[1]Kim TW, Lee SU, Park B, Jeon K, Park S, Suh GY, et al. Clinical effects of bacteremia in sepsis patients with community-acquired pneumonia. BMC Infectious Diseases. 2023; 23: 887. https://doi.org/10.1186/s12879-023-08887-5.
[2]Bush NG, Diez-Santos I, Abbott LR, Maxwell A. Quinolones: mechanism, lethality and their contributions to antibiotic resistance. Molecules. 2020; 25: 5662. https://doi.org/10.3390/molecules25235662.
[3]Jivcu C, Gotfried M. Gemifloxacin use in the treatment of acute bacterial exacerbation of chronic bronchitis. International Journal of Chronic Obstructive Pulmonary Disease. 2009; 4: 291–300. https://doi.org/10.2147/copd.s3903.
[7]Mu K, Jiang K, Wang Y, Zhao Z, Cang S, Bi K, et al. The biological fate of pharmaceutical excipient β-Cyclodextrin: pharmacokinetics, tissue distribution, excretion, and metabolism of β-Cyclodextrin in rats. Molecules. 2022; 27: 1138. https://doi.org/10.3390/molecules27031138.
[9]lshafie HS, Sadeek SA, Camele I, Mohamed AA. Biochemical characterization of new gemifloxacin schiff base (GMFX-o-phdn) metal complexes and evaluation of their antimicrobial activity against some phyto- or human pathogens. International Journal of Molecular Sciences. 2022; 23: 2110. https://doi.org/10.3390/ijms23042110.
[11]Munir R, Hadi A, Khan SUD, Asghar S, Irfan M, Khan IU, et al. Solubility and Dissolution Enhancement of Dexibuprofen with Hydroxypropylbetacyclodextrin (HPβCD) and Poloxamers (188/407) Inclusion Complexes: Preparation and In Vitro Characterization. Polymers. 2022; 14: 579. https://doi.org/10.3390/polym14030579.
[12]Mondal L, Mukherjee B, Chakraborty S, Bhattacharya S, Ehsan I, Sengupta S, et al. Comparison of enhanced solubility profiles, analysis of thermodynamic parameters and pharmacokinetic profile related to tamoxifen citrate solubilisation. Novel Approaches in Drug Design & Development. 2018; 3: 555624. https://doi.org/10.19080/NAPDD.2018.03.555624.
[13]Guo Y, Sun CC. Pharmaceutical Lauryl Sulfate Salts: Prevalence, Formation Rules, and Formulation Implications. Molecular Pharmaceutics. 2022; 19: 432–439. https://doi.org/10.1021/acs.molpharmaceut.1c00690.
[15]ElShaer A, Ouyang D, Hanson P, Mohammed AR. Preparation and evaluation of amino acid based salt forms of model zwitterionic drug ciprofloxacin. Journal of Pharmaceutics & Drug Delivery Research. 2013; 2: 1. http://dx.doi.org/10.4172/2325-9604.1000111.
[16]Pignatello R, Corsaro R, Bonaccorso A, Zingale E, Carbone C, Musumeci T. Soluplus® polymeric nanomicelles improve solubility of BCS-class II drugs. Drug Delivery and Translational Research. 2022; 12: 1991–2006. https://doi.org/10.1007/s13346-022-01182-x.
[17]Muhamad H, Bashir AB, Charlton-Harrison J, Abdulhussain R, Mawla N, Patel K, et al. Hot-melt extruded-FDM 3D-printed polyethylene oxide tablets: Dissolution imaging analysis of swelling and drug release. European Journal of Pharmaceutics and Biopharmaceutics. 2025; 208: 114636. https://doi.org/10.1016/j.ejpb.2025.114636.
[18]Loftsson T, Másson M, Brewster ME. Self-association of cyclodextrins and cyclodextrin complexes. Journal of Pharmaceutical Sciences. 2004; 93: 1091–1099. https://doi.org/10.1002/jps.20047.
[20]International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). ICH guideline M10 on bioanalytical method validation and study sample analysis. European Medicines Agency. 2022. Available at: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-m10-bioanalytical-method-validation-step-5_en.pdf (Accessed: 31 December 2025).
[21]Liu M, Higashi K, Ueda K, Moribe K. Supersaturation maintenance of carvedilol and chlorthalidone by cyclodextrin derivatives: Pronounced crystallization inhibition ability of methylated cyclodextrin. International Journal of Pharmaceutics. 2023; 637: 122876. https://doi.org/10.1016/j.ijpharm.2023.122876.
[22]Loftsson T. Drug permeation through biomembranes: cyclodextrins and the unstirred water layer. Die Pharmazie. 2012; 67: 363–370. https://doi.org/10.1691/ph.2012.1698.
[23]Sugano K, Kansy M, Artursson P, Avdeef A, Bendels S, Di L, et al. Coexistence of passive and carrier-mediated processes in drug transport. Nature Reviews Drug Discovery. 2010; 9: 597–614. https://doi.org/10.1038/nrd3187.
[24]Gharib R, Fourmentin S, Charcosset C, Greige-Gerges H. Effect of hydroxypropyl-β–cyclodextrin on lipid membrane fluidity, stability and freeze-drying of liposomes. Journal of Drug Delivery Science and Technology. 2018; 44: 101–107. https://doi.org/10.1016/j.jddst.2017.12.009.
[25]Lima BS, Campos CA, Santos ACRS, Santos VCN, Trindade GGG, Pereira EWM, et al. Development of morin/hydroxypropyl-β-cyclodextrin inclusion complex: Enhancement of bioavailability, antihyperalgesic and anti-inflammatory effects. Food and Chemical Toxicology. 2019; 126: 15–24. https://doi.org/10.1016/j.fct.2019.01.038.
[26]Chandrama Singh S, Choudhary M, Mourya A, Khatri DK, Singh PK, Madan J, et al. Acute and Subacute Toxicity Assessment of Andrographolide-2-hydroxypropyl-β-cyclodextrin Complex via Oral and Inhalation Route of Administration in Sprague-Dawley Rats. The Scientific World Journal. 2022; 2022: 6224107. https://doi.org/10.1155/2022/6224107.
[27]Allen A, Bygate E, Oliver S, Johnson M, Ward C, Cheon AJ, et al. Pharmacokinetics and tolerability of gemifloxacin (SB-265805) after administration of single oral doses to healthy volunteers. Antimicrobial Agents and Chemotherapy. 2000; 44: 1604–1608. https://doi.org/10.1128/AAC.44.6.1604-1608.2000.
[28]Su J, Zhang X, Cao S, Liu C, Fu X, Zhang R, et al. Pharmacokinetic studies of hyperoside-2-hydroxypropyl-β-cyclodextrin inclusion complex and ameliorated DSS-induced colitis in mice. Bioscience Reports. 2023; 43: BSR20230003. https://doi.org/10.1042/BSR20230003.
[29]Cheow WS, Hadinoto K. Factors affecting drug encapsulation and stability of lipid-polymer hybrid nanoparticles. Colloids and Surfaces. B, Biointerfaces. 2011; 85: 214–220. https://doi.org/10.1016/j.colsurfb.2011.02.033.
[30]Sun Y, Mao Y, He X, Zhao X. Development and evaluation of mPEG-PLLA polymeric micelles encapsulating enrofloxacin for enhanced solubility, bioavailability, and antibacterial performance. Frontiers in Veterinary Science. 2025; 12: 1595137. https://doi.org/10.3389/fvets.2025.1595137.
[31]Morales D, Pacurariu A, Slattery J, Pinheiro L, McGettigan P, Kurz X. Association Between Peripheral Neuropathy and Exposure to Oral Fluoroquinolone or Amoxicillin-Clavulanate Therapy. JAMA Neurology. 2019; 76: 827–833. https://doi.org/10.1001/jamaneurol.2019.0887.
[32]Schiele JT, Quinzler R, Klimm HD, Pruszydlo MG, Haefeli WE. Difficulties swallowing solid oral dosage forms in a general practice population: prevalence, causes, and relationship to dosage forms. European Journal of Clinical Pharmacology. 2013; 69: 937–948. https://doi.org/10.1007/s00228-012-1417-0.
