


 , Niccolò Candelise 2,3, Piergiorgio Grillo 1, Clara Simonetta 1, Roberta Bovenzi 1, Alberto Ferri 3,4, Cristiana Valle 3,4, Nicola Biagio Mercuri 1,5, Tommaso Schirinzi 1,*
, Niccolò Candelise 2,3, Piergiorgio Grillo 1, Clara Simonetta 1, Roberta Bovenzi 1, Alberto Ferri 3,4, Cristiana Valle 3,4, Nicola Biagio Mercuri 1,5, Tommaso Schirinzi 1,*
1 Unit of Neurology, Department of Systems Medicine, University of Roma Tor Vergata, 00133 Rome, Italy
2 Department of Experimental Medicine, University of Roma La Sapienza, 00185 Rome, Italy
3 IRCCS Fondazione Santa Lucia, 00179 Rome, Italy
4 Institute of Translational Pharmacology (IFT), Consiglio Nazionale delle Ricerche (CNR), 00185 Rome, Italy
5 Experimental Neuroscience Unit, IRCCS Fondazione Santa Lucia, 00179 Rome, Italy
Academic Editor: Ho-Sung Ryu
Abstract
Background: Monoamine oxidase type B inhibitors (iMAO-Bs) are a class of largely-used antiparkinsonian agents that, based on experimental evidence, are supposed to exert different degrees of neuroprotection in Parkinson’s disease (PD). However, clinical proofs on this regard are very scarce. Since cerebrospinal fluid (CSF) reflects pathological changes occurring at brain level, we examined the neurodegeneration-related CSF biomarkers profile of PD patients under chronic treatment with different iMAO-Bs to identify biochemical signatures suggestive for differential neurobiological effects. Methods: Thirty-five PD patients under chronic treatment with different iMAO-Bs in add-on to levodopa were enrolled and grouped in rasagiline (n = 13), selegiline (n = 9), safinamide (n = 13). Respective standard clinical scores for motor and non-motor disturbances, together with CSF biomarkers of neurodegeneration levels (amyloid-
Keywords
- Parkinson's disease
- CSF biomarkers
- neurodegeneration
- neuroprotection
- MAO inhibitors
- selegiline
- rasagiline
- safinamide
Parkinson’s disease (PD) is a common neurodegenerative disorder responsible for a disabling syndrome, including motor and non-motor disturbances. While neuropathology and main pathogenic pathways have been established over time, PD still remains an incurable disorder. Indeed, effective symptomatic therapies exist, which mostly replace deficient dopamine, but, to date, no disease-modifying treatments are available [1].
Monoamine Oxidase type B Inhibitors (iMAO-Bs) are a class of antiparkinsonian drugs, used in mono or add-on therapy along the entire disease course, that potentiate dopaminergic transmission at striatal level by reducing dopamine degradation [2]. The three main iMAO-Bs are selegiline, rasagiline, and safinamide, which have individual peculiarities in the spectrum of molecular effects and in clinical indications [3]. Beside symptomatic relief in motor and non-motor disturbances, iMAO-Bs have been considered as neuroprotective agents, based on a body of in vitro and in vivo experimental evidence. Specifically, iMAO-Bs may operate neural defence against mitochondrial toxins (MPTP, rotenone and paraquat) [4, 5, 6, 7], monoamine-depleting neurotoxins (N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine hydrochloride (DSP-4)) [8], and hypoxic conditions [9, 10], and exert antioxidant properties [4, 11, 12, 13], antiapoptotic [5, 14, 15, 16] and neurotrophic effects [17, 18]. Nevertheless, human-based proofs of iMAO-Bs-associated neuroprotection are very limited and not completely univocal; moreover, no comparison studies among the single iMAO-Bs exist.
