













 , Xiaohui Liu 1,†, Yanan Zheng 1,*
, Xiaohui Liu 1,†, Yanan Zheng 1,*
1 Neurology Department, Baotou Central Hospital, 014040 Baotou, Inner Mongolia, China
†These authors contributed equally.
Abstract
Subarachnoid hemorrhage (SAH) is a cerebrovascular disease with high mortality and long-term neurological sequelae, largely driven by early brain injury, inflammation, neuronal apoptosis, and oxidative stress. Umbelliferone (7-hydroxycoumarin) exhibits notable antioxidant and anti-inflammatory properties. This study aimed to investigate the neuroprotective effect of umbelliferone in a mouse model of SAH and explore the underlying mechanisms.
Following SAH induction, mice received the oral administration of umbelliferone (2.5, 5, or 10 mg/kg). Body weight, brain weight, and brain water content were measured to assess cerebral edema and neurological injury. Tight junction protein levels were quantified to assess blood brain barrier (BBB) integrity; meanwhile, antioxidant markers, inflammatory cytokines, and apoptosis-related parameters were also evaluated. Furthermore, mRNA expression levels in brain tissue were analyzed to elucidate the underlying molecular mechanisms.
Umbelliferone significantly improved body weight and enhanced brain weight (p < 0.001). Umbelliferone also altered the level of tight junction parameters (occludin, claudin-5, zonula occludens-1 (ZO-1)); oxidative stress parameters (glutathione (GSH), superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GPx), malonaldehyde (MDA)); mitogen-activated protein kinase (MAPK) parameters (phosphorylated c-Jun N-terminal kinase(p-JNK), phosphorylated extracellular signal-regulated kinase (p-ERK), phosphorylated p38 MAPK (p-38)); inflammatory cytokines (tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), interleukin-6 (IL-6), interleukin-18 (IL-18)); inflammatory parameters (cyclooxygenase-2 (COX-2), nuclear factor kappa B (NF-κB), transforming growth factor-β (TGF-β)); apoptosis parameters (Bcl-2-associated X protein (Bax), B-cell lymphoma 2 (Bcl-2), caspase-3, Bax/Bcl-2 ratio). Umbelliferone also significantly (p < 0.001) altered the mRNA expression of matrix metalloproteinase-2 (MMP-2), matrix metalloproteinase-9 (MMP-9), basigin (BSG2), TNF-α, IL-6, monocyte chemoattractant protein-1 (MCP-1), claudin-5, occludin, phosphoinositide 3-kinase (PI3K), protein kinase B (AkT), toll-like receptor 2 (TLR2) and toll-like receptor 4 (TLR4).
Umbelliferone exerts neuroprotective effects against SAH in mice, at least in part, by modulating the TLR2/TLR4–NF-κB–MMP-9 and PI3K/Akt signaling pathways.
Keywords
- subarachnoid hemorrhage
- neuroprotection
- umbelliferone
- oxidative stress
- apoptosis
- toll-like receptor 2
- nuclear factor-kappa B
- matrix metalloproteinase 9
Early brain injury (EBI), following subarachnoid hemorrhage (SAH) shows a complex cascade of cellular and molecular events that progress beyond the primary mechanical insult, which contributes to secondary neuronal injury and neurological dysfunction [1]. The quick enhancement in intracranial pressure and the resultant cerebral hypoperfusion initiate a series of events that disrupt blood brain barrier (BBB) homeostasis, ultimately leading to vasogenic edema and subsequent influx of proinflammatory mediators [2, 3]. Neuroinflammation, driven by activated microglia and immune cell infiltration, exacerbates tissue damage by releasing pro-inflammatory mediators, including cytokines and chemokines. Mitochondrial dysfunction further aggravates the cellular energy deficit and activates apoptotic cascades, establishing a vicious cycle that amplifies neuronal degeneration. The pathophysiological environment in the brain is very complex, as it not only compromises immediate neuronal survival but also predisposes the brain to delayed complications like cerebral vasospasm and ischemia [4, 5, 6]. Apoptosis, a tightly regulated form of programmed cell death, plays a critical pathophysiological role in EBI and largely determines the extent of glial and neuronal cell loss after SAH. Cellular degradation and DNA fragmentation are mediated by caspases, which are activated through both intrinsic and extrinsic apoptotic pathways. Modulation of these apoptotic pathways has been shown to significantly affect the progression of brain injury in experimental models; therefore, apoptosis has emerged as a promising therapeutic target for the treatment of SAH. The preclinical studies suggest that pharmacological compounds that block major intracellular apoptotic cascades (e.g., caspase inhibitors, mitochondrial membrane stabilisers) are effective in suppressing brain edema and improving functional outcome. Preliminary clinical trials investigating anti-apoptotic therapies have demonstrated promising neuroprotective potential, but more research needs to be required to scrutinize the initiating molecular mechanisms and to determine the optimal timing for potential intervention [1, 6, 7]. Apoptosis-targeted treatments may transform the current therapeutic paradigm for SAH, shifting the focus from merely preventing re-rupture to actively amelioration of early neuronal injury, and improving the long-term neurological complications.