Various solubilizing agents were used to improve the solubility of gemifloxacin, which has limited water solubility. Furthermore, the solubility enhancement and bioavailability of gemifloxacin were then assessed through in vitro solubility screening and in vivo pharmacokinetic studies in rats.
Materials and Methods:
Different solubilizing agents, including poloxamer 407, poloxamer 188, Soluplus®, Polyox N80, sodium lauryl sulfate, PEG 4000, L-arginine, L-lysine, β-cyclodextrin, and hydroxypropyl-β-cyclodextrin (HPCD), were evaluated for the associated solubilizing effects, and HPCD was selected as the optimal solubilizer. Gemifloxacin in samples collected from the in vitro and in vivo experiments was quantified using high-performance liquid chromatography (HPLC).
Results:
In vitro solubility screening showed that the solubility of gemifloxacin reached 69.37 ± 0.71 mg/mL with 4.1 g of HPCD, representing a 1.52-fold increase relative to the control group (45.68 ± 0.37 mg/mL). Further optimization revealed that 0.1 g of HPCD achieved a solubility of 66.27 ± 0.42 mg/mL, with minimal additional improvement at higher concentrations. The HPLC method exhibited excellent linearity (R2 = 0.9998) over the range of 0.03–45 μg/mL. The in vivo pharmacokinetic study demonstrated that the area under the curve (AUC0-∞) of the gemifloxacin–HPCD formulation (Group A: 7.527 ± 0.60 μg⋅h/mL) increased approximately 1.53-fold compared with that of the gemifloxacin-alone group (4.928 ± 0.85 μg⋅h/mL), significantly improving the bioavailability of gemifloxacin.
Conclusion:
The solubilization strategy using HPCD can effectively improve the solubility and bioavailability of gemifloxacin, representing a promising approach for the development of the associated oral formulations.
Graphical Abstract
Keywords
biological availability
gemifloxacin
2-hydroxypropyl-beta-cyclodextrin
pharmacokinetics
solubility
1. Introduction
Pathogens such as Streptococcus pneumoniae (S. pneumoniae),
which are easily transmitted in daily life, can cause community-acquired
pneumonia (CAP). The risk of bacteremia and sepsis increases if CAP remains
untreated; in severe cases, the long-term mortality risk remains high even after
treatment [1]. These causative organisms survive and proliferate through type II
topoisomerases (DNA gyrase) and topoisomerase IV, which are critical for
bacterial DNA replication, transcription, repair, and recombination, thereby
facilitating their easy transmission [2]. Therefore, the inhibition of DNA gyrase
and topoisomerase IV is essential for the treatment of CAP and acute exacerbation
of chronic bronchitis.
Gemifloxacin, moxifloxacin, and levofloxacin are new fluoroquinolone
antibiotics. Gemifloxacin primarily targets and inhibits both DNA gyrase and
topoisomerase IV, thereby inhibiting DNA synthesis and inducing cell death.
Furthermore, gemifloxacin exhibits lower minimum inhibitory concentrations (MICs)
against pathogens causing acute exacerbation of chronic bronchitis (AECB) than
other fluoroquinolone antibiotics. Clinically, gemifloxacin reduces
hospitalization duration and contributes to healthcare cost savings compared to
ceftriaxone and clarithromycin. Gemifloxacin has demonstrated higher clinical
success rates than levofloxacin over the long term, indicating that gemifloxacin
offers multiple advantages over other fluoroquinolone antibiotics [3].
Pharmacokinetic results from previous studies have demonstrated that an average
of 61 9.5% of the dose was excreted in feces following oral
administration to healthy subjects, whereas 36 9.3% was eliminated in
urine as an unchanged drug and metabolite. After repeated dosing of 320 mg, the
mean renal clearance was approximately 11.6 3.9 L/h (range 4.6–16 L/h),
suggesting that active secretion is involved in the renal excretion of
gemifloxacin [4].
Cyclodextrin (CD) is a cyclic oligosaccharide comprising glucose units linked by
-1,4-glycosidic bonds. Cyclodextrin is hydrophilic because of the
presence of hydroxyl groups on its exterior, whereas its interior is relatively
hydrophobic [5]. The application of -CD in formulation development is
severely limited by both its low intrinsic aqueous solubility due to strong
intramolecular hydrogen bonding and a strict acceptable daily intake (ADI) limit
of 0.35 g for humans. In contrast, hydroxypropyl--cyclodextrin (HPCD)
exhibits drastically improved aqueous solubility through hydroxypropyl
substitution and superior oral tolerability (up to 8 g/day) with minimal
gastrointestinal irritation [6]. Given these physicochemical and toxicological
advantages, HPCD is considered a safer and more effective oral excipient than
unmodified -CD. In addition, previous studies have demonstrated that CD
exhibits specific tissue distribution and clearance characteristics in rats,
suggesting that it not only aids absorption but also influences pharmacokinetic
changes [7].
Therefore, we identified solubilizing agents that affect the solubility of
gemifloxacin, a respiratory antibiotic with multiple advantages. In addition, we
used solubilizing agents to compare and evaluate the bioavailability and
pharmacokinetics of a new gemifloxacin formulation developed using a reduced
active ingredient content compared to a reference drug.
2. Materials and Methods
2.1 Samples and Reagents
The gemifloxacin, moxifloxacin, and physiological saline were purchased from
Sigma-Aldrich (St. Louis, MO, USA). Solubilizing agents, including HPCD, were
obtained from Roquette (Lestrem, France), and polyethylene glycol 4000 (PEG 4000)
was sourced from IC Chemical (Yeosu, Republic of Korea). -Cyclodextrin
and L-arginine were purchased from ES Food Ingredients Co., Ltd. (Gunpo, Republic
of Korea), whereas L-lysine monohydrochloride was obtained from Saewon Mulsan
Co., Ltd. (Seoul, Republic of Korea). Poloxamer 407, poloxamer 188, and Polyox
N80 were acquired from Colorcon (Gunpo, Republic of Korea), and sodium lauryl
sulfate (SLS) was purchased from Duksan (Ansan, Republic of Korea).
A Centrifuge 5804 R (Eppendorf; MA, USA), Evaporator Uuiequip Univapo 100H GA59
(UniEquip Laborgerätebau and Vertriebs GmbH; Munich, Germany), and Power
Sonic 520 sonicator (Hwashin Tech; Seoul, Republic of Korea) were used to prepare
the sample. High-performance liquid chromatography (HPLC) was performed using a
Shimadzu system (Kyoto, Japan) equipped with an LC-20AD pump, SIL-20AC
autosampler, CTO-20A column oven, and SPD-20A detector. Blood samples were
collected in EDTA tubes (Vacuette; Kremsmünster, Austria). Analytical-grade
trifluoroacetic acid (TFA) for mobile phase preparation was purchased from
Sigma-Aldrich (St. Louis, MO, USA). Acetonitrile (ACN), methanol (MeOH), and
monobasic sodium phosphate were obtained from Samchun Chemicals (Seoul, Republic
of Korea). Formic acid was purchased from Junsei Chemical (Tokyo, Japan). All the
solvents used were of extra-pure grade.
2.2 In Vitro Solubility Evaluation
In vitro solubility screening of gemifloxacin was performed using
non-ionic surfactants, anionic surfactants, and solubilizing agents. The
concentration of each excipient was determined by conducting preliminary
solubility experiments and was set to the maximum concentration that could be
completely dissolved in 4 mL of purified water. Accordingly, we used poloxamer
407 (0.37 g), poloxamer 188 (0.41 g), Soluplus® (0.41 g), Polyox
N80 (0.25 g), sodium lauryl sulfate (0.34 g), PEG 4000 (2.44 g), L-arginine (0.58
g), L-lysine (0.44 g), -cyclodextrin (0.03 g), and HPCD (4.1 g).
Additional solubility experiments were conducted with 0.05, 0.10, and 0.20 g HPCD
for further investigation. The control group was prepared using purified water
without solubilizing agents, following the same experimental procedures.
