
 , Bogdan Ovidiu Popescu 1,2
, Bogdan Ovidiu Popescu 1,21 Cell Biology, Neurosciences and Experimental Myology Laboratory, Victor Babeș Institute of Pathology, 050096 Bucharest, Romania
2 Department of Clinical Neurosciences, Carol Davila University of Medicine and Pharmacy, 050474 Bucharest, Romania
3 Department of Cellular and Molecular Biology and Histology, Carol Davila University of Medicine and Pharmacy, 050474 Bucharest, Romania
Academic Editor: Gernot Riedel
Abstract
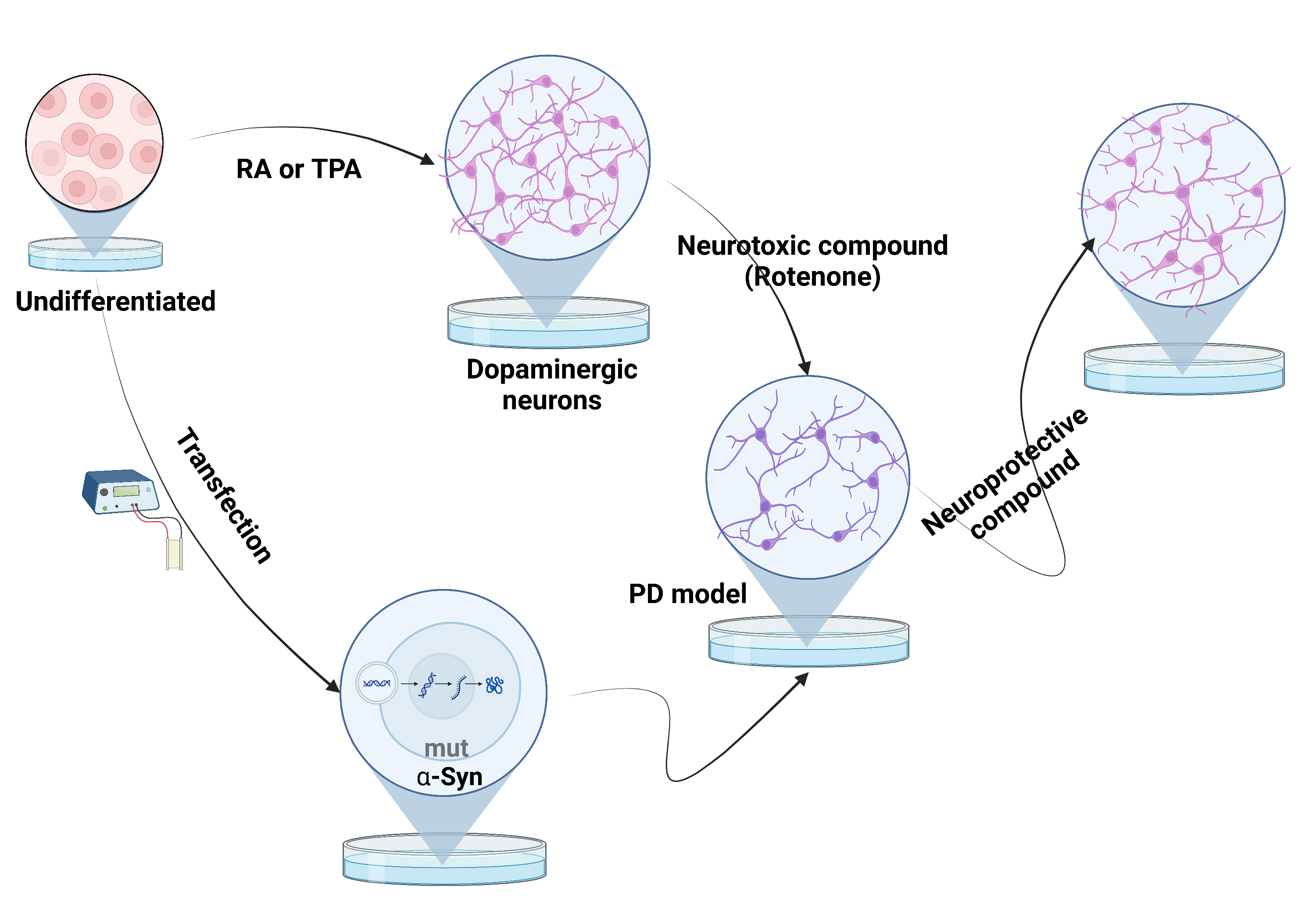
The SH-SY5Y cell line is a simple and inexpensive in vitro experimental model for studying Parkinson disease (PD). This experimental model is a useful tool for elucidating pathophysiological mechanisms of PD and in the development of new pharmacological therapies. In this review, we aim to summarize current protocols for SH-SY5Y cell culturing and differentiation and PD experimental designs derived from the SH-SY5Y cell line. The most efficient protocol for differentiation of the SH-SY5Y cell line into dopaminergic neurons seems to be the addition of retinoic acid to the growth medium, followed by 12-O-tetradecanoylphorbol-13-acetate (TPA) addition in a low concentration of fetal bovine serum. PD pathological changes, such as neuronal apoptosis and the intraneuronal alpha-synuclein aggregation, can be reproduced in the SH-SY5Y cell line either by the use of neurotoxic agents [such as rotenone, 1-methyl-4-phenylpyridinium (MPP+), 6-hydroxydopamine] or by genetic modification (transfection of the alpha-synuclein wild-type or mutant gene, genetic manipulation of other genes involved in PD). In addition, compounds with a potential neuroprotective role may be tested on neurotoxicity-induced SH-SY5Y models. The cell line can also be used for testing PD pathophysiological mechanisms such as the prion-like neuronal transmission of alpha-synuclein or the microbiota influence in PD. In conclusion, the use of the SH-SY5Y cell line represents a basic but consistent first step in experiments related to PD, but which must be followed by the confirmation of the results through more complex in vitro and in vivo experimental models.
Graphical Abstract
Keywords
- SH-SY5Y cell line
- Parkinson disease
- in vitro model
- pathophysiology
- microbiota
- autophagy
- therapeutic compounds
Parkinson’s disease (PD) is the second most common neurodegenerative disease, after Alzheimer’s disease, with an onset between the 5th and 6th decades. The main pathological characteristics of the disease are the degeneration of the dopaminergic neurons in the substantia nigra pars compacta and the intraneuronal deposition of the alpha-synuclein protein that forms Lewy bodies and Lewy neurites, affecting multiple areas of the central nervous system [1].
