelorism. This was performed using a frontal bone flap and strengthened with a titanium plate. 7 patients required a repeated medial canthopexy and one patient had a transient lumbar decompression. No patients required prolonged cerebrospinal fluid diversions, including patients with encephalocele containing ventricular protrusions [4].
The following case, involving the first reported open nasofrontal encephalocele intervention pursued within the first day of life, exemplifies the success of early surgical intervention and the benefit of utilizing a multidisciplinary surgical team of neurosurgeons and plastic surgeons for a single, two-stage operation in a newborn. The patient was born at full term and immediately transferred to the neonatal intensive care unit (NICU) where trans-facial and bi-coronal incisions were made to remove the herniated tissue and a fronto-orbital advancement was used to expose the intracranial defects and reshape the forehead. A bifrontal craniotomy was then used to allow further internal exposure, scissors were used to transect the cranial bones, and a central bandeau dissociation allowed for the frontal dural defect to be examined. Pericranial flaps were used to close the defect with DuraSeal and the patient’s dissected done was reconstructed to form a frontal bar. Resorbable fixation was not an option due to the frontal bone’s fragility, but the cranial and orbital incisions were able to be closed with absorbable sutures. The frontal and glabellar areas were then reconstructed, and the patient was stable upon being transferred back to the NICU. Two weeks post-operatively, there was dehiscence of the glabellar incision and CSF leakage from the wound. An external ventricular drain was put in place and antibiotic therapy administered. In retrospect, the patient may have benefited from external ventricular drainage during the initial intervention. Novosorb biodegradable temporizing matrices and ventriculoperitoneal shunts were used to guide the wound closed. The Novosorb matrix provided successful, expedited wound healing without the need for grafting due to a lack of adjacent tissue availability. No other complications were noted and at the one-year follow-up the patient displayed full range of motion and a 3–4-month developmental delay. The caregivers were informed that a cranioplasty and shunt revision may be beneficial in the future [8].
The surgical intervention of frontoethmoidal encephaloceles is similar to the approach taken with other anterior encephaloceles, involving mass removal to the exterior bony rim, periosteal flaps to reinforce the internal fossa, and calvarial bone grafts fixated to the external glabellar surface using plates and screws. Removal of the encephalocele specifically involves following the neck of the herniated glial tissue to the bony defect, excising the mass, approximating the dural edges, and use of a pericranium graft if necessary [13]. Some special considerations with frontoethmoidal encephalocele repairs are the cosmetic reconstruction of the nasal bone and orbital dysmorphia, while avoiding damage to the nasolacrimal structures [14].
Nasal reconstruction can be accomplished simultaneous to the primary encephalocele repair using resected nasal bone or conchal auricular cartilage grafts. A study of 52 cases of frontoethmoidal encephalocele corrections also reported the usefulness of using the meningocele itself to contribute to the soft tissue of the nasal bridge over the bone graft by deepithelization and invagination of the indurated hernia tissue. If the nose was prolonged, young patients were reported to benefit from telescoping. Rhinorrhea was the most common post-operative complication and was addressed with additional surgical dural closure or paramedian flaps, though it was self-resolving in all put one of the cases examined [14]. A postoperative concern with nasal reconstruction is the potential of a straight-line scar produced from the conventional elliptical excision used to excise frontoethmoidal encephaloceles. A purse-string closure and crisscross mattress sutures can be used to improve the cosmetic success of these repairs. The skin on top of the frontoethmoidal hernia should be pinched and marked vertically and horizontally to assure enough tissue is left to cover the space, following excision of the mass. Once the removal and bony reconstruction are accomplished, a nylon suture can be used to close the nasal area, starting superiorly, and moving inside-out from subcutaneous tissue to the dermis, and then outside-in in the opposite direction. This pattern is repeated until the suture reaches its initial position and then the two ends are pulled to approximate the wound’s edges. The edges can then be further stabilized by multiple vertical crisscross mattresses, which can be removed after roughly a week [15].
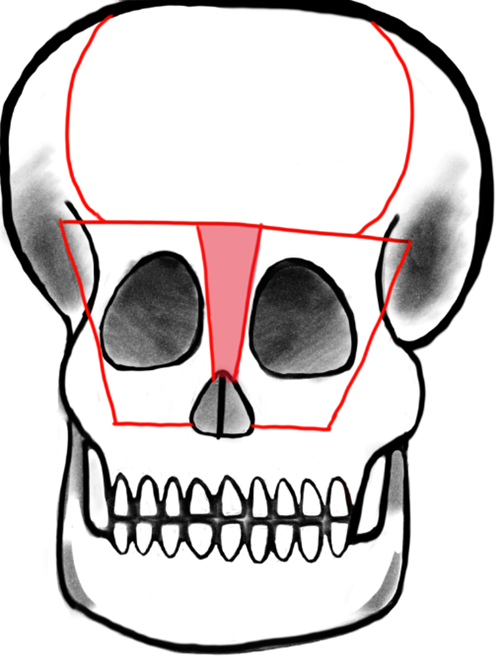
Telecanthus is defined by an increased inner canthal distance and is often also associated with frontoethmoidal meningoencephaloceles. Like the nasal reconstruction, telecanthus can be treated at the same time as the encephalocele. The necessity and success of telecanthus repair is measured with pre- and postoperative inner and outer canthal distance. The typical distances vary based on geographical origin and social standards. A study of 58 patients who were surgically treated for frontoethmoidal meningoencephaloceles showed that 45 of those patients presented with telecanthus prior to surgical intervention and 39 had successfully reduced inner and outer canthal distances postoperatively. The telecanthus repair involved bicoronal incisions, followed by movement of the dystrophic medial canthal ligaments and then either bifrontobasal trephination or bone replace between two burrholes above the supraorbital foramina. A T-shaped frontal orbital bone flap was created within the supraorbital margins and medial orbital walls (Fig. 4, Ref. [16]). The bone flap was remodeled externally while the encephalocele was excised. A bilateral medial canthopexy was done and then the carunculae were attached using absorbable sutures, which were then thread through the frontal bone so that the canthi could be repositioned superomedially. The bone flap was then placed and positioned using wires. Excess skin was removed if necessary and transnasal sutures were used on top of bolster dressings to fortify tissue to the lateral aspect of the nose [17].