Based on the reported saturation solubility of gemifloxacin (45.68 mg/mL
at pH 7.0, 37 °C) [8], 0.5 g of gemifloxacin was
added to ensure dissolution equilibrium. Next, the samples were vortex-mixed for
10 min at room temperature (25 2 °C), followed by a 5-min rest
period at room temperature to allow precipitation of undissolved drug. An aliquot
of 0.1 mL of the supernatant was transferred to a 100 mL volumetric flask,
dissolved with purified water, and filtered through a 0.45 µm regenerated cellulose (RC) filter (Sartorius AG; Goettingen, Germany). The filtered samples were analyzed according
to the HPLC conditions, and the concentrations were calculated using the external
standard method by applying the proportional relationship between the peak area
and concentration of a single-concentration standard solution. All in
vitro solubility experiments were performed five times for each condition
(n = 5).
2.3 In Vivo Pharmacokinetic Study
Male Sprague-Dawley (SD) rats (237.6 12.5 g) were used in this study.
The study was approved by the Institutional Animal Care and Use Committee (IACUC)
of Kyungsung University in accordance with the Animal Protection Act (Acts no.
4379 and no. 12681). Animals were obtained from Hyochang Science (Daegu, Korea)
and maintained under controlled environmental conditions with room temperature at
22 3 °C, relative humidity of 30–70%, and a 12-h light/dark
cycle. All rats were fasted overnight before the experiment, with free access to
water. Prior to each blood collection via the retro-orbital venous plexus, rats
were briefly anesthetized by isoflurane inhalation (2.5–3% for induction; 1–2%
for maintenance) using an anesthetic vaporizer with an induction chamber. At the
end of the study, all animals were euthanized by carbon dioxide (CO2)
inhalation (100% CO2; chamber fill rate 30–70% of chamber volume/min).
The in vivo dose of gemifloxacin was calculated based on body weight
proportion using a commercially available dose of 426.39 mg. The in
vitro solubility evaluation results demonstrated maximum solubility enhancement
at a gemifloxacin:HPCD weight ratio of 5:1. The samples for in vivo
evaluation were prepared using this composition. The formulation was prepared
freshly prior to use and administered immediately without storage. Fifteen rats
were randomly assigned to three groups (n = 5 per group) for the
gemifloxacin–HPCD complex study. The control group received 1.78 mg of
gemifloxacin dissolved in 3 mL of physiological saline via oral administration.
Group A received 1.0 mg of gemifloxacin with 0.2 mg of HPCD in 3 mL of
physiological saline, and Group B received 0.5 mg of gemifloxacin with 0.2 mg of
HPCD in 3 mL of physiological saline, both administered orally. For
pharmacokinetic evaluation, 1 mL blood samples were collected from the
retro-orbital venous plexus immediately before and at 0.25, 0.5, 0.75, 1, 1.5, 2,
2.5, 4, and 6 h of oral administration of gemifloxacin. All blood samples were
collected in EDTA tubes and centrifuged at 3000 rpm for 15 min at 4 °C
to separate plasma. The separated plasma samples were stored at –70 °C
until analysis. Pharmacokinetic parameters were calculated by non-compartmental
analysis using BA-Calc 2007 version 1.0 (KFDA, Osong, Republic of Korea). The
area under the plasma concentration–time curve from time zero to infinity
(AUC0-∞) was calculated using the linear trapezoidal rule. In
addition, other parameters, including maximum plasma concentration (Cmax), time
to reach maximum concentration (Tmax), and elimination half-life (t1/2) were
determined.
2.4 Plasma Sample Preparation
An aliquot of 200 µL of the plasma sample was transferred to a
microcentrifuge tube. To each sample, 10 µL of internal standard solution
(IS; 120 µg/mL) and 420 µL of ACN containing 0.1% (v/v) formic acid
were added. The tubes were sealed and vortex-mixed for 1 min, followed by
centrifugation at 3000 rpm for 10 min at 4 °C. After centrifugation,
200 µL of the supernatant was transferred to another microcentrifuge tube
and evaporated under reduced pressure using a speed vacuum at 40 °C. The
residue was reconstituted with 200 µL of methanol and analyzed by HPLC.
2.5 Preparation of Gemifloxacin Standard Solutions, Quality Control
(QC, and Internal Standard)
The diluent was prepared by mixing 80 mL of a solution containing 13.8 g of
monobasic sodium phosphate dissolved in 100 mL of purified water with 720 mL of
purified water and 200 mL of acetonitrile. This diluent was used to prepare a
0.36 mg/mL gemifloxacin stock solution and 120 µg/mL moxifloxacin (IS). The
stock solution was diluted in a single step using the above diluent to achieve
target concentrations of 0.03, 0.15, 0.3, 3, 6, 12, 24, and 45 µg/mL for
plasma calibration curve preparation. Quality control (QC) samples were prepared
at concentrations of 0.03, 0.3, 12, and 45 µg/mL.
2.6 Recovery
The recovery of gemifloxacin was evaluated using QC samples (0.03, 0.3, 12, and
45 µg/mL), with the requirement that the relative standard deviation (RSD)
should not exceed 15%.
2.7 HPLC–UV Analysis Conditions
The HPLC–UV method was adapted from the USP method for gemifloxacin. A Vision
HT C18-L column (4.6 250 mm, 5 µm particle size; Phenomenex,
USA) was employed. The mobile phase consisted of ACN:distilled water
(DW):trifluoroacetic acid at a ratio of 20:80:0.1 (v/v/v). The flow rate was set
at 1 mL/min with an injection volume of 20 µL. Both column and autosampler
temperatures were maintained at 25 °C, and detection was performed at
272 nm.
2.8 Statistical Analysis
All data are expressed as mean standard deviation (SD). Statistical
comparisons between groups were performed using Student’s t-test.
Statistical significance was set at p 0.05. All statistical analyses
were performed using BA-Calc 2007 (version 1.0, Biopharmaceutics Laboratory, College of Pharmacy, Kyung Hee University, Seoul, South Korea).
3. Results
3.1 In Vitro Solubility Studies
The in vitro solubility of gemifloxacin was screened using nonionic and
anionic surfactants and solubility enhancers. Differences in gemifloxacin
solubility were observed depending on the type of additive compared with the
control group, and the results are presented in Fig. 1. The solubility of
gemifloxacin was 69.37 0.71 mg/mL using 4.1 g of HPCD, representing the
greatest increase of 1.52-fold compared to the control group (45.68 0.37
mg/mL). This was followed by the Soluplus® (51.82 0.34
mg/mL) and -cyclodextrin (50.04 0.26 mg/mL) in descending order
of solubility enhancement. Based on these findings, an additional screening was
performed to determine the optimal HPCD concentration. The solubility of
gemifloxacin was measured as 50.30 0.81 mg/mL with 0.05 g of HPCD, 66.27
0.42 mg/mL with 0.1 g of HPCD, and 67.52 0.29 mg/mL with 0.2 g of
HPCD. The increase in gemifloxacin solubility was limited under conditions with
HPCD concentrations of 0.1 g or higher, suggesting a plateau effect. The phase
solubility profile (Fig. 2) shows an initial increase in solubility followed by a
plateau region, suggesting saturation of inclusion complex formation at higher
HPCD concentrations.
Fig. 1.
Dosage and results of surfactants used in gemifloxacin
solubility screening (n = 5 per group). All solubility analyses were
determined by high-performance liquid chromatography (HPLC). The solubility of
gemifloxacin ranged from 8.26 0.25 mg/mL (L-arginine) to 69.37
0.71 mg/mL (HPCD 4.1 g), and the control group (purified water) showed 45.68
0.37 mg/mL. PEG, polyethylene glycol; HPCD,
hydroxypropyl--cyclodextrin. Values are expressed as mean
standard deviation (n = 5 per group).
Fig. 2.
Phase solubility diagram of gemifloxacin as a function of HPCD
concentration. The solubility of gemifloxacin increased with increasing HPCD
concentration up to 0.1 g, followed by a plateau at higher concentrations,
indicating saturation of inclusion complex formation.