Extrinsic factors, among which the alterations of the intestinal microbiota is gaining increasing attention in the last few years, seem to have a much greater contribution to the development of neurodegenerative diseases, especially in the development of PD [2]. However, the pathophysiological mechanisms are not yet clearly understood. Thus, the proof-of-concept of these mechanisms could increase our understanding of this disease, and also open new perspectives for therapeutic strategies that mitigate or even eliminate this neurodegenerative phenomenon, a goal that has not yet been achieved by any current intervention.
In vitro models used for PD must reproduce the two main pathologic changes of the disease: the degeneration of the dopaminergic neurons and the intraneuronal deposition of alpha-synuclein [3].
The most used in vitro model for PD is the culturing of immortalized cell lines [4]. This in vitro model is widespread, easily available, and less expensive, and may represent the first step in testing pathomechanisms or interventions for this disease [3, 4]. To obtain the specific features of the diseased cells, the immortalized cell lines must be first differentiated towards a specific neuronal type that is affected in the pathophysiology of the disease. For PD, a differentiation in dopaminergic neuronal phenotype is usually achieved [4]. In addition, the cell culture may be either exposed to one of the several neurotoxins that induce cellular PD-specific changes, or be genetically modified by transfection to over-express the pathological protein [4].
The most common immortalized cell line used in this type of research is the human-derived SH-SY5Y cell line. The SH-SY5Y is a subline of cells isolated from the bone marrow from a metastatic neuroblastoma of a 4-year-old female [5]. The cell line has a catecholaminergic phenotype, equipped with tyrosine hydroxylase and dopamine-beta-hydroxylase enzymes, and is able to synthesize both dopamine and noradrenaline neurotransmitters [5]. In addition, SH-SY5Y can be further differentiated into a more mature dopaminergic phenotype [6]. Thus, these characteristics make this cell line a suitable in vitro model for the study of PD.
This review aims to summarize the most recent advancements of in vitro experimental models using the SH-SY5Y cell line to elucidate the components of the pathophysiological mechanisms of PD for the development of new therapeutic strategies.