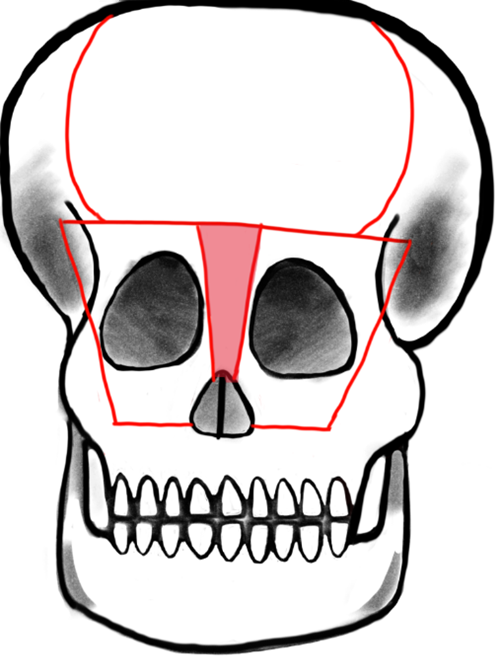 Fig. 4.
Fig. 4.Tessier’s original design: Box osteotomy. Shaded area represents bone to be removed for addressing hypertelorism or telecanthus by medial advancement of frontal bar and medial orbital osteotomies. Reproduced with permission from Raymond Cho, “Orbitofacial Developmental Anomalie - Meningoencephalocele.”, 2023 [16].
Endoscopic endonasal surgery (EES) is an emerging option for managing cases of fronto-naso-ethmoidal encephalocele in which open transcranial craniotomy is to be avoided for various reasons. Initially used primarily for intracranial neoplasms, microscopic transnasal surgery to repair encephaloceles would decrease the risk of infection, blood loss, and collateral injury to other tissues, in comparison to a standard, invasive open craniotomy. A systematic review performed in 2012 compared EES to open transcranial and transfacial approaches for managing encephaloceles, CSF leakages, intracranial abscesses, and pneumocephalus. The review found that there was no significant difference between repair success rates, but that complications and perioperative mortality were significantly lower in the EES cohort [18]. In adults, skull lesions are typically treated through endonasal approaches, but in pediatric patients EES is historically less preferred. The reasons for this are the limitation of the size of the patients’ nares, degree of severity of some congenital encephaloceles, difficulty in using the proper equipment in a smaller skull, inability to harvest a flap of the proper size, and possible implications on future craniofacial development. Several studies exist that show these restrictions are minimal in comparison to adults and that most pediatric patient’s nasal size is adequate for the use of standard endoscopic equipment [10].
There is concern of CSF leakage as a potential complication of EES. Multiple, watertight layers between the brain and the sinus cavity can help to prevent CSF leaks, infection, and abscess formation postoperatively. Traditionally, bone grafts and pedicle flaps are used during the repair of the skull base defect. Other options include bioabsorbable material such as dura mater substitutes and fibrin glue, as well as nonabsorbable equipment like titanium mesh and porous polyethylene. Another emerging option is bioabsorbable polydioxanone flexible plates, which have proven successful when used in nasal septal reconstruction and orbital floor repair [19]. One review identified 7 patients ranging from 26–86 years old who received endoscopic skull base defect repair for sinonasal tumors, meningoceles, and postoperative CSF leaks. A free graft multilayer approach was pursued, using polydioxanone plates as the inlay graft layer, mucosa, and other absorbable material as the onlay, and a supportive middle meatal spacer. Adequate healing and no recurrent CSF leaks were noted at the 6-week follow up. One patient had headaches and increased facial pressure, but no other adverse effects were reported [19].