3.2 Linearity, Accuracy, and Precision
The retention times of gemifloxacin and the IS were approximately 9.0 and 8.86
min, respectively. Plasma calibration curves were constructed using the same rat
plasma collected on different working days. The linear regression equation over
the concentration range of 0.03–45 µg/mL of gemifloxacin was determined to
be y = 0.1009x – 0.0073 (R2 = 0.9998, n = 5), demonstrating
excellent linearity.
The intra- and inter-day accuracy and precision were evaluated by analyzing QC
samples at four concentrations, each with five replicates. The accuracy was
assessed by calculating the percentage deviation of the measured values from the
theoretical concentrations, and the precision was evaluated as the coefficient of
variation (CV) of repeated measurements. The accuracy and precision of the
results are listed in Table 1. The intraday precision was confirmed to be within
a maximum of 5.40%, and the accuracy was within 1.14%. Inter-day
precision was below 5.71%, and accuracy was within 1.28%.
Table 1.
Intra- and inter-day accuracy and precision of gemifloxacin in
rat plasma (n = 5 per group).
LLOQ (0.03 µg/mL)
Low QC (0.30 µg/mL)
Middle QC (12 µg/mL)
High QC (45 µg/mL)
Intra-day accuracy and precision
Mean
0.03
0.30
12.13
44.83
SD
0.00162
0.0131
0.47
2.08
% CV
5.40
4.37
3.87
4.64
% Deviation
1.14
–0.87
1.08
–0.38
n
5
5
5
5
Inter-day accuracy and precision
Mean
0.03
0.30
12.11
45.25
SD
0.00171
0.0146
0.59
2.41
% CV
5.71
4.87
4.87
5.33
% Deviation
–0.91
1.28
0.92
0.56
n
5
5
5
5
CV, coefficient of variation; SD, standard deviation; LLOQ, Lower Limit of Quantitation; QC, Quality Control.
3.3 Recovery
The recovery of gemifloxacin from plasma samples was evaluated at QC sample
concentrations of 0.03, 0.3, 12, and 45 µg/mL, resulting in recovery rates
of 92.78%, 94.61%, 93.27%, and 97.89%, respectively. Each of the four QC
concentrations was analyzed in five replicates, and the maximum relative standard
deviation was 4.14%, indicating consistent and reliable extraction efficiency.
3.4 Pharmacokinetic Studies
Based on the solubility screening results of gemifloxacin, an in vivostudy was conducted using a formulation containing HPCD. The pharmacokinetic
parameters of the gemifloxacin–HPCD complex and gemifloxacin-alone formulations
are summarized in Table 2. The mean plasma concentration–time profiles of
gemifloxacin following oral administration to rats are shown in Fig. 3.
Table 2.
Pharmacokinetics of gemifloxacin in control, group A, and group
B in rats (n = 5 each group).
Parameters
Control
Group A
Group B
C max (µg/mL)
1.199 0.24
1.175 0.08
0.931 0.08
T max (h)
0.25
0.25
0.25
MRT (h)
3.91 0.2
4.57 0.1
4.27 0.2
t1/2 (h)
1.7 0.2
2.2 0.2
1.9 0.2
AUC (0–6) (µg·h/mL)
2.778 0.35
3.778 0.15
2.867 0.21
AUC (0–) (µg·h/mL)
4.928 0.85
7.527 0.60
5.582 0.55
AUC, area under the curve; MRT, mean residence time. Values are expressed as
mean standard deviation. Control group: 1.78 mg gemifloxacin in 3 mL
saline; Group A: 1.0 mg gemifloxacin + 0.2 mg HPCD in 3 mL saline; Group B: 0.5
mg gemifloxacin + 0.2 mg HPCD in 3 mL saline.
Fig. 3.
Plasma concentration of gemifloxacin in rat plasma time curve
(n = 5 each group). Gemifloxacin in plasma samples was analyzed by
HPLC. Statistical significance between groups was determined using Student’s
t-test (p 0.05). The groups are defined as follows: Control
group: 1.78 mg gemifloxacin in 3 mL saline; Group A: 1.0 mg gemifloxacin + 0.2 mg
HPCD in 3 mL saline; Group B: 0.5 mg gemifloxacin + 0.2 mg HPCD in 3 mL saline.
Notably, Group A showed the highest AUC0-∞ (7.527 0.60
µgh/mL), which was significantly higher than that of the control
group (4.928 0.85 µgh/mL). Values are expressed as mean
standard deviation (n = 5 per group).
The Cmax value was higher in the control (1.199 0.24 µg/mL)
compared to Group A (1.175 0.08 µg/mL) and Group B (0.931
0.08 µg/mL). However, the AUC0-∞ values were significantly
enhanced in Group A (7.527 0.60 µgh/mL) and Group B (5.582
0.55 µgh/mL), demonstrating increases of 1.53-fold and
1.13-fold, respectively, compared to the control group (4.928 0.85
µgh/mL). In addition, both Groups A and B demonstrated an
increasing trend in mean residence time (MRT) and elimination half-life
(t1/2) compared to the control group. These findings indicate that although
the maximum plasma concentration was slightly lower in the HPCD formulations, the
overall systemic exposure (as measured by AUC) was substantially improved,
suggesting enhanced absorption and bioavailability.
4. Discussion
Gemifloxacin is an amphoteric API possessing both carboxylic acid and amino
functional groups, and exhibits pH-dependent limited aqueous solubility, which
presents constraints in dissolution and absorption [9, 10]. We compared different
surfactants and solubility enhancers to improve the solubility of gemifloxacin.
Poloxamers are non-ionic surfactants composed of hydrophilic (ethylene oxide,
PEO)–hydrophobic (propylene oxide, PPO)–PEO block copolymers that form micelles
in aqueous solutions, thereby reducing the interfacial tension between the drug
and solvent and improving the wettability and dispersibility of drug particles
[11]. However, as depicted in Fig. 1, the solubility values of poloxamer 407 and
poloxamer 188 were 27.81 0.23 mg/mL and 22.62 0.73 mg/mL,
respectively, which were lower than those of the control group. This limited
enhancement was attributed to the fact that effective micellization is induced
only above the critical micelle concentration (CMC); micelle formation is
restricted at relatively low concentrations, preventing the solubility
enhancement effect from being fully manifested [12].
In addition, SLS demonstrated a solubility of 33.71 0.36 mg/mL, lower
than that of the control group. This effect is likely due to the strong
electrostatic interactions between the amphoteric drug gemifloxacin and SLS,
resulting in the precipitation of a poorly soluble lauryl sulfate salt instead of
micelle-mediated solubilization [13]. These results suggest that the application
of surfactants to amphoteric drugs requires consideration of physical stability
and drug–excipient interactions. Similarly, PEG 4000 exhibited a solubility of
19.19 0.82 mg/mL. PEG functions simply as a solvent modifier and cannot
effectively influence the polar or non-polar regions of the API, resulting in low
solubility [14]. L-lysine and L-arginine exhibited very low solubilities of 10.37
0.25 mg/mL and 8.26 0.19 mg/mL, respectively. Although amino
acids can enhance solubility via ionic and non-ionic interactions, the
improvement was limited in this study. This suggests that the interaction between
the drug and amino acids was insufficient to overcome the strong self-association
of the drug molecules [15]. Soluplus® demonstrated a relatively
high solubility increase of 51.82 0.34 mg/mL, which was attributed to its
function as a hydrophilic–hydrophobic graft copolymer that forms polymeric
micelles and functions as a solid dispersion matrix, thereby enhancing the
solubility of poorly soluble drugs. However, its effect has been reported to be
limited by the critical micellization concentration and plateau concentration
[16]. Polyox N80 displayed a solubility of 43.91 0.52 mg/mL, similar to
the control group. Polyox N80 is a high-molecular-weight polyethylene oxide
primarily used as a viscosity enhancer and release matrix; it contributes to
viscosity and release control rather than inclusion complex formation or strong
micellar solubilization, resulting in relatively low solubility enhancement
effects [17]. A comprehensive consideration of these results confirmed HPCD as
the solubilizing agent exhibiting the greatest solubility enhancement effect in
this study, and a solubility of 69.37 0.71 mg/mL at 4.1 g. HPCD possesses
high aqueous solubility due to hydroxypropyl substitution and significantly
increases equilibrium solubility by forming inclusion complexes with drugs
through its hydrophobic cavity. The plateau effect observed at HPCD
concentrations 0.1 g can be explained by the phase solubility behavior of
cyclodextrin inclusion complexes (Fig. 2). In general, drug–cyclodextrin systems
exhibit a limited complexation capacity, where the increase in drug solubility is
proportional to cyclodextrin concentration only up to a certain point [17].