In 2003, Heiko Braak [7] demonstrated in neuropathological studies that in sporadic PD there is a centripetal distribution of Lewy bodies in the nervous system in time and space, from the periphery to the central nervous system, from the medulla oblongata to the cortical structures. As the evolution of alpha-synuclein deposits progresses towards the center, the previously affected areas gradually become severely impacted with an extensive neurodegeneration process [7].
The manner in which Lewy bodies spread in the nervous system affecting neighboring structures in a centripetal way is similar to the spread of an infectious agent. The initial damage to the olfactory nuclei and the dorsal nucleus of the vagus, led to the genesis of a hypothesis that the onset of PD is due to an unknown pathogen from the nasal mucosa and/or the mucosa of the gastrointestinal tract [7, 8]. This hypothesis was also strengthened by the subsequent discovery of Lewy bodies in the enteric nervous system and the vagus nerve, as well as in the olfactory tract [9, 10].
A recent theory that is gaining momentum to explain this method of propagation of the neurodegenerative phenomena and the involvement of alpha-synuclein aggregates, is the spread of altered alpha-synuclein in a manner similar to a prion infection; transsynaptic, from neuron to neuron, inducing intraneuronal aggregation of normal endogenous alpha-synuclein, which subsequently leads to neuronal apoptosis [11]. Thus, according to this hypothesis, the origin of alpha-synuclein alteration and the origin of the prion-like spread of this protein is at the level of the nasal and/or digestive tract mucosa [8].
It is suggested that the microbiota (nasal, and especially intestinal) plays an important role in the initial stage of alpha-synuclein aggregation; since it has been observed that changes in the microbiota lead to the secretion of potentially toxic bacterial products to the neurons of the enteric wall [2]. These bacterial products cause changes in alpha-synuclein, leading to its aggregation [2].
In addition to the scientific importance of discovering a new and extremely interesting mechanism in a neurodegenerative disease, the involvement of the microbiota in the pathophysiology of PD can also have a therapeutic importance by eliminating or slowing down the phenomenon of neurodegeneration [2]. Various alternatives for modulating the microbiota, such as diet, administration of probiotics, prebiotics, and fecal transplantation, could prevent or retard these processes [2].
The autophagy-lysosome pathway is responsible for the clearance of dysfunctional proteins, and is one of the protein clearance systems [12]. It consists of three methods of clearance, macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA) [12]. Macroautophagy represents the fusion of the lysosome with the autophagosome, a de novo formed vesicle that sequesters the cargo but lacks proteolytic enzymes and needs the lysosome for its degradation [13]. Microautophagy represents direct invagination by the lysosome of the cytosolic components [13]. In CMA, specific proteins are targeted by molecular chaperones and translocated directly into the lysosome for degradation [14].
The autophagy-lysosome pathway plays a pivotal role in removing misfolded or damaged proteins and dysfunctional organelles from cells, especially at the level of neurons [15].
Malfunction of this pathway leads to accumulation and aggregation of various proteins, leading to cell death. Targeting genes for critical molecules involved in the induction of autophagy, such as autophagy related (Atg) 5 and Atg7, in mouse models and neuronal cell cultures, causes intraneuronal aggregation and induces a neurodegenerative process [16].
In the neuronal cells, the autophagy-lysosome pathway is an important route of degrading the pathological protein alpha-synuclein, along with the ubiquitin-proteasome system [17]. Monomeric and dimeric alpha-synuclein is degraded by CMA, while oligomeric alpha-synuclein is degraded by macroautophagy [18].
In PD, autophagy impairment appears to be a major mechanism that leads to alpha-synuclein aggregation, and mitochondrial dysfunction resulting in oxidative stress [19]. Genes associated with PD are strongly linked to autophagy-lysosome pathway dysfunction [19]. PTEN-induced kinase (PINK1) and Parkin play an important role in the process of mitophagy, where the damaged mitochondria are marked to be engulfed by the autophagosomes [20]. PINK1 fixates on the outer membrane of the dysfunctional mitochondria, while Parkin is activated and ubiquitinates the marked mitochondria. The mutations of the genes increases the mitochondrial damage that contributes to the pathogenesis of PD [20]. PARK9 is an ATPase which has an important role in the maintenance of a certain level of pH inside the lysosome [21]. Mutations in PARK9 dysregulates the lysosomal activity and impedes the fusion between the lysosome and the autophagosome, thus leading to the accumulation of intraneuronal alpha-synuclein [21]. Evidence suggests that glucocerebrosidase (GBA) and leucine rich repeat kinase 2 (LRRK-2) are involved in autophagy and the mutation of the genes increases the risk of PD by disrupting the autophagy-lysosome system [19].