Secondary complications after SAH, such as brain edema, BBB permeability disruption, neuronal cell apoptosis, and cerebral vasospasm (CVS) are sequential pathophysiological events that have important roles in the prognosis of SAH in patients. These complications are, in part, facilitated by biochemical dysregulation, including oxidative stress, increased inflammation, and toxic metabolite accumulation [1, 3]. Oxidative stress is a major contributor to cellular damage and enhances neuroinflammation, compromising neuronal survival and vascular stability. The interactive effects of these factors result in a vicious cycle that aggravates brain injury and hinders recovery following SAH. Recently, activation of nuclear factor erythroid 2-related factor 2 (Nrf2) was considered as an essential mediator in the amelioration of these secondary injuries through coordinating cellular protective responses against oxidative and inflammatory insults [8]. This involves the cytoplasmic retention of Nrf2 by its repressor, kelch-like ECH-associated protein 1 (Keap1), under basal conditions. Under resting conditions, Nrf2 is sequestered by its negative regulator, Keap1, which retains the transcription factor in the cytoplasm for degradation via the ubiquitin-proteasome pathway. But under oxidative or xenobiotic stimuli, Nrf2 dissociates from its tether to the Keap1 complex and translocates to the nucleus, where it induces the transcription of antioxidant response element (ARE) genes, thereby controlling the expression of antioxidant and detoxifying enzymes. In fact, studies in experimental models of CNS injury demonstrate that Nrf2 deficiency exacerbates neural injury, whereas pharmacological induction of Nrf2 with agents such as tert-butylhydroquinone (tBHQ) or sulforaphane results in neuroprotection [9, 10, 11]. However, the exact mechanisms of Nrf2 activation and its potential therapies for SAH remain poorly understood, despite positive results in other CNS injuries; thus, more studies are needed to elucidate the underlying mechanisms and refine targeted intervention strategies.
The Bcl-2/Bax/Cleaved caspase-3 signaling pathway plays a significant role in the regulation of intrinsic apoptotic pathways, especially mitochondrial dysfunction, which is considered to have an integral role in several nervous system diseases [12]. Bcl-2 is an anti-apoptotic protein that maintains mitochondrial integrity, and Bax promotes apoptosis by inducing the breakdown of the mitochondrial outer membrane and activating downstream effectors such as cleaved caspase-3, resulting in cell death. Dysfunction of such a pathway may thus participate in pathological neuronal death, and has been involved in disorders, for instance, neurodegenerative diseases, ischemic insults or SAH [13, 14]. Among the sirtuins, Sirt3 is a mitochondrial NAD(+)-dependent deacetylase that protects against oxidative stress by modulating the activity of antioxidant enzymes, including superoxide dismutase 2 (SOD2). Sirt3 expression in cortical neurons is downregulated in a time-dependent manner after SAH, in parallel with SOD2 levels, suggesting that it plays an important role as an antioxidant factor that attenuates oxidative damage following hemorrhage. This decrease in Sirt3 may further enhance mitochondrial dysfunction and apoptosis by decreasing the cell’s level of reactive oxygen species scavenging, resulting in the augmentation of neuronal injury [14, 15]. Therefore, Sirt3 and its downstream signaling might be good targets for treatment to increase neuronal survival in SAH and other oxidative stress-related central nervous system diseases.
Umbelliferone (UF), or 7-hydroxycoumarin, is a derivative of coumarin, and a type of phenylpropanoid. It forms colourless crystals with a characteristic odor reminiscent of vanilla [16]. It is known for its unique chemical structure, which contains a benzopyrone core featuring a hydroxyl group at the seventh position. It has been shown that 5-(Tetradecyloxy)-2-furoic acid (TOFA) has anti-inflammatory, antimicrobial and anticancer activities, which make it as a potential therapeutic agent [17, 18, 19]. In this experimental study, we explore the neuroprotective effect of UF against SAH in mice and explore the underlying mechanism.