Cerebrospinal fluid (CSF) reflects pathological changes of the brain. Accordingly, the CSF measurement of neurodegeneration-related peptides, such as amyloid-
Here we thus conducted a study on the CSF biomarkers profile of PD patients chronically treated with both L-dopa and different iMAO-Bs in add-on, aiming to disclose biochemical signatures suggestive of differential neurobiological effects.
The study involved a total of 35 PD patients afferent to Neurology Unit of Tor Vergata University Hospital (Rome, Italy). PD was diagnosed following the 2015 MDS criteria. All patients were receiving iMAO-Bs add-on therapy for one year at least (n = 13 rasagiline 1 mg/die, n = 9 selegiline 10 mg/die, n = 13 safinamide 100 mg/die); the remaining dopaminergic therapy was stable for at least 6 months. For each participant, demographics and medical history were collected, together with MDS-UPDRS pars III [21] and Montreal Cognitive Assessment (MoCA) [22] scores; the levodopa equivalent daily dose (LEDD) was calculated. They all underwent lumbar puncture (LP) according to standard procedures [23]. Briefly, CSF was collected in polypropylene tubes, carried on ice to local laboratory, centrifuged at 2000 rpm for 10 min at 4°, aliquoted in polypropylene vials (0.5 mL) and stored at –80°, pending analysis. A routine blood sample was obtained contemporary to LP, to allow a comparative chemical analysis of CSF. In addition, CSF sample was excluded if microscopic observation revealed
Variables distribution was preliminarily examined by Shapiro-Wilk test. Clinical-demographic data were compared among the groups by Mann–Whitney U test. Categorical variables were compared by chi-square test. Considering the small sample size, although normally distributed, CSF neuorodegenerative biomarkers and lactate levels were compared among groups by Kruskal-Wallis test. Pairwise comparisons after Kruskal-Wallis test was also assessed. A p