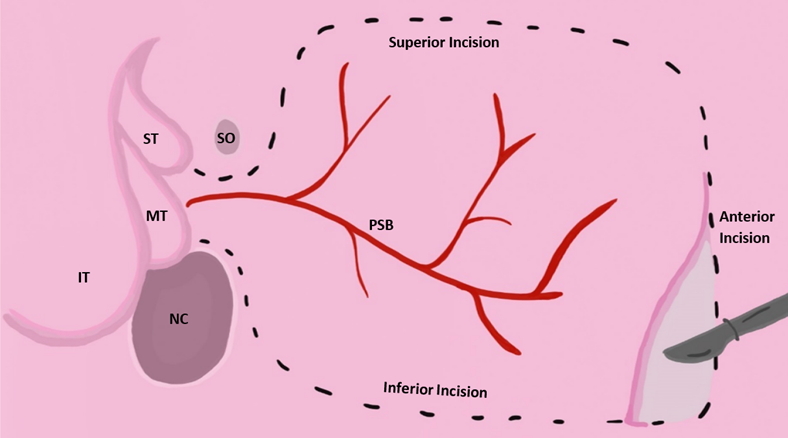
Some studies suggest pairing EES with reconstruction using nasoseptal skin flaps to treat anterior and basal encephaloceles. The nasoseptal skin flap technique involves using mucoperiosteum and mucoperichondrium from the patient’s own nasal septum to close congenital clefts. There is limitation with this strategy in pediatric patients because many do not possess enough septal flap tissue to provide complete closure of the cleft [10]. There is evidence that suggests a vascularized pedicled nasoseptal flap, a procedure sometimes referred to as the Hadad-Bassagasteguy flap approach, can provide the appropriate amount of vascularized tissue to help reduce the incidence of postoperative CSF leakage by up to 15% [20]. In the Hadad approach, a flap of the nasal septum is vascularized using the sphenopalatine artery. While the nasoseptal flap is the “gold standard”, flaps from the pericranium, palate, occiputs, buccinator, and temporoparietal fascia may be used as well [21]. One study reviewed four cases of infants with a mean age of 7 months who received surgical intervention for meningoceles and meningoencephaloceles using Hadad flaps [22]. While each patient had unique clinical manifestations, the general trend of their surgeries was similar. The procedures were performed under general anesthesia, a 2.7-mm Hopkins rod endoscope was used to visualize the defects, and hemostasis was accomplished by endoscopic bipolar forceps. The defects were resected and covered with a Hadad flap. The flap was supported by tissue glue, surgicel, and gel foam, while the area on the septum where the flap was derived was covered with a silastic splint. All four infants had CSF leakage prior to surgical intervention. Two of the patients received a transcranial repair, one a transpalatal repair, and the fourth a primary endonasal surgery. All cases represent successful use of a Hadad flap in managing encephaloceles in infants, with no major complications [22]. Another study reviewed 12 children under the age of 18 with anterior skull base lesions that were successfully treated with the endoscopic endonasal technique and vascularized nasoseptal flaps [23]. In summary, there is evidence that suggests EES paired with Hadad flap reconstruction are viable options for treating pediatric patients with craniofacial neural tissue herniations and may be a safer alternative to open cranial surgery (Fig. 5, Ref. [24]).
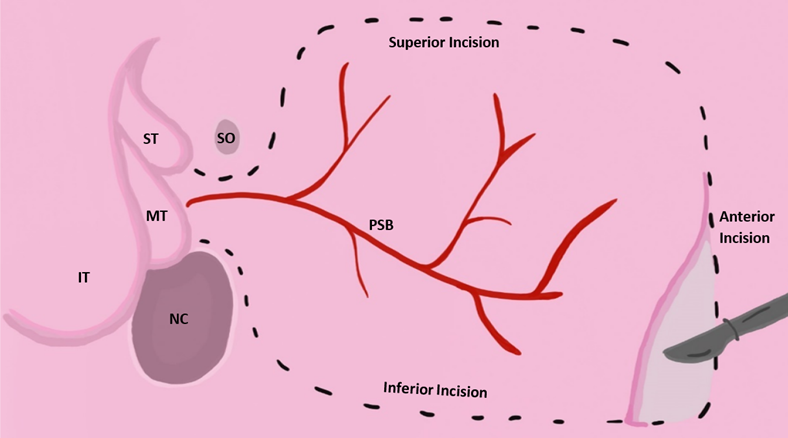 Fig. 5.
Fig. 5.Vascularized nasoseptal flap excision approach adapted from [24]. Reproduced with permission from Wade R. Gutierrez BS, Vascular pedicled flaps for skull base defect reconstruction; published by Wiley, 2020 [24]. Acronyms for Fig. 5: IT, inferior turbinate; MT, middle turbinate; ST, superior turbinate; SO, sphenoid ostium; NC, nasal choana; PSB, posterior septal branch of palatine artery.
The HULA procedure is another surgical technique that has shown to be safe and effective in managing encephaloceles in pediatric patients, while avoiding full craniotomy. The HULA method is a modified form of the “Chula” repair and stands for: H = hard tissue sealant, U = undermine and excise encephalocele, L = lower supraorbital bar, and A = augment nasal dorsum. With this technique, the herniated mass is removed, skeletal defects repaired, and facial reconstruction are accomplished simultaneously and without open frontal craniotomy. Instead, a T-shaped osteotomy is performed bilaterally over the superomedial orbital region. A study performed in 2009 reviewed twelve patients with frontonasoethmoidal encephalomeningoceles that were treated with the HULA method. This study assessed the postoperative successful of the procedure and found that patients did not experience CSF leakage, infection, or increased intracranial pressure.
Postoperative developmental surveys, parental satisfaction, and Whitaker scoring were also within a desirable range [25]. Another study performed a review of 108 HULA operations performed from 1986 to 1999 and found similar results [26].