Beyond this concentration, the system reaches a saturation state in which most of
the drug molecules are already complexed, and further addition of HPCD does not
significantly enhance solubility. Although the phase solubility profile suggests
the formation of a drug–HPCD inclusion complex, this interpretation is based on
indirect evidence. Advanced physicochemical characterization techniques such as
differential scanning calorimetry (DSC), Fourier-transform infrared spectroscopy
(FTIR), and nuclear magnetic resonance (NMR) were not employed in this study.
Therefore, the molecular-level interactions between gemifloxacin and HPCD could
not be directly confirmed. This behavior is consistent with the formation of a
1:1 inclusion complex and the transition from a linear phase to a plateau region
in the phase solubility diagram. Additionally, the self-association of
cyclodextrin molecules and the formation of aggregates at higher concentrations
can lead to a deviation from ideal solubilization behavior, thereby limiting the
solubilization efficiency [18]. Based on these results, HPCD was determined to be
a highly applicable solubility enhancer in terms of physical stability and
formulation design. Considering its applicability to tablet formulations, the
HPCD concentration was increased in a stepwise manner. The highest solubility of
66.27 0.42 mg/mL was observed at 0.1 g, suggesting that the solubility
increase did not follow a simple dose-dependent pattern. Although a complete
concentration–response curve was not established, the observed plateau in
solubility above 0.1 g HPCD suggests that further increases in cyclodextrin
concentration do not significantly enhance complexation efficiency under the
tested conditions (Fig. 2). Previous studies have reported that
cyclodextrin–drug complexes exhibit diverse phase–solubility curves and
non-linear characteristics in the concentration–solubility relationship [19].
Therefore, 0.1 g of HPCD was determined to be the optimal solubilization
condition considering both the solubility enhancement effect and practical
formulation applicability.
The bioanalytical method was validated according to the ICH M10 guidelines
before conducting in vivo experiments to ensure the reliability and
validity of the analytical method for the quantitative analysis of gemifloxacin
in plasma. The linear regression equation of the calibration curve established in
rat plasma over the range of 0.03–45 µg/mL was y = 0.1009x – 0.0073
(R2 = 0.9998, n = 5), demonstrating appropriate linearity. The
lower limit of quantification (LLOQ) of gemifloxacin was set at 0.03 µg/mL;
the accuracy was within 1.14%, and precision was up to 5.71% at this
concentration, meeting the criteria (20%) presented in the ICH M10
guidelines. The three QC sample concentrations excluding LLOQ demonstrated
accuracy within 1.28% and precision up to 5.33%, all meeting the
acceptance criteria (15%) presented in the ICH M10 guidelines [20]. Thus,
the analytical method established in this study has sufficient reliability and
reproducibility for quantitative analysis of gemifloxacin in rat plasma.
In vitro solubility evaluation results confirmed that HPCD significantly
improved gemifloxacin solubility. Pharmacokinetic experiments were conducted in
rats to evaluate whether this in vitro solubility enhancement translates
into improved in vivo absorption. Although the absolute Cmax values were
higher in the control group than in Groups A and B, the dose-corrected Cmax/dose
values of Group A (1.175 0.08 µg/mL) and Group B (1.862
0.08 µg/mL) increased by 1.75-fold and 2.77-fold, respectively, compared to
the control group (0.673 0.13 µg/mL). To clarify the calculation of
dose-normalized exposure, the Cmax values were normalized by the administered
dose (Cmax/dose). The calculated values were 0.673, 1.175, and 1.862 for the
control, Group A, and Group B, respectively. Accordingly, the dose-normalized
Cmax values in Group A and Group B were increased by 1.75-fold and 2.77-fold,
respectively, compared to the control group. The AUC0-∞ values of
Group A (7.527 0.60 µgh/mL) and Group B (5.582
0.55 µgh/mL) increased by 1.53-fold and 1.13-fold, respectively,
compared to the control group (4.928 0.85 µgh/mL).
Notably, Group B received half the dose of gemifloxacin compared to Group A,
which was intentionally designed to evaluate whether the HPCD formulation could
maintain or enhance drug absorption efficiency at reduced doses. Despite the
lower administered dose, Group B exhibited a higher dose-normalized Cmax compared
to both the control and Group A. This indicates that the HPCD complex improved
the absorption efficiency of gemifloxacin, rather than simply increasing systemic
exposure due to higher dosing. These findings suggest that the formulation may
enable dose reduction while maintaining effective drug absorption. Despite the
low administered dose, the observed improvement in absorption cannot be explained
solely by an increase in solubility. Rather, the formation of an inclusion
complex with HPCD likely inhibited the crystallization of gemifloxacin, thereby
reducing precipitation and maintaining a supersaturated state within the
intestinal lumen [21]. In particular, to overcome the unstirred water layer
(UWL), which is recognized as a primary absorption barrier for poorly
water-soluble drugs, the inclusion complex maintains a steep concentration
gradient across the entire UWL. As a result, the inclusion complex efficiently
transports the drug across the UWL to the lipophilic epithelial surface. Upon
reaching the membrane surface, the complex reversibly dissociates, releasing free
drug molecules and thereby enhancing mucosal drug permeation [22]. This suggests
that the increased solubility of the gemifloxacin–HPCD complex increased the
drug concentration in the intestinal lumen, resulting in enhanced absorption by
passive diffusion [23]. The interaction between HPCD and phospholipids increases
acyl chain disorder, leading to enhanced membrane fluidity and structural
perturbation, which in turn increases membrane permeability [24]. This increase
in membrane permeability may have partially contributed to the enhanced
gemifloxacin absorption observed in this study. These results were consistent
with those of previous studies demonstrating that the bioavailability of poorly
soluble drugs can be improved by HPCD inclusion complex [25]. HPCD is a widely
used pharmaceutical excipient with a well-established safety profile. Previous
studies have reported a no-observed-adverse-effect level (NOAEL) of approximately
600 mg/kg/day in rats and low acute toxicity with oral LD50 values exceeding
2000 mg/kg. In the present study, the administered amount of HPCD was
substantially lower than these safety thresholds, suggesting minimal risk of
toxicity [26]. The increased bioavailability of gemifloxacin is attributed to its
improved solubility and enhanced intestinal absorption induced by complexation
with HPCD. The plasma concentration of gemifloxacin was the highest at the first
sampling time point (1.199 0.24 µg/mL) and decreased by
approximately 48.04% by the third sampling time point (0.623 0.09
µg/mL) in the control group. Thereafter, no change was observed in the
concentration magnitude from the fourth sampling time point (0.621 0.11
µg/mL), and a biphasic decline pattern was observed where the rate of
decrease gradually became more gradual (Fig. 3). This pattern is consistent with
that mentioned in previous reports, suggesting that the experimental design and
data derived in this study are highly reliable [27]. Plasma concentrations were
also the highest at the first sampling time point, at 1.175 0.08
µg/mL and 0.931 0.08 µg/mL, respectively, in Groups A and B.
The plasma concentrations of Group A (0.905 0.06 µg/mL) and Group B
(0.727 0.08 µg/mL) decreased by 23% and 22%, respectively, at the
third sampling time point, and thereafter demonstrated a gradual decline pattern
from subsequent sampling points (Fig. 3). Unlike the rapid plasma concentration
decline observed in the control group, the gradual and sustained decline pattern
observed in Groups A and B was consistent with that reported in previous
pharmacokinetic studies, demonstrating an increased systemic exposure time to
drugs in inclusion complexes using HPCD [28]. Various solubilization strategies
have been explored to improve the bioavailability of poorly soluble
fluoroquinolone antibiotics. However, previously reported approaches have often
faced technical limitations. For example, lipid–polymer hybrid nanoparticles
have exhibited formulation failure or physical instability due to uncontrolled
electrostatic interactions [29]. In addition, polymeric micelle systems that
require complex multistep preparation processes and the use of organic solvents
have shown only limited improvements in bioavailability, with increases of
approximately 1.6-fold [30]. In contrast, the HPCD inclusion strategy employed in
the present study significantly improved the Cmax/dose by up to 2.77-fold
compared with the control group using a simple preparation process without
organic solvents. These results suggest that HPCD complexation represents a
highly effective, practical, and promising strategy for improving the oral
delivery of gemifloxacin. The improvement of gemifloxacin bioavailability
observed in this study is expected to reduce the required amount of API,
ultimately enhancing dosing safety and significantly improving patient compliance
through smaller tablet sizes [31, 32]. In addition, the reduced API usage may
contribute to lowering the drug cost.