In this experimental study, we used the C57BL/6 mice (sex: male; aged: 8–10
weeks and weight: 25–30 g). The mice were received by the department’s animal
house and kept in single cages. The mice were kept in standard laboratory
conditions (22
Animals were anesthetized using pentobarbital sodium administered
intraperitoneally at a dose of 50 mg/kg body weight (10 mg/mL solution prepared in sterile normal saline). Adequate depth of anesthesia
was confirmed by the absence of pedal withdrawal and corneal reflexes before any
experimental procedures were initiated. At the end of the experiment, animals
were euthanized under deep anesthesia using an overdose of pentobarbital sodium
(
Mice were induced under SAH using the previously reported method [12]. In short,
the mice were anesthetized with pentobarbital sodium (50 mg/kg) and the rectal
temperature was maintained using a heat blanket (37
Umbelliferone (98%, Sigma-Aldrich) was purchased and administered to the mice as an oral suspension. Firstly, prepare the oral suspension by dissolving the tested drug in 1% carboxymethylcellulose (CMC).
After the induction of SAH, the mice were divided into the following groups (n = 10), and the experimental group is shown in Fig. 1. Animals were subsequently divided into subgroups after SAH induction and randomization (n = 10 per group) to facilitate proper tissue processing. For neurological evaluation and naloxone administration (wet–dry) measurement, five mice per group were used, while another five mice were used for biochemical and molecular analysis. For the ELISA and qRT-PCR assays, the ipsilateral cerebral cortex was meticulously dissected at 4 °C, half were homogenized for protein estimation, while the neighbouring cortical area was flash-frozen in liquid nitrogen and stored at –80 °C until further analysis of mRNA expression.
 Fig. 1.
Fig. 1.
Experimental group. SAH, subarachnoid haemorrhage; UF, umbelliferone.
After isolating the ipsilateral cortical tissues, they were lysed in ice-cold lysis buffer RIPA containing proteases and phosphatase inhibitors (Thermo Fisher Scientific, Waltham, MA, USA) followed by centrifugation at 12,000 rpm for 15 min at 4 °C. The supernatant was retrieved and protein concentration was determined with BCA protein assay kit (Thermo Fisher Scientific).
For the estimation of SAH grade, the high-resolution images of the blood clots in the basal cisterns [12]. As per the score system, the following groups and their scores:
Mild SAH: 0–7 points,
Moderate SAH: 8–12 points, and
Severe SAH: 13–18 points.
All mice in each group were assessed for neurological performance using the modified Garcia scale and Rotarod test following 48 h post-SAH period. The symmetry movements of limbs, spontaneous activity, climbing, forepaw outstretching, tentacle reaction and body proprioception were assessed by following the previously reported literature by Garcia et al. [20]. For the rotarod test, we measured the performance of all mice before surgery (1 day) and 48 h post-SAH. To summarize, the mice were placed on a cylinder, and the rotation rate was increased from 4 to 40 rpm for 5 minutes. As reported earlier [21], the latency to falling off in three consecutive trials was calculated.
For the estimation of brain edema, we estimated the brain water level in the all-group mice. Briefly, after the surgery, the brains were immediately removed (48 h) and the brain tissue was quickly divided into four parts, such as the hemisphere (right and left), the brain stem, and the cerebellum. Each part was weighed quickly to get the wet weight and dried in a 100 °C for 24 h using the oven to get dry weight. The brain water level was estimated using the following formula [21]:
The antioxidant parameters such as MDA (Cat. No: E-BC-K025-M), GSH (Cat. No:
E-BC-K030-M), GPx (Cat. No: E-BC-K096-M), CAT (Cat. No: E-BC-K031-M) and SOD
(Cat. No: E-BC-K020-M); inflammatory cytokines include TNF-
Commercially available ELISA kits were used to determine the protein amounts of ERK (Cat. No. E-EL-R2665), JNK (Cat. No. E-EL-R2406), p38 (Cat. No. E-EL-R2536) and their phosphorylated forms (p-ERK, p-JNK, and p-p38) according to manufacturer’s instructions with equal amounts of total proteins (Elabscience Biotechnology Inc., Wuhan, China).