Historically, treatment by removing the encephalocele and performing a temporal resection and amygdalohippocampectomy, followed by dural repair has been shown to be successful in treating seizures of a temporal origin [27]. One study reported that as many as 24% of patients with encephaloceles may experience seizures at some point in their life. This is especially true of patients with secondary encephaloceles that develop later in life and may go undetected [28]. There is increasing interest in successful surgical intervention of temporal encephaloceles because there is an increased frequency of these encephaloceles being identified as the causative agent of adult temporal lobe epilepsy. Surgery is often considered when patients experience drug-resistant seizures associated with craniofacial encephaloceles. Surgical success in treating these seizures depends on the origin of the epileptic waves and the expendability of the associated brain tissue incase surgical removal of the tissue is considered. Fibrous dysplasia involves connective tissue displacing normal bone development and can lead to encephaloceles. Two cases of patients with fibrous dysplasia of craniofacial bones were reviewed to understand what approach to take with patients experiencing multiple drug-resistant focal epilepsy, caused by temporal encephaloceles ipsilateral to the site of seizure onset [29]. In the two patients examined, polyostotic fibrous dysplasia of the temporal and sphenoid bones resulted in temporal encephaloceles, which led to both focal and focal to bilateral tonic-clonic seizures. The first patient was a 19-year-old male with seizure onset at 10-years-old. Surgical intervention involved reducing his encephaloceles by temporal resection and reconstructing the middle fossa floor. He continued having seizures following the initial surgery, so then received a right anterior temporal lobectomy and amygdalohippocampectomy. At his one-year follow-up he reported no persisting seizures. The second patient was a 30-year-old-man with an onset of seizures at 21-years-old, who was treated with removal of the encephalocele and then a temporal tip resection. At his six-week follow-up the patient was no longer experiencing seizures [29].
A study of 23 adult patients experiencing temporal epileptogenic seizures who underwent surgical treatment of temporal encephaloceles was also reviewed. The patients were both males and females with ages ranging from 16–70 years old. 12 of the patients underwent surgical intervention. 5 of those patients underwent temporal lobe resection and amygdalohippocampectomy, with modified neocortical resection to spare the upper temporal gyrus. The resections measured between 3.5–4.0 centimeters. The extraosseal portion of the encephaloceles were removed with the temporal resection by excising the gliotic stems near the defective bone at the base of the skull. Another 7 patients were treated with local disconnection of the encephalocele and temporal pole resection. The remaining patients were deemed not suitable for surgery due to other active psychiatric comorbidities that needed to be addressed first, because their seizures were responsive to antiepileptic drugs, there was major risk due to other large skull defects, the likelihood of frontal seizure onset, or the likelihood of bitemporal encephaloceles. There was an average follow-up duration of 2.8 years. 9 of the 12 patients that received surgical intervention were seizure-free following the operations. 3 of the patients who underwent local disconnection and temporal pole resection were not completely seizure-free but did report a reduced frequency of seizures. The worst outcome was seen in one patient with bilateral encephaloceles, who was treated with local disconnection and temporal pole resection but did not experience any improvement in the severity of seizures experienced [28]. This suggests that the success of surgical intervention in treating seizures caused by temporal encephaloceles depends on the origin of the epileptiform abnormalities and encephaloceles, with bilateral origination of both the seizure and herniation leading to least effective surgical outcomes.
More recently, magnetic resonance-guided laser interstitial thermal therapy has been introduced as a minimally invasive method to treat intractable temporal lobe epilepsy. A prospective study examined the outcomes of 20 patients with drug-resistant temporal lobe epilepsy who underwent stereotactic laser interstitial thermal therapy between 2011 and 2014 and concluded it is a safe alternative to anterior temporal lobectomy, though there was a reduced odd of complete seizure freedom [30]. Laser ablation involves a shorter operation period, time spent in the hospital, and pain-reliever requirement compared to more invasive procedures [31]. The use of MRI-guided laser ablation in treating intractable pediatric epilepsy secondary to temporal epilepsy has also been shown to have positive outcomes in alleviating seizures in children [32].
There is also evidence of the benefit of using stereo-electroencephalography (SEEG) to target the seizure origin and guide surgical intervention. SEEG utilizes small incisions to implant depth electrodes within brain tissue to record specific and concentrated waves in patients with unclear MRI readings. One report describes SEEG use in an 18-year-old woman with seizure onset at 10-years-old [33]. Initial neurological examination was unremarkable, but the patient continued experiencing persistent seizures that were nonresponsive to antiepileptic drugs. Typical scalp EEG showed sharp waves originating from the left temporal mesial area and intermittent slow waves from the left frontotemporal area. Three-tesla magnetic resonance imaging (3-T MRI) studies then showed abnormalities within the temporal horns of the lateral ventricles. A positron emission tomography (PET) scan showed reduced activity in the anterior and mesial temporal regions on both sides. Extra-operative invasive evaluation was used due to an uncertainty as to what specific structures were epileptogenic and should be focused on during surgical intervention. SEEG showed sharp waves in the left temporal lobe, amygdala, and hippocampus but, more specifically, that the activity began in the temporal pole and spread primarily to the amygdala. These findings helped guide the surgical plan to resect the temporal lobe and amygdala, sparing the hippocampus. The patient underwent a left temporal craniotomy 6 weeks after the SEEG evaluation and reported no seizures 1-year postoperatively. This case exemplifies the benefit of using a more specific, invasive imaging modality when targeting epileptogenic neural tissue, especially with the increasing incidence of temporal seizures and uncertain etiology of the seizures [33].
To understand the best surgical approaches to take in treating encephalopathies, an understanding of the development of the neurological and osteological anatomy is necessary. Skeletal cleft formations may occur as early as gastrulation and begins as a deficiency in matrix synthesis which results in insufficient bone growth and soft tissue displacement. This abnormal anatomical displacement leads to a disruption of the normal forces exerted on neighboring tissue, resulting in the open cleft. The neuromeric theory suggests an intimate relationship between the genetic control of an embryo’s neuroanatomy and skeletal development. Neural crest cells can be divided into transverse developmental zones called neuromeres that contain unique protein expression and supply specific areas of ectoderm and mesoderm. Understanding the implications of neuromeric levels can also lead to a better understanding of somite formation and subsequently dysfunction of skeletal development, such as neural tube closure defects (NTD) [34].