Intraneuronal alpha-synuclein inclusions leads to dysfunction of autophagy, which in turn leads to more alpha-synuclein aggregation and accumulation [22]. Rather than being a nonspecific process, overexpression of alpha-synuclein exerts a specific action that inhibits autophagy, by inhibiting Rab1a with further mislocalization of Atg9, blocking the formation of the autophagosomes precursors [23].
These studies demonstrate that the dysfunction of the autophagy-lysosome system is involved in the pathogenesis of PD and may lead to the possibility of discovering another class of drugs that could target the direct cellular mechanisms in an attempt to prevent this neurodegenerative process.
The SH-SY5Y is usually cultured with DMEM or a mixture of DMEM/F12 growth mediums [24]. The medium is usually supplemented with 10% fetal bovine serum. The growth properties are mixed, some cells are adherent and some grow in suspension [24]. Undifferentiated cells have a high proliferation rate, tend to grow in clumps, have a rounded body and develop short neurites, suggesting a phenotype that is closer to a neoplastic state [25]. In contrast, the differentiated cells have a pyramidal body with long neurites and a low proliferation rate [25]. The cell line can exhibit a neuron-like phenotype (N-type) and an epithelial-like phenotype (S-type) with epithelial morphological features such as adherent, flat, rounded cells, without any projections [26, 27]. The switch between one phenotype to another occurs spontaneously and the ratio between the N-type cells and the S-type cells can influence the experimental models because only N-type cells are needed for the study of PD [26].
The induction of differentiation of the SH-SY5Y cell line into dopaminergic
neurons is usually the first step for developing in vitro experimental
designs to study PD. The morphological aspects of the neurons and the low
proliferation rate makes the differentiated cells resemble the primary
mesencephalic dopaminergic neurons, while the undifferentiated cells are much
closer to a cancerous type of cell, related to their neuroblastoma origin [25].
The differentiation into dopaminergic neurons is most frequently obtained by
adding several compounds to the growth medium such as retinoic acids and phorbol
esters, in parallel with a low concentration of fetal bovine serum [6]. Retinoic
acid is a derivative of vitamin A, which is known to have a significant role in
cell differentiation and in the control of proliferation [28]. Nevertheless, it
must be taken into account that the addition of retinoic acid only in the process
of differentiation promotes an induction of a cholinergic phenotype [6]. A
dopaminergic phenotype is obtained by further treatment with a phorbol ester such
as 2-O-tetradecanoyl-phorbol-13 acetate (TPA) [29]. Thus, the approach that seems
to induce a more efficient dopaminergic phenotype is the addition in the growth
medium of retinoic acid, usually in a concentration of 10
Other differentiation protocols use only the addition of 10
To validate the differentiation protocol toward a dopaminergic phenotype, the expression of several specific markers for dopaminergic neurons such as tyrosine hydroxylase, D2 type dopamine receptor, D3 type dopamine receptor, and dopamine transporter should also be tested. Although these markers can also be detected in the undifferentiated SH-SY5Y cells due to the original catecholaminergic phenotype, an increase in their expression confirms the dopaminergic differentiation [25, 31].
An important step in the SH-SY5Y cell line in vitro modulation is to simulate the neuronal degeneration. To date there are multiple protocols to induce changes that mimic PD pathological features in the SH-SY5Y cell line.
The treatment with a neurotoxic compound that induces apoptosis of the neurons, such as rotenone, 1-methyl-4-phenylpyridinium (MPP+) or 6-hydroxydopamine (6-OHDA) is critical especially in searching for new drug therapies, by testing molecules that prevent the neurotoxicity induced by these compounds. Rotenone is a lipophilic insecticide that easily crosses the membrane and accumulates inside the cell [32]. MPP+ is a metabolite of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) that has a high affinity for the dopamine transporter and accumulates inside the dopaminergic neurons [33]. 6-OHDA has a similar structure to that of the catecholamines and enters the cells by using their transporters [33]. All three neurotoxic compounds induce mitochondrial dysfunction, increase oxidative stress and produce neuronal cell death [3, 24, 33]. These models also induce an increased expression and aggregation of alpha-synuclein, although usually these effects are not primarily investigated [32, 34]. These neurotoxins are also administered in mice in order to obtain a PD in vivo model, by inducing the cell death of the dopaminergic neurons from the substantia nigra pars compacta [35].