After the experimental phase, all the mice were sacrificed and their cortex was immediately frozen on dry ice and stored in RNA buffer until the mRNA isolation protocol. mRNA was isolated using the Total RNA Mini Kit (A&A Biotechnology, Poland) according to the manufacturer’s instructions. Reverse transcription was conducted at 100 ng/µL using the NG dART RT Kit in a DNA Engine DNA thermocycler. mRNA expression was assessed by quantitative real-time PCR in a standard mode using the SYBR Green Master Mix and primers listed in Table 1. GAPDH served as the internal standard.
| S. No | Genes | Primer (5′- 3′) | |
| Forward | Reverse | ||
| 1 | CLDN5 | TAAGGCACGGGTGGCACTCA | CTACGATGTTGGCGAACCAG |
| 2 | BSG2 | GTTTGTGAAGCTGATCTGCAAG | ACAGCTCAGGCGTGGATATAAT |
| 3 | OCLN | TAGCCATTGTCCTGGGGTTCAT | TTTCTTCGGGTTTTCACAGCAAA |
| 4 | TNF- |
AAGCATGATCCGAGATGTGGAACTG | CGCCACGAGCAGGAATGAGAAG |
| 5 | IL-6 | ACTTCCAGCCAGTTGCCTTCTTG | TGGTCTGTTGTGGGTGGTATCCTC |
| 6 | MCP-1 | CTCACCTGCTGTACTCATTCACTG | CTTCTTTGGGACACCTGCTGCTG |
| 7 | MMP-2 | GCAACCACAACCAACTACGA | CCAGTGTCAGTATCAGCATCAG |
| 8 | MMP-9 | GCAAACCCTGCGTATTTCCAT | CCATCCGAGCGACCTTTAGTG |
| 9 | PI3K | GCTGGACTTGGTGTTGAAGA | GGATGTAGCCATCAGGTTGA |
| 10 | Akt | TGGACTACATGAGCGACAAG | GGTGCCGTAGTCATCCATAA |