The incidence of NTD, like craniofacial encephaloceles, has been reduced by oral supplementation of folic acid in expecting mothers. Despite this, understanding the underlying genetic mechanisms of NTDs is necessary for further prevention, successful surgical intervention, and optimal prognosis. Cranial neural crest cells contribute to the formation of the craniofacial skeleton and nervous tissue. Neural crest cell development and migration is regulated by multiple pathways, including Sonic Hedgehog (Shh), Wnt, Bone morphogenetic proteins (BMP), Fibroblast growth factors, and Retinoic acid. Shh and BMP are integral to the growth and morphology of the neuroepithelium comprising the neural tube. Wnt and BMP signaling from the roof of the neural tube establishes its dorsal aspect, while Shh signals from the notochord establish the ventral portion [35]. Shh dysregulation has been associated with defective fetal neurogenesis, including neural tube and craniofacial malformation, such as the craniofacial encephaloceles seen in diseases like holoprosencephaly. Shh is key in fetal morphogenesis, especially early forebrain regionalization and skull bone development [36]. The gradient of Shh expression is imperative to proper neural tube patterning, with more activation of the pathway in the ventral portion and more suppression in the dorsal. The lack of Shh suppression leads to aberrant growth of the ventral portion of the neural tube and disrupts the dorsal formation [35]. Some NTDs may also be associated with abnormal upregulation of the Shh pathway repressors. One of these negative regulators includes the terminal effector GLI transcription factors, which bind DNA via zinc finger domains and are normally tightly regulated by epigenetic modifications. There is evidence suggesting that GLI transcription factors are involved with ciliopathies that can lead to aberrant Shh function and abnormal fetal neurogenesis. Primary cilia formation is necessary for Shh pathway operation, specifically, for functional signal reception and transduction during neural tube development. Primary cilia found on neuroepithelium contain morphogens and growth factors which allow successful intercellular local transduction and neuronal migration [36].
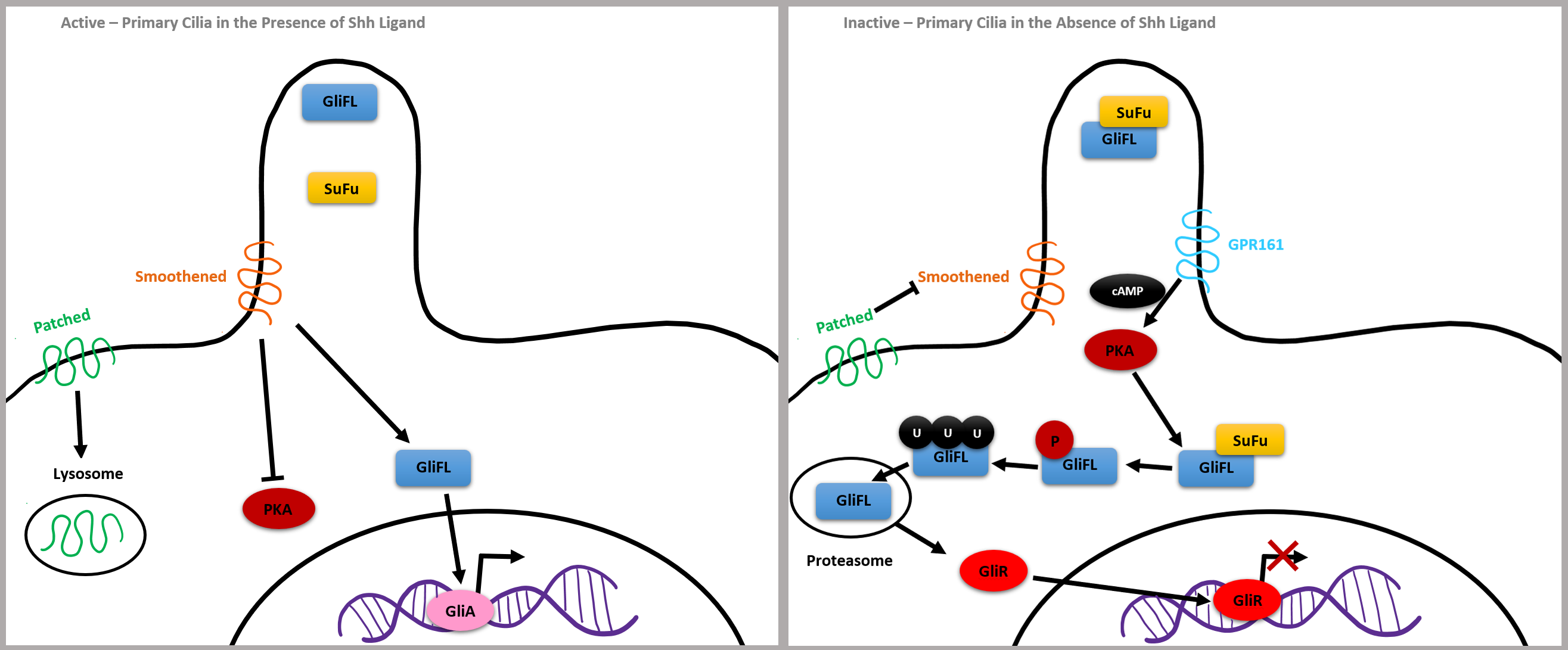
G-protein-coupled receptors (GPCR) activate secondary messengers critical to gene expression in humans and have been associated with NTDs as well [35]. G protein-coupled receptor 161 (GPR161) is involved with the regulation of the Shh pathway and may be involved in abnormal neural tube closure. GPR161 is expressed in primary cilia and represses Shh by increasing cyclic adenosine monophosphate (cAMP) levels, which activates protein kinase A (PKA). This activation leads to the increased cleavage of full length GLI3 (GLI-FL) into GLIR, which then induces the off state of the Shh pathway. Mice with partial loss of GPR161 function developed spina bifida and mice with complete GPR161 knockout developed encephalocele, cranial and spinal NTDs, midbrain protrusion, microcephaly, and malformed pharyngeal arches. Mice with ciliary defects and functional cAMP also developed craniofacial abnormalities [37].
