


 , Junya Kobayashi 2, Ryo Sugaya 1, Imaharu Nakano 3,‡, Shigeru Fujimoto 1
, Junya Kobayashi 2, Ryo Sugaya 1, Imaharu Nakano 3,‡, Shigeru Fujimoto 11 Division of Neurology, Department of Internal Medicine, Jichi Medical University School of Medicine, Tochigi, 329-0498, Japan
2 Department of Genome Repair Dynamics, Radiation Biology Center, Kyoto University, Kyoto, 606-8501, Japan
3 Department of Neurology, Tokyo Metropolitan Neurological Hospital, Tokyo, 183-0042, Japan
‡: Deceased July 16, 2014
Abstract
Autosomal recessive cerebellar ataxias comprise many types of diseases. The most frequent autosomal recessive cerebellar ataxias are Friedreich ataxia, but other types are relatively rare. We encountered a consanguineous family with two cases of late-onset cerebellar ataxia with neuropathy. We performed whole-exome sequencing in one patient and confirmed by Sanger sequencing in other family members. Neurological examination revealed cerebellar ataxia, hand tremor, and neck dystonia, distal muscle wasting, and diminished tendon reflexes. The patients had no conjunctival telangiectasia or immunodeficiency. Blood examination revealed slightly elevated α-fetoprotein. Brain MRI demonstrated marked cerebellar atrophy and mild brainstem atrophy. The electrophysiologic study and nerve biopsy showed axonal neuropathy. Whole-exome sequencing revealed a novel homozygous missense variant (NM_000051.3: c.496G > C) in the ataxia-telangiectasia mutated gene. This homozygous variant was found in another patient, co-segregated within the family members—this variant results in aberrant splicing (skipping exon 5) on RT-PCR analysis. We identified the ataxia-telangiectasia mutated variant in an adult, late-onset autosomal recessive cerebellar ataxias family. We should consider ataxia-telangiectasia even in late-onset autosomal recessive cerebellar ataxias without telangiectasia or immunodeficiency.
Keywords
- Autosomal recessive cerebellar ataxia
- muscle atrophy
- neuropathy
- ataxia-telangiectasia mutated
- splicing mutation
- whole-exome sequencing
Autosomal recessive cerebellar ataxias (ARCA) comprise numerous types of diseases that are characterized by early-onset cerebellar ataxia with additional clinical features, including peripheral neuropathy, oculomotor apraxia, involuntary movements, and retinal abnormalities. The most frequent ARCA in the worldwide is Friedreich ataxia (Anheim et al., 2012), whereas we could not find any reports about Japanese ARCA cases with frataxin (FXN) gene mutations to date (Tsuji et al., 2008). The RFC1 expansion causes cerebellar ataxia, neuropathy, and vestibular areflexia syndrome (CANVAS), which is a relatively common cause of late-onset ARCA (10%) (Cortese et al., 2019).
In Japan, the most frequent ARCA is early-onset ataxia with oculomotor apraxia and hypoalbuminemia/ataxia with oculomotor apraxia type 1 (EAOH/AOA1) (Yokoseki et al., 2011), followed by autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) (Shimazaki and Takiyama, 2012), ataxia with oculomotor apraxia type 2 (AOA2) (Asaka et al., 2006; Ichikawa et al., 2013; Nakamura et al., 2009), and α-tocopherol transfer protein deficiency (Gotoda et al., 1995; Yokota et al., 1997). Ataxia-telangiectasia is relatively rare in Japan (Morio et al., 2009), as well as autosomal recessive cerebellar ataxia type 1 (SYNE1 mutation) (Izumi et al., 2013), ANO10 (Maruyama et al., 2014), and TTC19 (Morino et al., 2014).
Ataxia-telangiectasia (AT) usually develops before five years of age. AT is characterized by ataxia, conjunctival telangiectasia, susceptibility to infections and malignancy, neuropathy, oculomotor apraxia, and elevated α-fetoprotein (AFP). Ataxia-Telangiectasia Mutated (ATM) is the causative gene for AT (Savitsky et al., 1995). Late-onset Ataxia-telangiectasia, so-called ‘AT variant’, is known to show atypical neurological features (Verhagen et al., 2009).
Here, we encountered two siblings having AT variant with adult-onset, cerebellar ataxia, involuntary movement, and neuropathy with a novel homozygous splicing variant in the ATM gene. They showed atypical clinical manifestations such as marked distal muscle wasting and prolonged survival.
The case report includes two patients and one healthy sibling in a Japanese consanguineous family. The family tree is shown in Fig. 1A. The parents (III-1 and III-2) were second cousins. Their mother (III-2) died from gastric cancer, and their brothers (IV-3 and IV-4) suffered from prostatic cancer and died from esophageal cancer, respectively.