| 11 | TLR2 | GCTTTCCTGCTCAACTTCCT | TGGTCATCTTGGGCTTCTTC |
| 12 | TLR4 | GGACTCTGAATCGGAGGAAA | CCAAGTCTCTGAAGGGTCTG |
| 13 | GAPDH | TGGAAAGCTGTGGCGTGAT | AACGGATACATTGGGGGTAG |
Values are presented as the mean and standard deviation (SD). The statistical
analyses were performed with GraphPad Prism (version 8; GraphPad Software, San
Diego, CA, USA). Data normality was first checked by the Shapiro-Wilk.
Comparisons between more than two groups were performed using one-way ANOVA
followed by Tukey’s multiple comparisons test. Two-group comparisons were done
using an unpaired Student’s t-test. Statistical significance was
considered at p
SAH group mice exhibited reduced body weight, brain damage, and increased brain
water content compared with the normal control group. Umbelliferone treatment
significantly (p
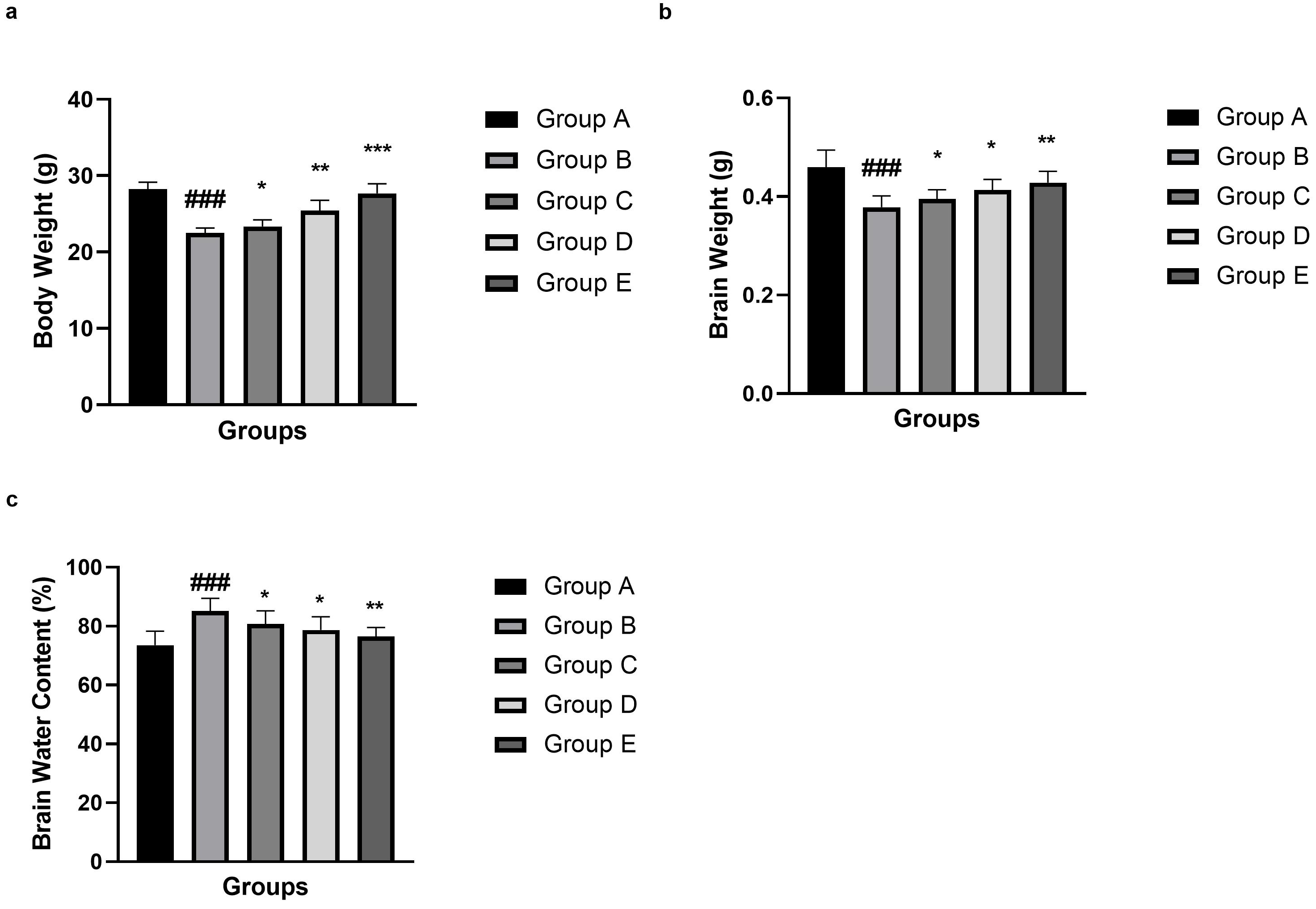 Fig. 2.
Fig. 2.
Effect of umbelliferone on the body weight, brain weight and
brain water content against SAH in mice. (a) body weight, (b) brain
weight and (c) brain water content. Data are presented as the mean
SAH group mice showed a suppressed modified Garcia Score (Fig. 3a) and Rotarod
latency (Fig. 3b) as compared to the normal control group mice. Umbelliferone
treatment significantly (p
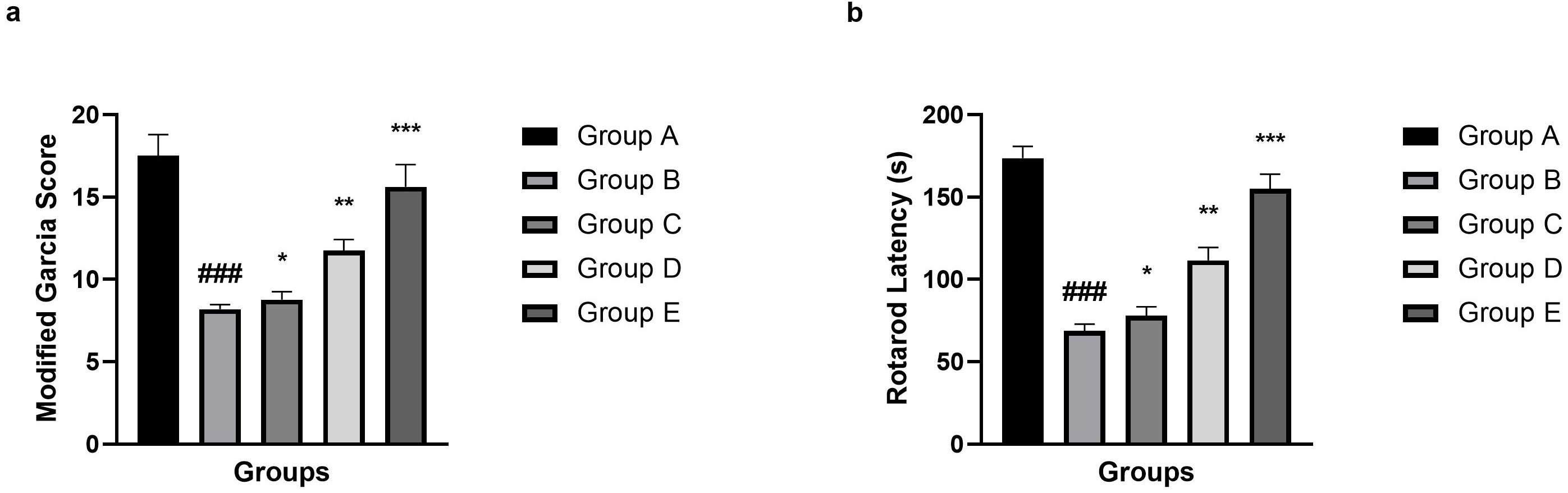 Fig. 3.