Mutations in flagellar transport genes contribute to a reduction of Shh target genes, such as a membrane receptor Patched and a G protein-coupled receptor called Smoothened, which are critical to intracellular signaling. If the Shh ligand is not bound to Patched, it inhibits Smoothened and GLI3 forms a complex with chaperone molecule SuFu, which renders GLI3 inactive. GPR161 membrane protein activates adenylyl cyclase, increasing cAMP, leading to post-translational phosphorylation of GLI by PKA. GLI2 is degraded by proteasomes, and GLI3 is cleaved into a potent repressor, now called GLIR, which is capable of translocating into the nucleus and repressing Shh target gene expression (Fig. 6, Ref. [36]).
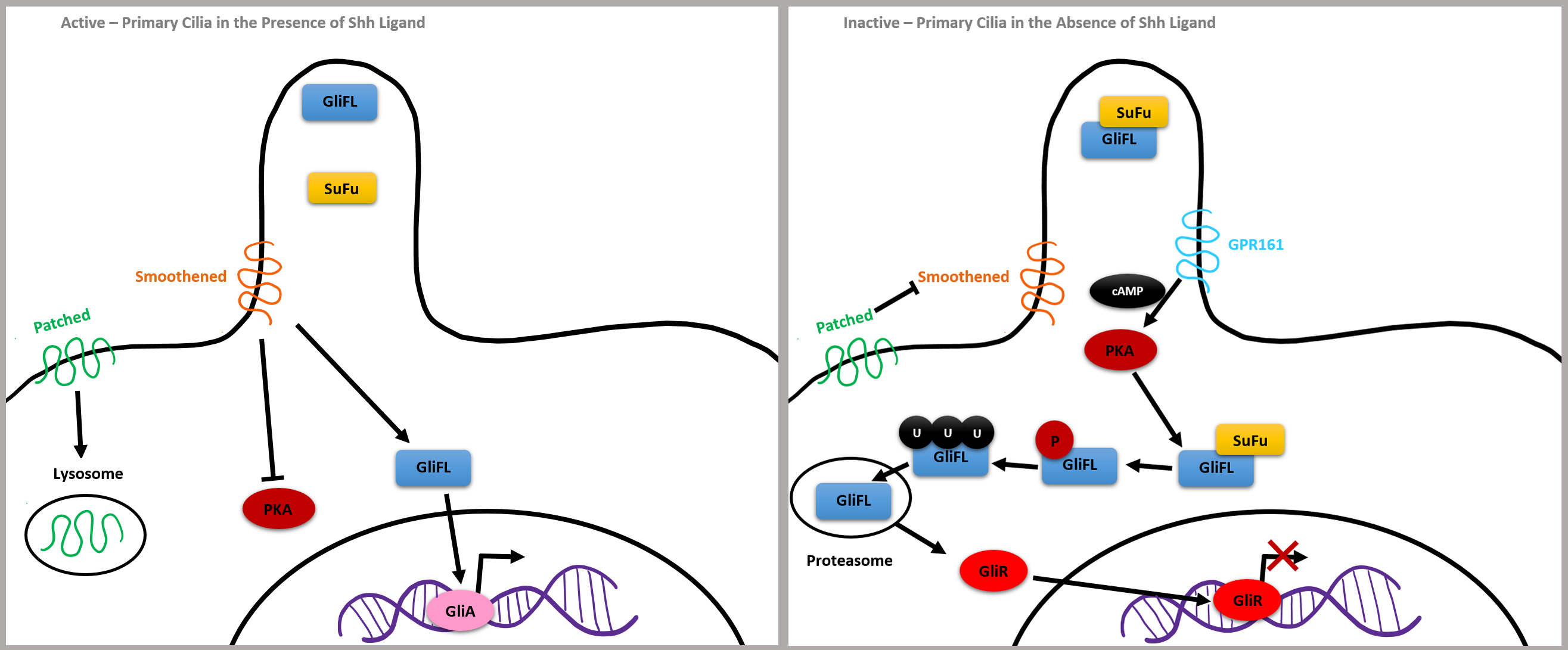 Fig. 6.
Fig. 6.Simplified Shh Pathway adapted from [36]. Reproduced with permission from Abraham Andreu-Cervera, Cilia, ciliopathies and hedgehog-related forebrain developmental disorders; published by Elsevier, 2021 [36].