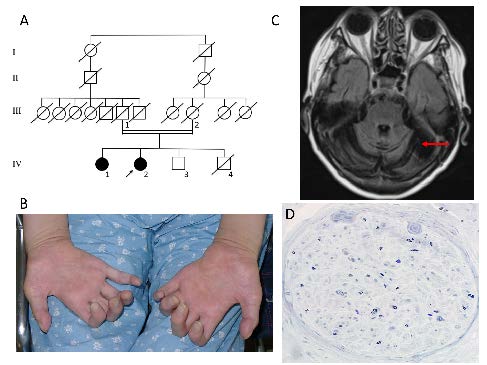 Figure 1.
Figure 1.Clinical findings in patient IV-2. A. Family tree. The proband (IV-2) and his older sister (IV-1) show almost the same clinical phenotype. Other members are all healthy. The parents (II-1 and 2), and a sibling (IV-3) were neurologically asymptomatic. Patient IV-1 died at age 71 of gallbladder cancer, and IV-2 died at age 76 of chronic lymphocytic leukemia. Their mother (III-2) died of gastric cancer. IV-4 died with esophageal cancer. IV-3 suffered from prostatic cancer recently. B. Bilateral intrinsic hand muscle atrophies were noted. C. Brain MRI showed marked cerebellar atrophy (arrow) and mild brainstem atrophy. D. Sural nerve biopsy revealed severe loss of axons and myelin.
The patients (IV-1 and IV-2) suffered from gait disturbance and hand action tremor at age 17. They noticed diminished foot sensation at age 24. They could walk without assistance at age 37. At age 38, tremulous cervical dystonia appeared and gradually worsened. Their neck torticollis-like movement and hand tremors were so severe that we carried out stereotaxic operations on their Vim thalami. The operations (thalamotomy) alleviated their tremor and torticollis-like movement to some extent. The ataxia was gradually progressive, and their distal muscle power and muscle bulks decreased. The younger sister (IV-2) was admitted to our hospital at age 66. Neurological examination revealed marked slurred speech and bilateral gaze nystagmus, but no oculomotor apraxia. The distal muscle weakness and atrophy were prominent in both the upper (Fig. 1B) and lower extremities. Tendon reflexes were all absent, and glove and stocking type hypoesthesia was observed at all sensory modalities.
Nerve conduction studies revealed that compound muscle action potentials were reduced in the upper extremities, and absent in the lower extremities. Sensory nerve action potentials were absent in any extremities. Needle EMG showed chronic neurogenic changes in all examined muscles. Brain MRI showed cerebellar atrophy and mild brainstem atrophy (Fig. 1C). Sural nerve biopsy revealed marked myelinated axonal loss (Fig. 1D).
The two patients did not exhibit conjunctival telangiectasia or immunodeficiency. Blood examinations disclosed mild AFP elevation (IV-1: 21 ng/ml, IV-2: 37 ng/ml (normal < 10 ng/ml)). After that, the two patients (IV-1 and IV-2) died from gallbladder cancer at age 70 and chronic lymphocytic leukemia at age 76, respectively.
DNA analyses were performed for both patients (IV-1 and 2), and the healthy sibling (IV-3). We performed whole-exome sequencing in one patient (IV-2).
Whole-exome sequencing (WES) was performed using DNA extracted from the proband’s (IV-2) leukocytes, as previously described (Shimazaki et al., 2014). DNA was sheared and captured with an Agilent SureSelect XT Human All Exon V4 kit. Paired-end sequencing was carried out on an Illumina Hiseq 2000. The coverage of the target region (read depth 10X) was 99.79%. We aligned and mapped the sequences using the Burrows-Wheeler Aligner (BWA), and carried out single nucleotide variants (SNVs) and Insertion/deletions (Indels) calling with a Genome Analysis Toolkit (GATK). We performed PCR to detect GAA repeat expansions of the frataxin gene (Campuzano et al., 1996).
We confirmed the variant by Sanger sequencing of the family members. To confirm the nucleotide substitution of exon 5 in the ATM gene observed on WES analysis, Sanger sequencing was performed with an ABI 3730 utilizing DNA from the proband (IV-2), older sister (IV-1), and healthy brother (IV-3). The primer sequences used for PCR and sequence were as follows: ATM-ex5F: 5’-agt tgc cat tcc aag tgt ctt-3’, ATM-ex5R: 5’-gtc agg tca ctt ggg gga ta-3’.
We extracted total RNA from the proband’s (IV-2) leukocytes with informed consent. First-strand cDNA was generated with a SuperscriptTM III First-Strand Synthesis System (Invitrogen) according to the manufacturer’s recommendations and was used as a template for PCR.
The primer sequences used for RT-PCR were as follows: ATMcDNA-4F: 5’-agg cag aaa aag atg cag ga -3’, ATMcDNA-7R: 5’-ttc gaa agt tga cag cca aa-3’. The PCR products were directly sequenced for both strands using the above primers.
Whole-exome sequencing (WES) revealed a novel homozygous single nucleotide variant (NM_000051.3: c.496G > C) in the last nucleotide of exon 5 in the ATM gene (Fig. 2A). This homozygous variant was found in another patient (IV-1), was heterozygous in IV-3, indicating co-segregation within the family members (Fig. 2A). Moreover, this substitution was not found in the 1000 Genomes project, Human Genome Variation Database, or Exome Aggregation Consortium (ExAC) database. WES and sequencing analyses did not reveal any pathologic substitutions in the APTX, SETX, PIK3R5, PNKP, MRE11, and TDP1 genes. We did not detect GAA repeat expansions of the FXN gene.