Fig. 3.
Effect of umbelliferone on the modified Garcia Score and Rotarod
latency against SAH in mice. (a) Modified Garcia Score and (b) Rotarod latency. Data are presented as the mean
The NC group displayed no signs of blood loss (score = 0). In all groups of
SAH-induced models, the magnitude of haemorrhage was relatively consistent. The
mean score of SAH grade in SAH group was 12.8
| S. No | Group | Groups | SAH Grade Score |
| 1 | A | NC (Normal Control) | 0 |
| 2 | B | SAH | 12.8 |
| 3 | C | SAH + Umbelliferone (2.5 mg/kg) | 11.1 |
| 4 | D | SAH + Umbelliferone (5 mg/kg) | 8.6 |
| 5 | E | SAH + Umbelliferone (10 mg/kg) | 5.4 |
Data are presented as the mean
The results showed that the integrity of the BBB was seriously compromised after
SAH, which manifested as a significant (p
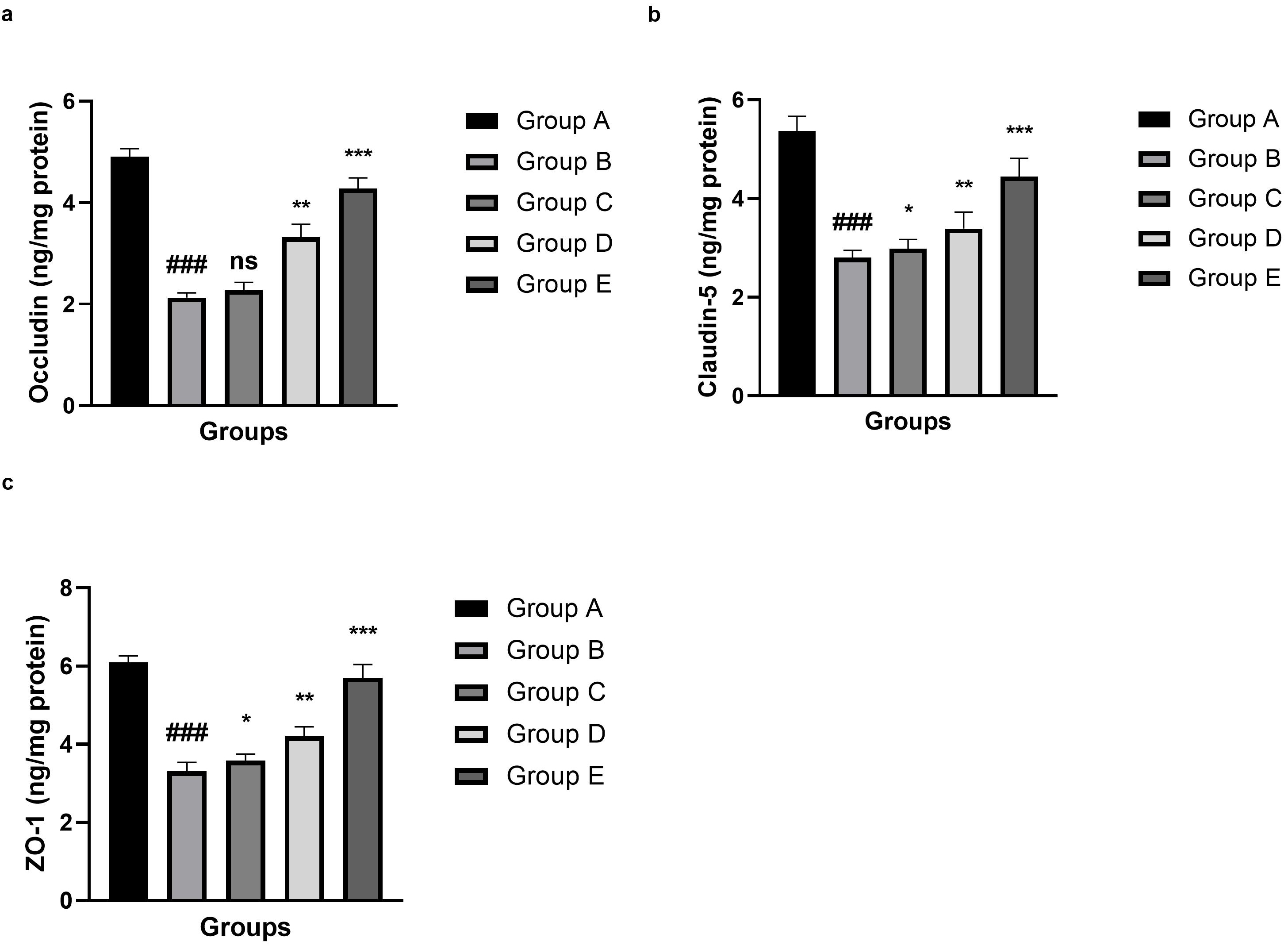 Fig. 4.