These results suggest that HPCD complexation can improve the absorption rate of
gemifloxacin, demonstrating its potential to effectively improve the oral
delivery of gemifloxacin.
5. Strengths and Limitations of This Study
This study has several strengths. First, a systematic and comprehensive
screening of ten structurally diverse solubilizing agents including non-ionic
surfactants, an anionic surfactant, amino acids, and cyclodextrins was performed
under identical experimental conditions, enabling a direct and objective
comparison of solubilization capacity for gemifloxacin. Second, the selected
HPCD-based formulation was evaluated through both in vitro solubility
studies and in vivo pharmacokinetic studies in rats, providing an
integrated assessment that bridges bench-level observations with physiologically
relevant outcomes. Third, the bioanalytical HPLC method was rigorously validated
in accordance with ICH M10 guidelines, demonstrating excellent linearity (R2
= 0.9998), precision (CV 5.71%), and accuracy (within 1.28%),
which supports the reliability of the pharmacokinetic data reported herein.
However, several limitations should be acknowledged. First, the in vivo
pharmacokinetic study was conducted using a small sample size (n = 5 per
group), which may limit the statistical power and generalizability of the
pharmacokinetic findings; future studies with larger cohorts are warranted to
confirm these results. Second, the study was restricted to male Sprague-Dawley
rats, and it remains unknown whether the observed solubility and bioavailability
improvements would translate to the same extent in female animals or in human
subjects; further preclinical and clinical studies are needed to evaluate
species- and sex-dependent differences. Third, long-term stability testing of the
HPCD formulation under various storage conditions was not conducted, and its
feasibility for scale-up manufacturing into a solid dosage form (e.g., tablet)
remains to be demonstrated. Another limitation of this study is the lack of
direct physicochemical characterization of the gemifloxacin–HPCD inclusion
complex. Techniques such as DSC, FTIR, NMR, and determination of stability
(binding) constants were not performed, which limits the ability to conclusively
confirm inclusion complex formation and quantify the strength of interaction.
Therefore, the proposed mechanism of solubility enhancement is based on indirect
evidence, including phase solubility analysis and pharmacokinetic outcomes.
Future studies are warranted to perform detailed structural and thermodynamic
characterization of the complex.
6. Conclusion
This study successfully demonstrated that HPCD effectively enhanced the
solubility and bioavailability of gemifloxacin through a systematic evaluation
combining in vitro solubility screening and in vivo
pharmacokinetic studies. Among the different solubilizing agents evaluated,
including non-ionic surfactants (poloxamers, PEG 4000, Soluplus®,
Polyox N80), anionic surfactants (SLS), amino acids (L-arginine, L-lysine), and
cyclodextrins (-cyclodextrin, HPCD), HPCD exhibited superior solubility
enhancement, achieving a maximum solubility of 69.37 0.71 mg/mL at a high
concentration (4.1 g), representing a 1.52-fold increase over the control. The
optimized HPCD concentration of 0.1 g achieved 66.27 0.42 mg/mL
solubility, demonstrating its practical applicability for tablet formulations.
The validated HPLC method provided reliable quantification with excellent
linearity (R2 = 0.9998), precision (CV 5.71%), and accuracy (within
1.28%), meeting the ICH M10 guidelines. In vivo pharmacokinetic
studies revealed that HPCD formulations significantly improved bioavailability,
with AUC0-∞ increasing by 1.53-fold (Group A) and 1.13-fold (Group
B) compared to gemifloxacin alone. The dose-normalized Cmax/dose values increased
by 1.75-fold and 2.77-fold, respectively, indicating enhanced absorption
efficiency. The sustained plasma concentration profiles observed for the HPCD
formulations, characterized by a gradual rather than rapid decline, suggest
prolonged systemic exposure and improved pharmacokinetic profiles. These results
established HPCD-based solubilization as a promising and practical strategy for
the development of improved oral gemifloxacin formulations, potentially leading
to enhanced therapeutic outcomes, improved patient compliance, and reduced dosing
requirements for clinical applications. Future investigations are warranted to
clinically validate, optimize manufacturing processes for commercial development,
and explore the underlying molecular mechanisms of drug-CD interactions.
Availability of Data and Materials
The datasets used and analyzed during the current study are available from
corresponding author on reasonable request.
Author Contributions
JWK, GEP, JSK, YHK and KMK conceptualized and designed the study and performed the experiments. YHK, KMK and JSK supervised the study and performed data acquisition, analysis, and interpretation. GEP and JWK wrote the manuscript and prepared the figures and tables. YHK and KMK performed the reference search and confirmed the authenticity of all data. All authors contributed to editorial revisions of the manuscript. All authors read and approved the final manuscript and have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Ethics Approval and Consent to Participate
All animal experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The study was approved by the Institutional Animal Care and Use Committee of Kyungsung University (approval number: study-2025-001A).
Acknowledgment
Not applicable.
Funding
This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea [grant number: RS-2020-KH088726 (HR20C0026)].
Conflicts of Interest
The authors declare no conflicts of interest. We confirm that YHK’s affiliation with Kolon Pharm did not influence data interpretation, the writing of the manuscript, or the scientific judgments made in this study.
References
[1] Kim TW, Lee SU, Park B, Jeon K, Park S, Suh GY, et al. Clinical effects of bacteremia in sepsis patients with community-acquired pneumonia. BMC Infectious Diseases. 2023; 23: 887. https://doi.org/10.1186/s12879-023-08887-5.
[2] Bush NG, Diez-Santos I, Abbott LR, Maxwell A. Quinolones: mechanism, lethality and their contributions to antibiotic resistance. Molecules. 2020; 25: 5662. https://doi.org/10.3390/molecules25235662.
[3] Jivcu C, Gotfried M. Gemifloxacin use in the treatment of acute bacterial exacerbation of chronic bronchitis. International Journal of Chronic Obstructive Pulmonary Disease. 2009; 4: 291–300. https://doi.org/10.2147/copd.s3903.
[7] Mu K, Jiang K, Wang Y, Zhao Z, Cang S, Bi K, et al. The biological fate of pharmaceutical excipient β-Cyclodextrin: pharmacokinetics, tissue distribution, excretion, and metabolism of β-Cyclodextrin in rats. Molecules. 2022; 27: 1138. https://doi.org/10.3390/molecules27031138.
[8] LG Chem, Ltd. FACTIVE® (gemifloxacin) tablets, for oral use. 2019. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2003/021158lbl.pdf (Accessed: 28 December 2025).
[9] lshafie HS, Sadeek SA, Camele I, Mohamed AA. Biochemical characterization of new gemifloxacin schiff base (GMFX-o-phdn) metal complexes and evaluation of their antimicrobial activity against some phyto- or human pathogens. International Journal of Molecular Sciences. 2022; 23: 2110. https://doi.org/10.3390/ijms23042110.
[11] Munir R, Hadi A, Khan SUD, Asghar S, Irfan M, Khan IU, et al. Solubility and Dissolution Enhancement of Dexibuprofen with Hydroxypropylbetacyclodextrin (HPβCD) and Poloxamers (188/407) Inclusion Complexes: Preparation and In Vitro Characterization. Polymers. 2022; 14: 579. https://doi.org/10.3390/polym14030579.