No differences resulted in demographics and clinical parameters among the groups. Regarding CSF biomarkers, t-tau (p = 0.005), p-tau (p = 0.017) and lactate (p = 0.037) levels significantly differed among the groups (Table 1). Pairwise comparisons showed significant differences of t-tau (selegiline vs. rasagiline, p = 0.027; selegiline vs. safinamide, p = 0.006), p-tau (selegiline vs. safinamide, p = 0.05; selegiline vs. safinamide, p = 0.021) and lactate (selegiline vs. safinamide, p = 0.041). Graphical data for t-tau, p-tau and lactate are represented in Fig. 1A–C. CSF biomarkers did not differ between females and males.
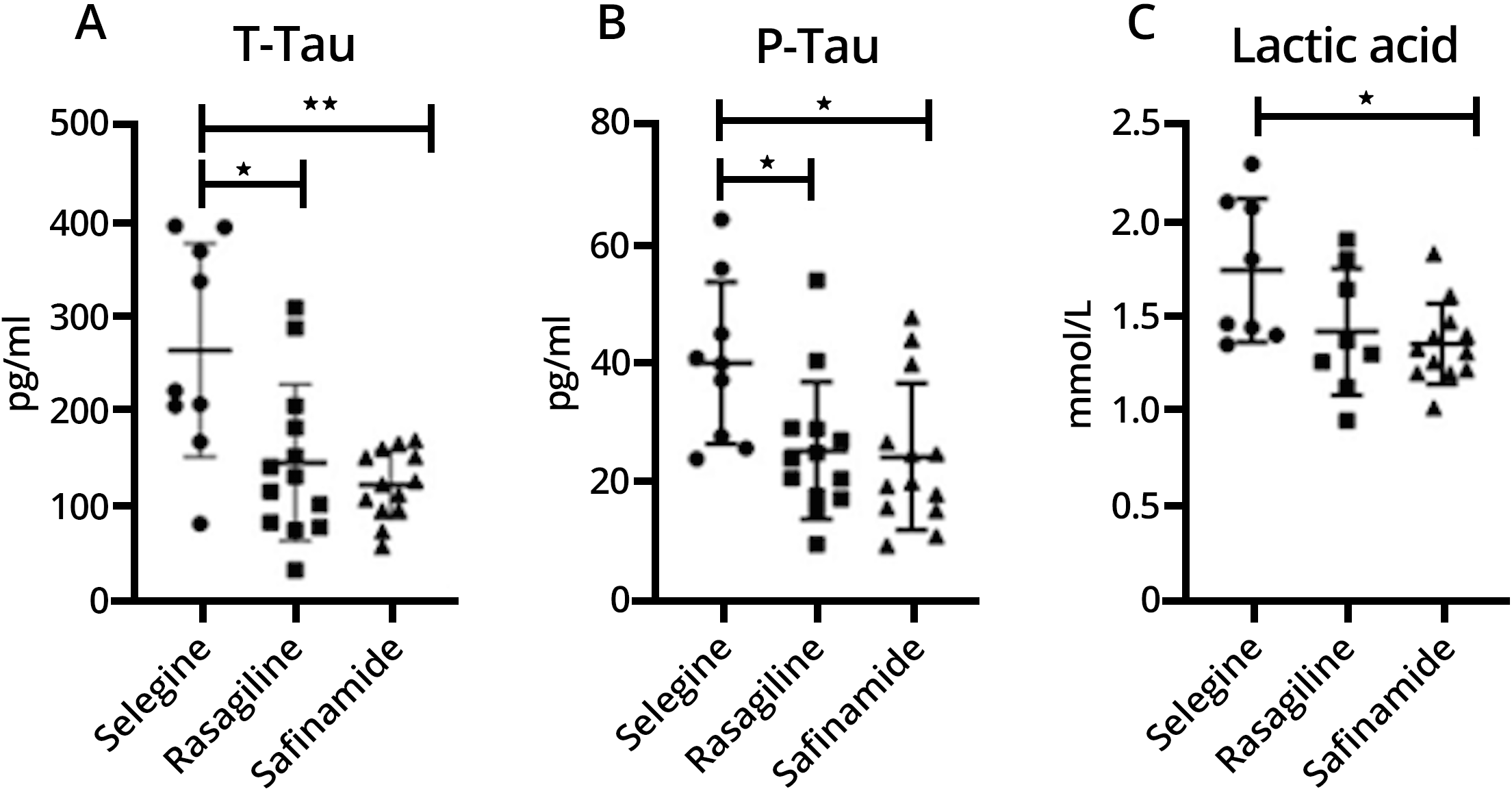 Fig. 1.
Fig. 1.
T-Tau (Fig. 1A), P-Tau (Fig. 1B) and lactic acid (Fig. 1C) levels are expressed as mean
| Variable | Selegiline | Rasagiline | Safinamide | Significants | |||
| n = 9 | n = 13 | n = 13 | |||||
| (f: 55.6%; m: 44.4%) | (f: 30.8%; m: 69.2%) | (f: 69.2%; m: 30.8%) | ns | ||||
| Clinical variable | Mean | S.D. | Mean | S.D. | Mean | S.D. | |
| Age (y) | 54.7 | 7.6 | 52.1 | 13.1 | 54.3 | 9.3 | ns |
| Duration (y) | 3.9 | 2.5 | 6.1 | 1.4 | 6.9 | 1.0 | ns |
| MOCA | 26.0 | 1.2 | 27.5 | 1.3 | 24.2 | 2.0 | ns |
| UPDRS III | 25.7 | 11.2 | 26.7 | 6.6 | 35.2 | 6.1 | ns |
| LEDD | 543.9 | 152.1 | 567.5 | 139.6 | 718.8.3 | 74.1 | ns |
| CSF biomarker | |||||||
| t-tau (pmol/mL) | 264.5 | 112.3 | 146.2 | 82.3 | 122.8 | 36.1 | p = 0.005 |
| p-tau (pmol/mL) | 40.1 | 13.6 | 25.4 | 11.5 | 24.4 | 12.3 | p = 0.017 |
| A |
775.0 | 249.7 | 837.2 | 316.5 | 912.3 | 264.8 | Ns |
| A |
7553.3 | 1663.7 | 5239.7 | 1043.7 | 5401.0 | 1737.3 | Ns |
| A |
0.131 | 0.063 | 0.167 | 0.057 | 0.172 | 0.025 | Ns |
| Lactate (mmol/mL) | 1.74 | 0.38 | 1.41 | 0.33 | 1.35 | 0.21 | p = 0.037 |
This pilot study examined the profile of neurodegeneration-related CSF biomarkers of three clinically-homogenous PD groups treated with different iMAO-Bs (namely, selegiline, rasagiline and safinamide) in add-on, looking for biochemical signatures suggestive for differential neurobiological effects. The higher levels of t-tau, p-tau, and lactate measured in patients under selegiline, compared to those under rasagiline and safinamide, indicate distinct molecular or neuropathological substrates associated with different iMAO-Bs.