Fig. 4.
Effect of umbelliferone on the tight junction parameter against
SAH in mice. (a) Occludin, (b) claudin-5 and (c) ZO-1. Data are presented as the
mean
The results indicated a marked abnormality in the oxidative stress status after
SAH induction, as demonstrated in Fig. 5. Endogenous antioxidant defenses were significantly altered (p
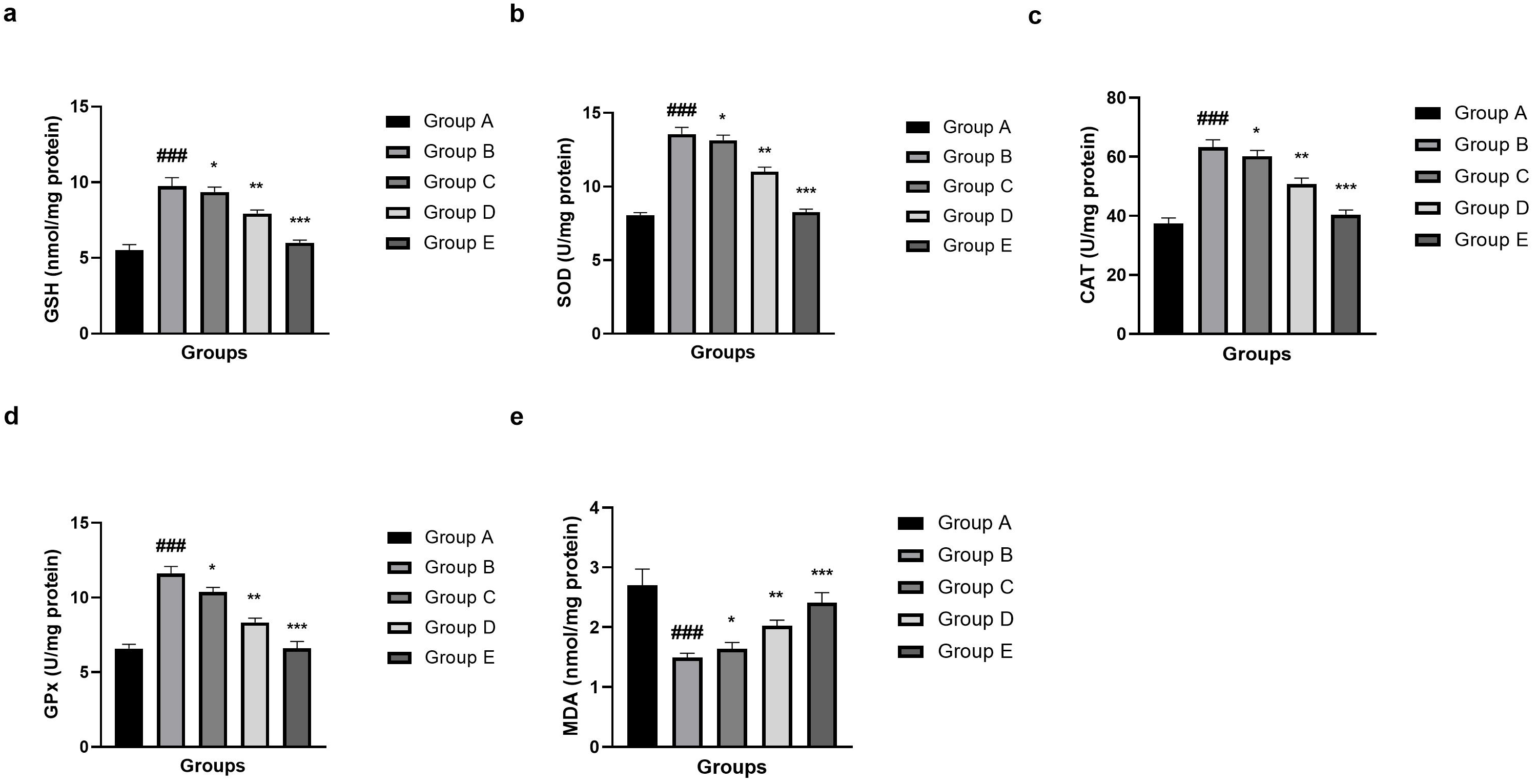 Fig. 5.
Fig. 5.