A novel method for obtaining an in vitro PD model is to expose the neuronal cells to alpha-synuclein preformed fibrils (PFF) [36]. PFF are recombinant alpha-synuclein molecules subjected to a process of sonication that induce the formation of small fibrils [36, 37]. Added in cell culture, the PFF favors the aggregation of endogenous alpha-synuclein which subsequently leads to synaptic dysfunction and cell death [36]. This process imitates the formation of Lewy bodies and Lewy neurites which are discovered in the brain of the human patients with PD [36, 37]. PFF use the normal level of endogenous alpha-synuclein, thus there is needless to use an in vitro model of alpha-synuclein overexpression [36]. Although this in vitro model was initially used in hippocampal primary neuronal cultures, it was subsequently applied to the SH-SY5Y cell line [36, 38, 39].
To simulate the pathological change of the intraneuronal aggregation of alpha-synuclein, the SH-SY5Y cell line can be genetically modified by transfection with the gene that encodes for alpha-synuclein, either a wild-type or a mutant gene [3]. The transfection of the alpha-synuclein wild-type or mutant gene drives an over-expression of alpha-synuclein, that favors intracellular accumulation [3]. This process ultimately produces aggregates of the protein with a neurotoxic effect that leads to cellular death [3].
Other genes that appear to have a role in the pathophysiology of PD, especially the genes involved in the genetic forms of PD, such as LRRK-2, DJ-1, Parkin, PINK1 can be manipulated in three different ways; to over-express the protein, knock-down the gene leading to the lack of protein, or gene mutation leading to an altered type of protein; but these models are used less frequently [24].
The search for neuroprotective treatments in PD is of great interest since there is lack of compounds that have had any clinical effect. Neurotoxicity-induced SH-SY5Y models serve as a starting point for studying the protective effect of different compounds. The mechanism by which these compounds exert the neuroprotective effect has also been investigated. In recent years, the modulation of autophagy has gained attention as a neuroprotective mechanism in PD. Many of the compounds with in vitro neuroprotective effects appear to stimulate the neuronal autophagic process, thus decreasing the aggregation of the alpha-synuclein [40, 41].
Erythropoietin (EPO), a molecule secreted by the kidney that has a significant role in modulating erythropoiesis, is also produced by neurons and has an anti-apoptotic effect [42]. In a rotenone-induced neurotoxicity model of SH-SY5Y cells, EPO increased cell viability and decreased the expression and aggregation of alpha-synuclein [40]. This appears to be the effects of enhanced autophagy [40]. Lithium, an inductor of autophagy that uses an mTOR-independent pathway, prevented the cellular apoptosis in rotenone-exposed SH-SY5Y cells [43]. An induction of autophagy and a decreased apoptosis and alpha-synuclein expression was observed after the treatment with sirtuin 3 (SIRT3) in rotenone-exposed SH-SY5Y cells [41]. SIRT3 is a molecule which modulates mitochondrial function that has been associated with the pathogenesis of carcinogenesis and aging [41]. Vitamin D3 has also been proposed as a possible neuroprotective therapy, having in vitro results similar to the previously mentioned compounds [44].
Several studies indicate that natural phenols, which are found mostly in fruits and vegetables, have a certain antioxidant effect. This effect of the phenols was also tested in PD models with the aim of finding molecules that prevent the neurodegenerative phenomenon.
Curcumin, a known phenolic compound with possible antioxidant and anti-inflammatory effects, was seen to confer cellular protection against the toxic injury of MPP+ in a SH-SY5Y model [45]. Polydatin, a derivative of resveratrol with a strong antioxidant effect, protects the SH-SY5Y cells against rotenone toxicity, by promoting autophagy, reducing oxidative stress and mitochondrial damage [46]. This effect was also seen in a Parkinson knockdown SH-SY5Y model [46]. Naringenin, a natural flavonoid which has a possible effect of counteracting the neurodegenerative process in Alzheimer disease, increased cell viability due to its antioxidative properties in a neurotoxicity-induced model of SH-SY5Y cells [47]. Baicalein, a flavonoid used in traditional medicine herbs, reduced the MPTP-induced neurotoxicity of the SH-SY5Y cells [48]. There was also an effect of reduced oxidative stress and neuroprotection in a 6-OHDA induced neurotoxicity SH-SY5Y model with maackiain, a flavonoid extracted from the plant named Sophora flavescens [49]. Quercetin is another phenolic compound that decreases apoptosis in a neurotoxicity induced in vitro model by modulating the autophagic pathway [50]. Green tea phenols exerted antioxidative effects in 6-OHDA or rotenone induced neurotoxicity SH-SY5Y models [51].