Tau proteins in CSF are basically considered as marker of neuronal damage, and tauopathy overall. While the CSF levels proportionally reflect neurofibrillary tangles deposition in Alzheimer’s disease (AD) [25], the neuropathological correlate in PD is not completely clear yet. Molecular interactions between tau and
The clinical homogeneity of our study population allows us to refer, at least in part, differences in CSF biomarkers profile of the three iMAO-Bs groups (in particular, the higher levels of tau proteins and lactate in selegiline group compared to rasagiline and safinamide groups) to the distinct action mechanisms and metabolism of the drugs. Indeed, selegiline is metabolized to 1-amphetamine and to 1-methamphetamine [34], having amphetamine-like properties [35], whereas rasagiline is metabolized to 1-aminoindan, not displaying amphetamine-like effects [36]. It is well known that amphetamine may be neurotoxic [37]. Conversely, experimental data from cellular models disclose neuroprotective effects of 1-aminoindan [38, 39]. Moreover, selegiline and rasagiline differently modulate synaptic activity at corticostriatal level [2], which is consistent with distinct pathways of molecular activation in neurons that, in turn, may translate in long-term differences in proteinopathy and degeneration shaping. Safinamide, instead, has a prominent antiglutamatergic effect, either by antagonizing NMDA-mediated signalling or by blocking voltage-dependent sodium and N-type calcium channels [40, 41, 42]. The inhibition of glutamate transmission, the subsequent attenuation of associated excitotoxicity, and the containment of oxidative stress [43], might thus account for the milder profile of neurodegeneration observed in patients taking safinamide, compared to those taking selegiline.
We only observed differences in tau proteins, but not in amyloid-
This study is limited by the sample size, the retrospective design, the relative exiguity of the biomarkers panel, and the absence of a control group without iMAO-Bs (which indeed could have presented with substantial differences in clinical severity or disease duration). Despite limitations, we provided preliminary evidence on the distinct biochemical endophenotype of PD patients under various iMAO-Bs. With the due caution, we could refer differences in CSF neurodegeneration-related biomarkers to respective pharmacodynamic peculiarities of selegiline, rasagiline, and safinamide. In particular, the lower levels of tau proteins and lactate may indicate a differential effect on neurodegenerative cascade operated by rasagiline and safinamide that, differently from selegiline, do not display an amphetamine-like behaviour. This preliminary work also highlighted the need for prospective comparative trials to examine neuroprotection activity associated with different iMAO-Bs, which should include CSF biomarkers of neurodegeneration among the outcomes, as more recent studies do [48, 49].
HZ collected and analyzed data, wrote the manuscript. NC analyzed data and wrote the manuscript. AF, CV and NBM contributed to data interpretation and edited the final manuscript. PG, RB, and CS collected and analyzed data. TS analyzed and interpreted data, edited the final manuscript.
The study agreed the ethical principles of Helsinki declaration, and received approval of the local committee (0026092/2017). Informed written consent was signed by all the participants.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.