[12] Mondal L, Mukherjee B, Chakraborty S, Bhattacharya S, Ehsan I, Sengupta S, et al. Comparison of enhanced solubility profiles, analysis of thermodynamic parameters and pharmacokinetic profile related to tamoxifen citrate solubilisation. Novel Approaches in Drug Design & Development. 2018; 3: 555624. https://doi.org/10.19080/NAPDD.2018.03.555624.
[15] ElShaer A, Ouyang D, Hanson P, Mohammed AR. Preparation and evaluation of amino acid based salt forms of model zwitterionic drug ciprofloxacin. Journal of Pharmaceutics & Drug Delivery Research. 2013; 2: 1. http://dx.doi.org/10.4172/2325-9604.1000111.
[16] Pignatello R, Corsaro R, Bonaccorso A, Zingale E, Carbone C, Musumeci T. Soluplus® polymeric nanomicelles improve solubility of BCS-class II drugs. Drug Delivery and Translational Research. 2022; 12: 1991–2006. https://doi.org/10.1007/s13346-022-01182-x.
[17] Muhamad H, Bashir AB, Charlton-Harrison J, Abdulhussain R, Mawla N, Patel K, et al. Hot-melt extruded-FDM 3D-printed polyethylene oxide tablets: Dissolution imaging analysis of swelling and drug release. European Journal of Pharmaceutics and Biopharmaceutics. 2025; 208: 114636. https://doi.org/10.1016/j.ejpb.2025.114636.
[20] International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). ICH guideline M10 on bioanalytical method validation and study sample analysis. European Medicines Agency. 2022. Available at: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-m10-bioanalytical-method-validation-step-5_en.pdf (Accessed: 31 December 2025).
[21] Liu M, Higashi K, Ueda K, Moribe K. Supersaturation maintenance of carvedilol and chlorthalidone by cyclodextrin derivatives: Pronounced crystallization inhibition ability of methylated cyclodextrin. International Journal of Pharmaceutics. 2023; 637: 122876. https://doi.org/10.1016/j.ijpharm.2023.122876.
[22] Loftsson T. Drug permeation through biomembranes: cyclodextrins and the unstirred water layer. Die Pharmazie. 2012; 67: 363–370. https://doi.org/10.1691/ph.2012.1698.
[23] Sugano K, Kansy M, Artursson P, Avdeef A, Bendels S, Di L, et al. Coexistence of passive and carrier-mediated processes in drug transport. Nature Reviews Drug Discovery. 2010; 9: 597–614. https://doi.org/10.1038/nrd3187.
[24] Gharib R, Fourmentin S, Charcosset C, Greige-Gerges H. Effect of hydroxypropyl-β–cyclodextrin on lipid membrane fluidity, stability and freeze-drying of liposomes. Journal of Drug Delivery Science and Technology. 2018; 44: 101–107. https://doi.org/10.1016/j.jddst.2017.12.009.