On the other hand, bound Shh ligands cause GLI2 to become a strong activator, leading to the expression of the genes encoding Patched and GLI1, which further upregulates the Shh pathway. Shh binds the Patched receptor causing Smoothened to move inside of the cilia. Smoothened inactivates GPR161 and GLI-FL transcription factors are not bound by SuFu, meaning they will not be cleaved and will remain full length. GLIA is then created and translocates to the nucleus to activate the expression of genes, such as the Kif7 kinesin gene which contributes to the proper formation of cilia microtubules. In summary, the balance between GLIA and GLIR expression via the Shh pathway is critical for the closing pattern of the neural tube and can contribute to congenital neuroanatomical abnormalities (Fig. 6, Ref. [36]).
The genetic implications of some craniofacial encephaloceles seems to surround the formation of primary cilia and subsequently the aberrant functioning of critical development pathways within, such as Wnt and Shh. The malformation of primary cilia could be due to many different genetic mutations on several chromosomes, so more insight needs to be established to develop focused gene therapies that may be beneficial to patients. There are several syndromes associated with encephalocele formation which can give some insight to the genetic implications of the condition.
Chiari malformation is a collection of conditions that results in brain tissue herniated into the spinal canal, which can result in an occipital myelomeningocele. There are three subtypes, the first in which the malformation is a product of developmental skull dysplasia during late childhood and adulthood. Chiari malformation types 2 and 3 are congenital and present at birth [38]. Chiari malformation type 3 is the rarest version and, while the definition of the condition varies, it is commonly associated with occipital encephaloceles. Chiari 3 can be diagnosed prenatally via a single shot fast spin echo sequence MRI and this diagnosis can aid in preparation for possible complications following birth, such as hydrocephalus and CSF leakage. While prognosis of Chiari 3 patients is typically considered poor, a review of 17 case reports presenting 27 cases found that early surgical intervention and preparation for possible postoperative complications can improve prognosis [39].
In dog models, mutations in specific genes associated with the height and volume of the cranial fossa, as well as cadherin cell adhesion proteins in the brain and spinal cord, were found to be associated with cranial malformations like Chiari. A chimpanzee was also discovered with a Chiari-like disorder. Upon genome sequencing, a missense mutation was found in the gene encoding LRP5 protein, which is associated with the Wnt pathway. A 2019 study investigated the genetic associations of Chiari malformation type 1 using whole genome and exome sequencing and linkage analysis using animal and human models. While specific genes could not be identified in direct association with a predisposition for Chiari malformation type 1 due to the genetic heterogeneity of the samples used, the study supports that there is an underlying genetic component. A study of 23 families with 71 individuals with Chiari showed linkage between chromosomes 9 and 15. Chromosome 15 contains a Fibrillin 1 gene which is associated with other syndromes of craniosynostosis and brain malformation. Linkage on chromosomes 8 and 12 was also evident in the studies. These chromosomes contain growth differentiation genes GDF6 and GDF3, which are involved with Klippel-Feil syndrome, which is associated with up to 5% of Chiari cases. In twin studies, there is a higher concordance of the malformation between monozygotic compared to dizygotic twins, suggesting a genetic linkage. Familial aggregation also suggests potential genetic inheritance because, while most cases of Chiari malformation type 1 is sporadic, there has been evidence of families with autosomal dominant and recessive inheritance patterns. In the 2019 study, next generation sequencing was used to target rare variants seen within families that had multiple members diagnosed with Chiari. Additionally, co-inheritance of Chiari with many other syndromes that also affect skeletal formation supports the hypothesis that Chiari is associated with genetic abnormalities in paraxial mesodermal migration and subsequently occipital somite development [40].
Hydrocephalus can be diagnosed by an increase in head size, filling of fontanelles, and distension of ventricles on CT/MRI. Initial treatment of hydrocephalus typically consists of ventricular drainage. If hydrocephalus persists after this drainage, craniovertebral decompression may be pursued [41]. A 2021 retrospective study of posterior encephalocele cases found that 60–90% were associated with hydrocephalus [42]. Of the 25 patients the study observed with hydrocephalus, 23 were treated with ventriculoperitoneal shunts and 2 received an endoscopic third ventriculostomy. In 2 of the patients that received a ventriculoperitoneal shunt, 2 of the patients’ shunts failed either due to infection or obstruction. In the two patients who received endoscopic third ventriculostomy, 1 failed and received a secondary ventriculoperitoneal shunt and the other was successfully up to the time of the study being published. Dandy-Walker malformation was present in 20% of patients and Chiari 3 malformation in 4% [42].
Dandy-Walker malformation is a disorder associated with occipital encephalocele and holoprosencephaly due to a failure of the fourth ventricle to close, the posterior fossa enlarging, and cerebellar vermis displacing superiorly. The initial evidence that Dandy-Walker could have genetic linkage was determined when first-degree relatives of individuals with the malformation were seen to have an increased risk of the disease compared to an unrelated individual. Triploidy, trisomies of chromosomes 9, 13, 18, and 21, and deletions on chromosomes 6 and 3 are found to be associated with Dandy-Walker. Evidence suggests that long-term management and outcomes are favorable in 30%–42% of Dandy-Walker cases if the patients had no other anomalies [43].
Meckel-Gruber syndrome (MKS) is an autosomal dominant congenital defect which is characterized by occipital encephaloceles, cystic kidneys, and liver defects. There are 14 genes that have been found to be associated with the onset of MKS. MKS is considered a lethal ciliopathy because the core issue is the development and function of primary cilia, leading to dysregulation of the Wnt and Shh pathways and improper neural tube formation [44].