Numerous other molecules have been tested using SH-SY5Y in vitro models of induced neurotoxicity, but the results need to be further confirmed by more complex experimental models. These experiments open new perspectives and the hopes for the prevention or mitigating this neurodegenerative phenomenon.
The prion-like neuronal transmission of pathological alpha-synuclein is one of the current research topics in the pathophysiology of PD. The SH-SY5Y in vitro models are a powerful tool for the proof-of-concept of the proposed mechanism on which this hypothesis is based. Exosomes are extracellular vesicles with a diameter of 30–150 nm secreted by the cells that carry different small molecules such as proteins or microRNAs [52]. The exosomes are taken up by a recipient cell and the cargo may modulate cell behavior, thus having a significant role in intercellular communication [52]. The role of interneuronal transmission of the alpha-synuclein via the exosomes has gained increased interest [53]. In a SH-SY5Y -derived cell model that overexpresses wild-type alpha-synuclein, exosomes that carry alpha-synuclein were identified in the cell culture supernatant [54]. After incubating normal SH-SY5Y cells with the supernatant enriched in exosomes carrying alpha-synuclein, the presence of alpha-synuclein was also detected in the recipient cells, similar to the prion-like neuronal transmission via exosomes [54].
The SH-SY5Y cell line is an appropriate starting point for in vitro experiments that study the role of the microbiota in the pathophysiology of PD. Short chain fatty acids (SCFA) such as butyrate, acetate or propionate are produced by the gut microbiota and have a role in the maintenance of gut inflammatory homeostasis [55]. A decrease of the SCFA in the gut has been observed in PD patients [56]. The protective role of the SCFA in PD was tested in vitro by using models of SH-SY5Y neurotoxicity, in which a decrease of toxicity-induced apoptosis was seen when SCFA were administered [57]. Another study revealed that the supernatant of bacteria that grow in the gut microbiota (containing the metabolites secreted in culture, including SCFA) exerted a neuroprotective effect in neurotoxic-exposed SH-SY5Y cells [58].
In another in vitro experiment, the interaction between the three key players that are involved in the mechanisms in which microbiota favors the pathological changes of PD (the bacteria, the intestinal cells and the neurons) was studied [59]. Intestinal epithelial CaCo-2 cells were treated with a bacterial strain that reside in the intestinal microbiota, Ruminococcus albus, and then the conditioned media was administered in neurotoxicity-induced SH-SY5Y cells [59]. This study showed that the conditioned media had a protective effect and prevented oxidative stress and apoptosis in the SH-SY5Y cells [59].
In contrast to those studies in which the lack of certain molecules favors the neurodegenerative phenomenon and the experimental models of cellular neurotoxicity show the protective role of these molecules in preventing cellular death, there is also those instances in which the exposure to a certain compound causes pathological changes. Curli protein is an amyloid protein secreted by the commensal bacteria from the gut and is able to induce misfolding of other amyloidogenic proteins [60]. An in vitro model of transfected SH-SY5Y cells with over-expression of alpha-synuclein was established and the cells were exposed to the major curlin subunit (CsgA), the major subunit of the curli protein [61]. A significant increase in alpha-synuclein aggregation was noticed in the presence of CsgA, suggesting the role of this bacterial peptide in the promotion of aggregation of the pathological protein, a process that, from a pathophysiological point of view, can represent the initiation of PD [61].
The SH-SY5Y cell line was also successfully used to study autophagy. Studies demonstrating the involvement of autophagy in neurotoxicity models of SH-SY5Y and showing that neuroprotective compounds modulate the autophagy-lysosome system have been published [62, 63].