[25] Lima BS, Campos CA, Santos ACRS, Santos VCN, Trindade GGG, Pereira EWM, et al. Development of morin/hydroxypropyl-β-cyclodextrin inclusion complex: Enhancement of bioavailability, antihyperalgesic and anti-inflammatory effects. Food and Chemical Toxicology. 2019; 126: 15–24. https://doi.org/10.1016/j.fct.2019.01.038.
[26] Chandrama Singh S, Choudhary M, Mourya A, Khatri DK, Singh PK, Madan J, et al. Acute and Subacute Toxicity Assessment of Andrographolide-2-hydroxypropyl-β-cyclodextrin Complex via Oral and Inhalation Route of Administration in Sprague-Dawley Rats. The Scientific World Journal. 2022; 2022: 6224107. https://doi.org/10.1155/2022/6224107.
[27] Allen A, Bygate E, Oliver S, Johnson M, Ward C, Cheon AJ, et al. Pharmacokinetics and tolerability of gemifloxacin (SB-265805) after administration of single oral doses to healthy volunteers. Antimicrobial Agents and Chemotherapy. 2000; 44: 1604–1608. https://doi.org/10.1128/AAC.44.6.1604-1608.2000.
[28] Su J, Zhang X, Cao S, Liu C, Fu X, Zhang R, et al. Pharmacokinetic studies of hyperoside-2-hydroxypropyl-β-cyclodextrin inclusion complex and ameliorated DSS-induced colitis in mice. Bioscience Reports. 2023; 43: BSR20230003. https://doi.org/10.1042/BSR20230003.
[29] Cheow WS, Hadinoto K. Factors affecting drug encapsulation and stability of lipid-polymer hybrid nanoparticles. Colloids and Surfaces. B, Biointerfaces. 2011; 85: 214–220. https://doi.org/10.1016/j.colsurfb.2011.02.033.
[30] Sun Y, Mao Y, He X, Zhao X. Development and evaluation of mPEG-PLLA polymeric micelles encapsulating enrofloxacin for enhanced solubility, bioavailability, and antibacterial performance. Frontiers in Veterinary Science. 2025; 12: 1595137. https://doi.org/10.3389/fvets.2025.1595137.
[31] Morales D, Pacurariu A, Slattery J, Pinheiro L, McGettigan P, Kurz X. Association Between Peripheral Neuropathy and Exposure to Oral Fluoroquinolone or Amoxicillin-Clavulanate Therapy. JAMA Neurology. 2019; 76: 827–833. https://doi.org/10.1001/jamaneurol.2019.0887.
[32] Schiele JT, Quinzler R, Klimm HD, Pruszydlo MG, Haefeli WE. Difficulties swallowing solid oral dosage forms in a general practice population: prevalence, causes, and relationship to dosage forms. European Journal of Clinical Pharmacology. 2013; 69: 937–948. https://doi.org/10.1007/s00228-012-1417-0.




 , Jae Seon Kang 2,†
, Jae Seon Kang 2,†
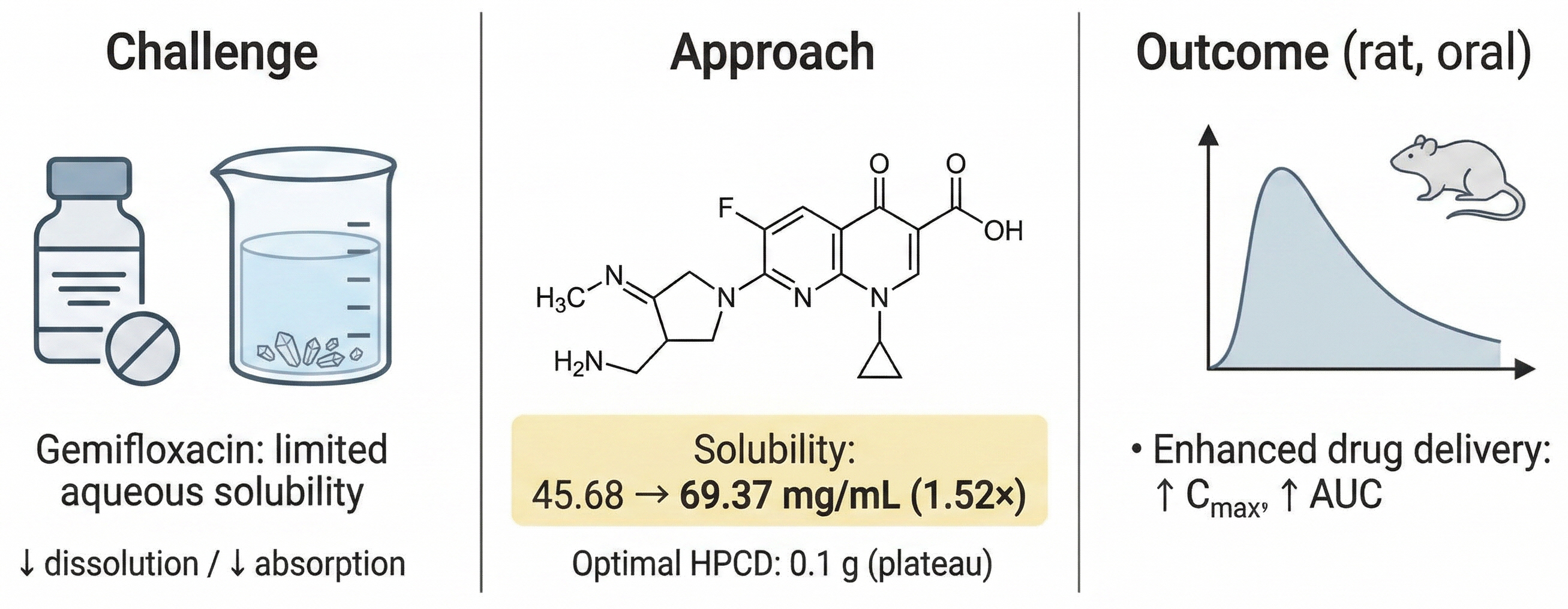
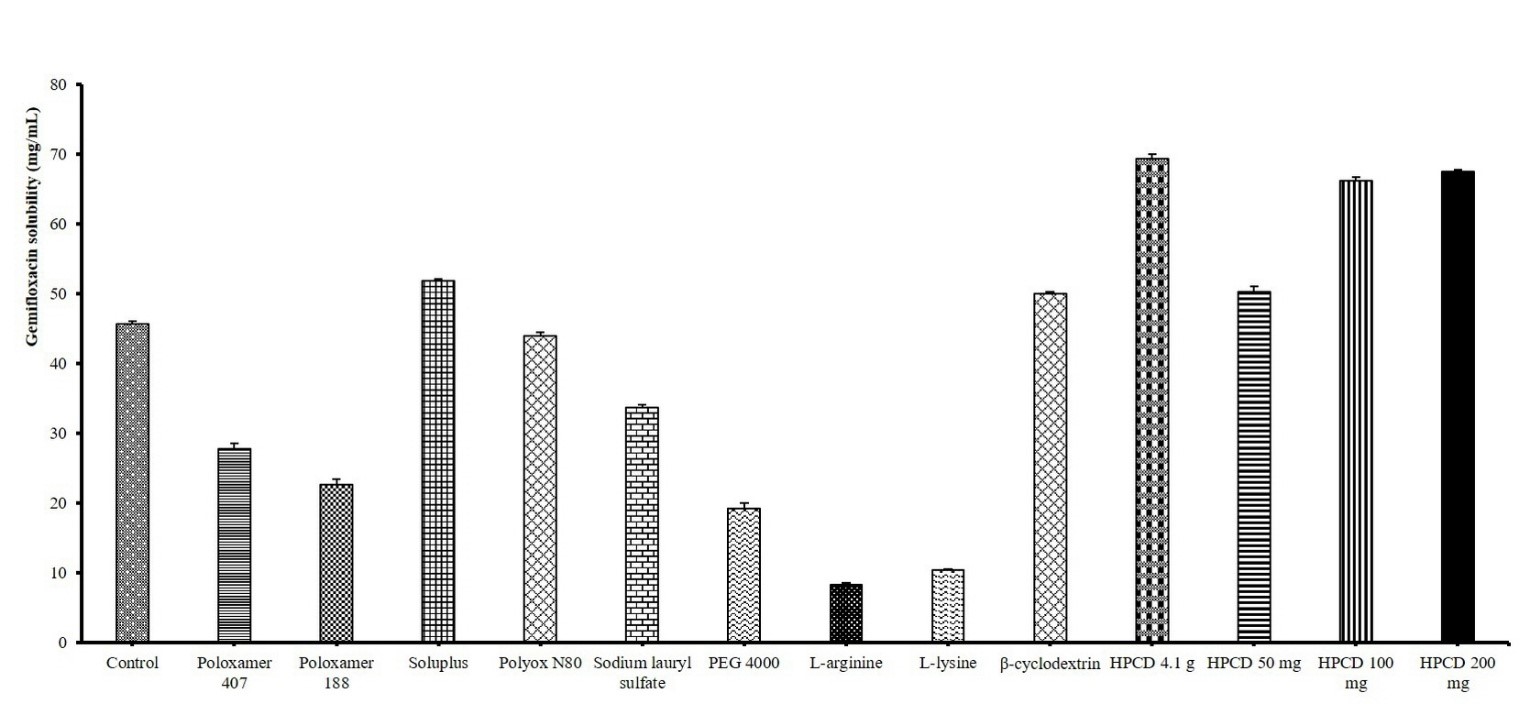 Fig. 1.
Fig. 1.
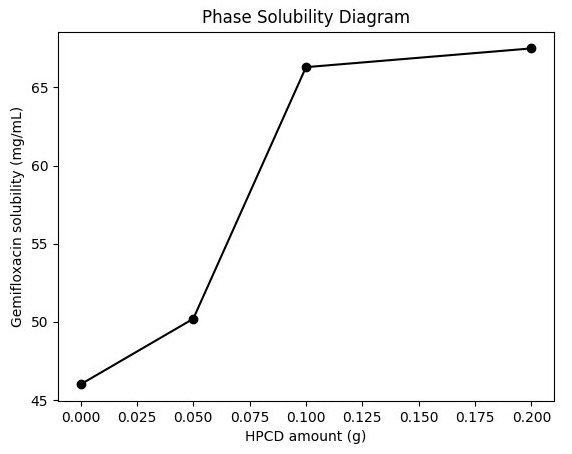 Fig. 2.
Fig. 2.
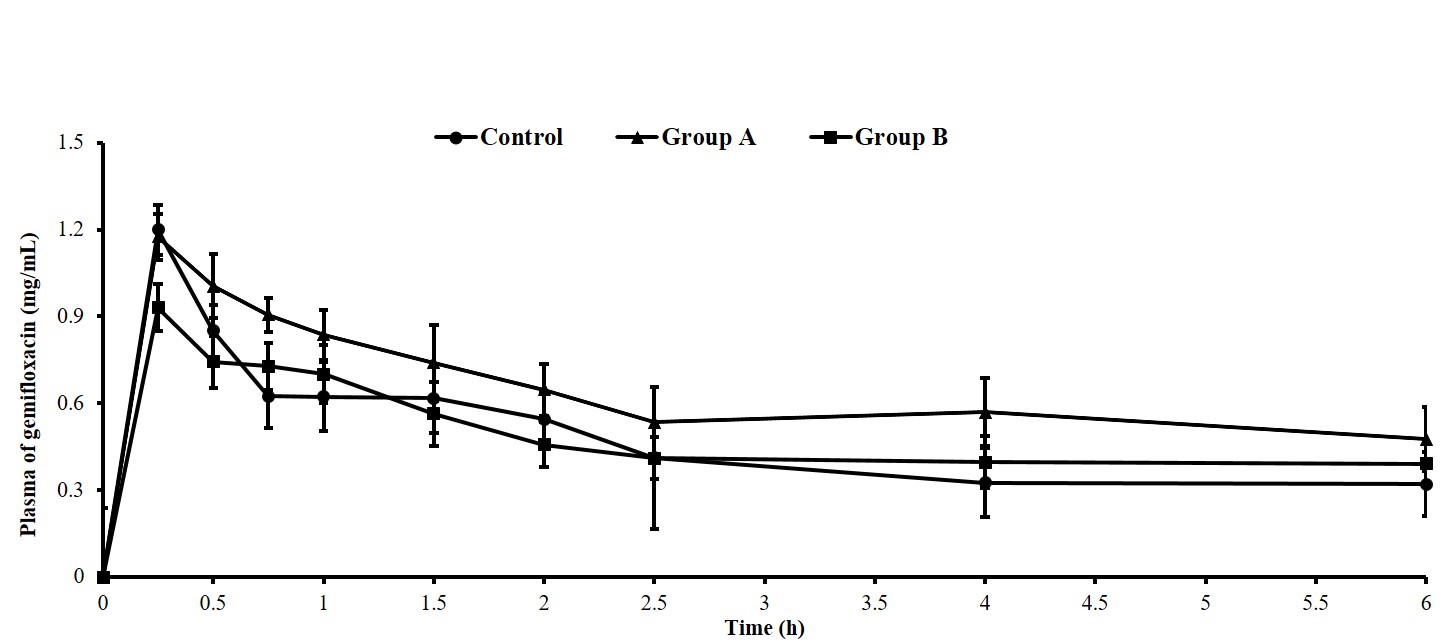 Fig. 3.
Fig. 3.