Long-term follow-up of patients with craniofacial encephaloceles are often limited due to their rarity and commonly focus on reoccurrence of herniation, infection, cosmetic satisfaction, neurological development, CSF leak, increased intracranial pressure, and incidence of seizures. Early surgical intervention is important to prevent morbidity in critical cases but delaying surgical intervention in cases that are not imminently life threatening can still result in long-term success postoperatively. The location and the contents of the encephalocele is the best predictive factor for prognosis, with occipital bone defects and herniations containing large amounts of cortical brain tissue being associated with worse neurologic signs and outcomes, compared to more rostrally located herniations containing smaller amounts of tissue. Some of the major concerns postoperatively include infection, CSF leakage, infringement of craniofacial development, elevated intracranial pressure, wound dehiscence, and developmental delay. Depending on the severity of encephalocele prior to surgery, the surgical approach taken, any post-operative complications, and the age of the patient, rehabilitation approaches may vary. Prophylactic intravenous antibiotics are often used in young patients with open wounds and high risk of infection. Shunts and additional dural repairs are considered for patients with CSF leakage and prolonged rhinorrhea.
One case report followed a patient with frontoethmoidal encephalomeningocele, who initially presented at 9-months old in northern Thailand, where the incidence of these encephaloceles is relatively high. The patient’s encephalocele was corrected using the modified Chula’s technique, which involved removing the herniated sac, repairing the dura, and repositioning the nasal bone. A titanium mesh was used to prevent future reoccurrence of herniation and gel foam was used to rebuild the orbital floor. Postoperatively, the patient was doing well and at the 3-week follow-up the wound had fully healed with no infection or increase of intracranial pressure. The patient was followed-up annually for 7 years. At the latest follow-up, the patient was 8-years old and had been attending school with no limitations in daily activities or social integration. Upon physical examination, the patient had a fine-line scar and no visible infraorbital bone abnormalities. The patient and his parents reported no issues with the cosmetic success of the surgery. Telecanthus was improved with a medial intercanthal distance of 2.9 centimeters. CT scan indicated that the titanium mesh was still in place and that there was no recurrence of the mass, CSF leak, or elevated intracranial pressure [45].
Even with limited resources and more invasive approaches to surgical intervention, successful treatment and long-term follow up of craniofacial encephaloceles is possible. A study followed 21 patients in the Philippines who had nasoethmoidal encephaloceles treated by a combination of transcranial and direct anterior wedge frontal craniotomies [46]. Due to a limitation of resources and surgical expertise, a less invasive technique like EES or Chula was not pursued, and mobilization of the orbital walls and fixation of bone grafts with absorbable sutures was not done. Some surgical complications like hemorrhaging, infection, CSF leakage, wound dehiscence, and postoperative seizures were noted in 9.5% of the patients. Patients were followed-up at 1 month, quarterly, and semiannually, with an average follow-up period of 22 months. No major complications were noted and none of the patients were recommended for additional revisionary operations up to the date of the study being published [46].
Recent clinical trials were identified on the National Institutes of Health’s ClinicalTrials.gov database and give an idea of the direction current research is taking on better management of craniofacial encephaloceles and other neural tube closure disorders of infants. One observational cohort that began in April 2017 and was completed in October 2018 aimed to standardize the measurements of the brain stem and posterior fossa on both prenatal and postnatal MRI to differentiate fetuses with spinal and hindbrain dysplasia and track the success of fetal surgery. This lays the foundation for more studies to be completed to further demonstrate the usefulness of in utero repair of myelomeningoceles [47].
A prospective randomized study that began in March 2019 and was estimated to be completed in March 2021 aims to compare the cognitive outcomes of adult patients with brain herniation receiving “early” cranioplasty within 3 months of receiving a decompressive craniectomy versus “late” cranioplasty performed 3 months after the initial craniectomy. This is important for balancing the risk of hemorrhage and infection associated with cranioplasty against the risk of trephined, which may result from lack of bony support, leading to decreased cerebral perfusion and CSF flow, and can be alleviated by cranioplasty. By establishing the ideal timing of cranioplasty, optimal neurological function can be assured following brain herniation repair via craniectomy [48].
While taking a preventative folic acid supplement during reproductive years is becoming more commonplace all over the world, tertiary interventions to prevent neural tube defects are currently building upon our knowledge of what hematological levels should be monitored and maintained to provide for the best developmental outcomes in growing fetuses. A single-blind cluster, randomized controlled trial that began in October 2018 and is estimated to be completed in December 2023 aims to study the benefit of treating preconception folate deficiency of individuals with reduced red blood cell folate levels and elevated homocysteine during pregnancy, to prevent birth defects [49].
Proper measurement of intracranial pressure is critical in the pre- and postoperative monitoring of patients with encephaloceles to prevent morbidity, mortality, and cognitive decline. Currently, intracranial pressure is typically measured at a frontal level, but there is little known about the usefulness of measuring posterior intracranial pressure. An observational, prospective cohort study that began in July 2019 and is estimated to be completed in May 2023 aims to compare posterior fossa pressure measurements in postoperative patients who received posterior fossa repairs with their frontal intracranial pressure measurements to help guide postoperative management and rehab [50].
Conceptualization and overview by BLW. Section writing and figure design by AA. Editing by AA. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest. Brandon Lucke-Wold is serving as one of the Guest editors of this journal. We declare that Brandon Lucke-Wold had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Gernot Riedel.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.