Autophagy was analyzed in a rotenone-induced neurotoxicity model of SH-SY5Y and showed that after exposure to rotenone, autophagic vacuoles form in the cytoplasm of the neurons and autophagic markers, such as Microtubule-associated protein 1A/1B-light chain 3 (LC3) or tumor protein p63 (p63) [62]. This suggests that the autophagy-lysosome system has a certain role in the induction of the PD cellular model [62].
Another experiment using SH-SY5Y cells assessed the temporal relation between alpha-synuclein inclusions and the degree of activation of autophagy [63]. Exogenous alpha-synuclein fibrils were administered in differentiated SH-SY5Y cell culture and the level of alpha-synuclein and autophagic markers, such as p62, LC3 and lysosome-associated membrane protein 2 (LAMP-2), were measured [63]. The exogenous fibrils induced the aggregation of endogenous alpha-synuclein and an increase in the autophagic markers was noticed with a similar temporal profile [63]. Moreover, the activation of autophagy with rapamycin lowered the apha-synuclein inclusions, while the inhibition of autophagy with chloroquine increased the inclusions [63].
Studies in which the potential therapeutic compounds in neurotoxicity models of SH-SY5Y cells have been performed to analyze their effect on the autophagy-lysosome system and have shown that neuroprotective molecules also induce a stimulation of the autophagy-lysosome system.
From a physiological point of view, primary neuronal cultures are currently the most relevant in PD research [3, 64]. Primary neuronal cultures are especially difficult to obtain from PD patients, thus, they are usually obtained from the embryonic or post-natal midbrain of rodents [3, 64]. Nevertheless, when compared to primary neuronal cultures, the most important advantage of using the SH-SY5Y cell line is that the cells are easier to maintain in culture than primary neurons [3]. The main disadvantage is that differentiation to dopaminergic neurons may prove to be a difficult and variable process [3].
Other popular cell lines for the study of PD are: the hybrid MN9D immortalized dopaminergic neuronal cell line, which was obtained from the fusion of mouse embryonic ventral mesencephalic cells with neuroblastoma cells, the pheochromocytoma cell line PC12, Lund human mesencephalic cells (LUHMES), and rat immortalized neuronal progenitor cell line CSM14.1. Among these cell lines, SH-SY5Y is the only cell line which is entirely of human origin [3, 65].
In conclusion, the SH-SY5Y cell line represent a simple, easy, inexpensive in vitro experimental model for studying PD that can bring extremely valuable data to this field of research. The cell line has some disadvantages such as the possibility of turning into an epithelial phenotype and the presence of genetic instability, considering the origin of the cell line is from a neuroblastoma. Nevertheless, it is useful as a first step when testing for pathomechanisms and potential interventions in experimental PD. This may be further expanded to more complex in vitro models such as primary neurons, induced pluripotent stem cells or to in vivo murine models. The SH-SY5Y cell line is well known for in vitro models of induced neurotoxicity in searching for new drugs in PD. In addition, this cellular line can be easily used as an in vitro model for the less explored topic of pathophysiological mechanisms in PD, especially for expanding our knowledge in the role of microbiota in the initiation and propagation of the disease, of which there is rapidly growing interest.
PD, Parkinson disease; CMA, chaperone-mediated autophagy; Atg, as autophagy related; PINK1, PTEN-induced kinase; GBA, glucocerebrosidase; LRRK-2, leucine rich repeat kinase 2; TPA, 2-O-tetradecanoyl-phorbol-13 acetate; BDNF, Brain-derived neurotrophic factor; dbcAMP, Dibutyryl cyclic adenosine monophosphate; 6-OHDA, 6-hydroxydopamine; MPP+, 4-phenylpyridinium; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; EPO, Erythropoietin; SCFA, Short chain fatty acids; LUHMES, lund human mesencephalic cells; PFF, preformed fibrils; sirtuin 3, SIRT3; CsgA, major curlin subunit; LC3, Microtubule-associated protein 1A/1B-light chain 3; p63, tumor protein p63; LAMP-2, lysosome-associated membrane protein 2.
Writing (original draft preparation) of the manuscript—OCI; Review and editing of the initial draft—LCC, BOP; Supervision—LCC, BOP.
Not applicable.
Not applicable.
This work was funded by Ministry of Research, Innovation and Digitalization in Romania, under Program 1—The Improvement of the National System of Research and Development, Subprogram 1.2—Institutional Excellence-Projects of Excellence Funding in RDI, Contract No. 31PFE/30.12.2021 and the National Program 1N/2019/PN19.29.02.01.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.