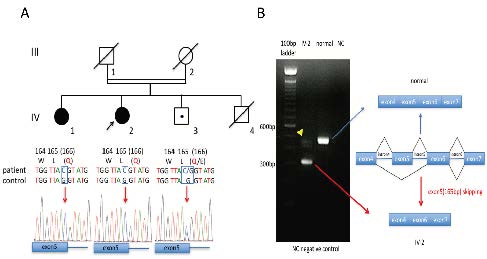 Figure 2.
Figure 2.Results of the ATM gene and mRNA sequence analyses. A.Whole-exome sequencing revealed a novel homozygous single nucleotide variant in the last nucleotide of ATM exon 5. Sanger sequencing confirmed the homozygous missense variant (c.496G > C) of the ATM gene in the proband (IV-2) and the affected sister (IV-1). An asymptomatic brother (IV-3) was a heterozygous carrier. This mutation was co-segregated with the disease in this family. B. RT-PCR analysis of IV-2 mRNA. RT-PCR was performed with mRNA from the proband’s leukocytes. The results showed short and normal (arrowhead) PCR products. Sequence analysis of the short product disclosed it had exon 5 (165bp) completely deleted. So, this mutation was a leaky splice site mutation.
RT-PCR of the patient’s cDNA and its sequencing showed aberrant splicing of exon 5 in the ATM gene (Fig. 2B). RT-PCR products indicated that the mutant mRNA was shorter than the wild-type ATM mRNA (Fig. 2B, left). Sequencing analysis of this short RT-PCR product disclosed a complete 165 nucleotide loss of exon 5 at the cDNA level. We demonstrated this splicing variant (NM_000051.3: c.496G > C) generated a product of skipping exon 5 (p.[R111_L165del; E166K]) (Fig. 2B, right).
We could score this variant using ACMG criteria: PM1 (Located in a mutational hot spot and/or critical and well-established functional domain without benign variation.), PM2 (Absent from controls in Exome Sequencing Project, 1000 Genomes Project, or Exome Aggregation Consortium.), PP3 (Multiple lines of computational evidence support a deleterious effect on the gene or gene product.), PP5 (Reputable source recently reports variant as pathogenic, but the evidence is not available to the laboratory to perform an independent evaluation.). We could interpret this variant as likely pathogenic, even without functional evidence (Hampel et al., 2015).
We report 2 cases of the late-onset variant ataxia telangiectasia, with no telangiectasia, nor immunodeficiency. Both cases exhibited cervical dystonia, hand tremor, cerebellar ataxia, peripheral neuropathy, and distal muscle atrophy. Distal muscle atrophy is known as an atypical feature of AT and was a prominent symptom in these patients. Focal dystonia of neck or upper limbs is a relatively common feature in adult AT.
In these cases, prolonged survival was observed, both dying their seventies, compared to the median Japanese AT survival age of 26 years (Morio et al., 2009). These atypical and milder phenotypes might result from partially preserved ATM kinase activities in these patients, but unfortunately, cell lines were not available from either A-T patient to test the level and size of their ATM as well as its activity. In contrast, those with mild or late-onset symptoms had missense or leaky splice site variants that showed some ATM kinase activity (Verhagen et al., 2012).
Ataxia-telangiectasia is characterized by increased cancer susceptibility (Lavin, 2008). Previous reports showed that patients exhibited high frequencies of lymphoid tumors and breast cancer (Reiman et al., 2011). Our two patients with a homozygous ATM mutation died of chronic lymphocytic leukemia and gallbladder cancer, respectively. Also, the heterozygous carrier developed prostate cancer, while patients' mother (III-2) and brother (III-4), who might have been carriers, suffered from gastric and esophageal cancer, respectively. Carriers with ATM heterozygous mutations are also susceptible to develop cancer and should be followed carefully.
We identified a novel homozygous single nucleotide variant in the ATM gene. This variant is co-segregated within the family, and not found in normal databases. Moreover, this nucleotide substitution is located at the 3’ end of exon 5, resulting in aberrant splicing of the ATM mRNA and in-frame deletion of exon 5 comprising 165bp. In summary, AT should be considered even in late-onset ARCA without telangiectasia or immunodeficiency.
H.S. and J.K. conceived and designed the experiments; H.S. and J.K. performed the experiments; H.S., J.K., R.S., and I.N. analyzed the data; H.S. and R.S. wrote the paper. S.F. supervised the study.
Peripheral blood leukocytes were obtained with the informed consent of all participants. The institutional review board of the Jichi Medical University approved genetic analyses.
This study was supported by a Grant-in-Aid for Scientific Research (C) (23591253 to HS) from The Ministry of Education, Culture, Sports, Science, and Technology in Japan.
The authors declare no conflict of interest. The deceased author had no conflict of interest via his wife.