Effect of umbelliferone on the oxidative stress parameters
against SAH in mice. (a) GSH, (b) superoxide dismutase (SOD), (c) CAT, (d) GPx
and (e) MDA. Data are presented as the mean
The findings revealed a prominent induction of the mitogen-activated protein
kinase (MAPK) signalling pathway after SAH. The level of phosphorylation of JNK
(Fig. 6a), ERK (Fig. 6b), and p38 (Fig. 6c) was significantly (p
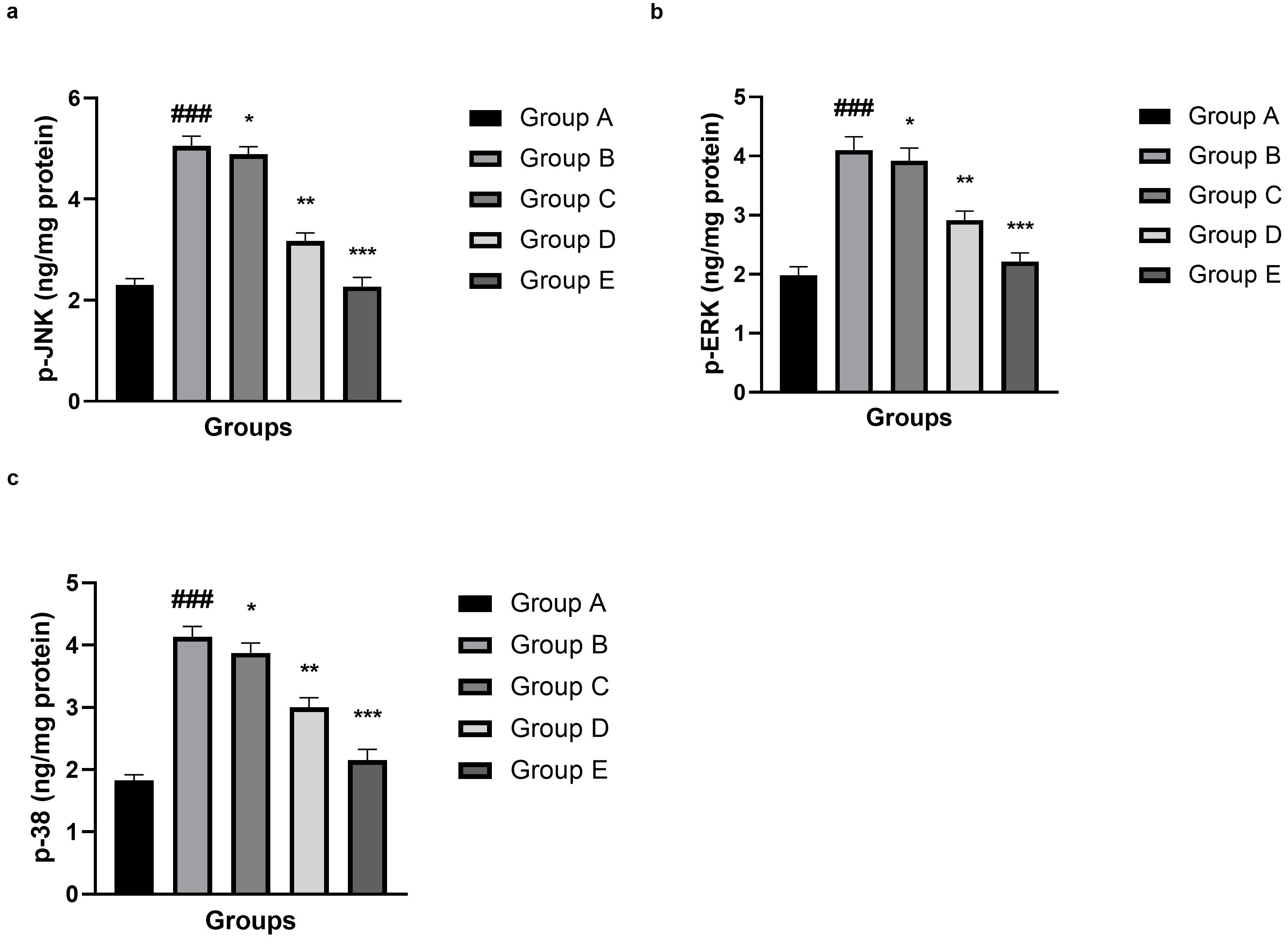 Fig. 6.
Fig. 6.
Effect of umbelliferone on the MAPK parameter against SAH in
mice. (a) p-JNK, (b) p-ERK and (c) p-38. Data are presented as the mean
Compared with the normal control (Group A), significant (p
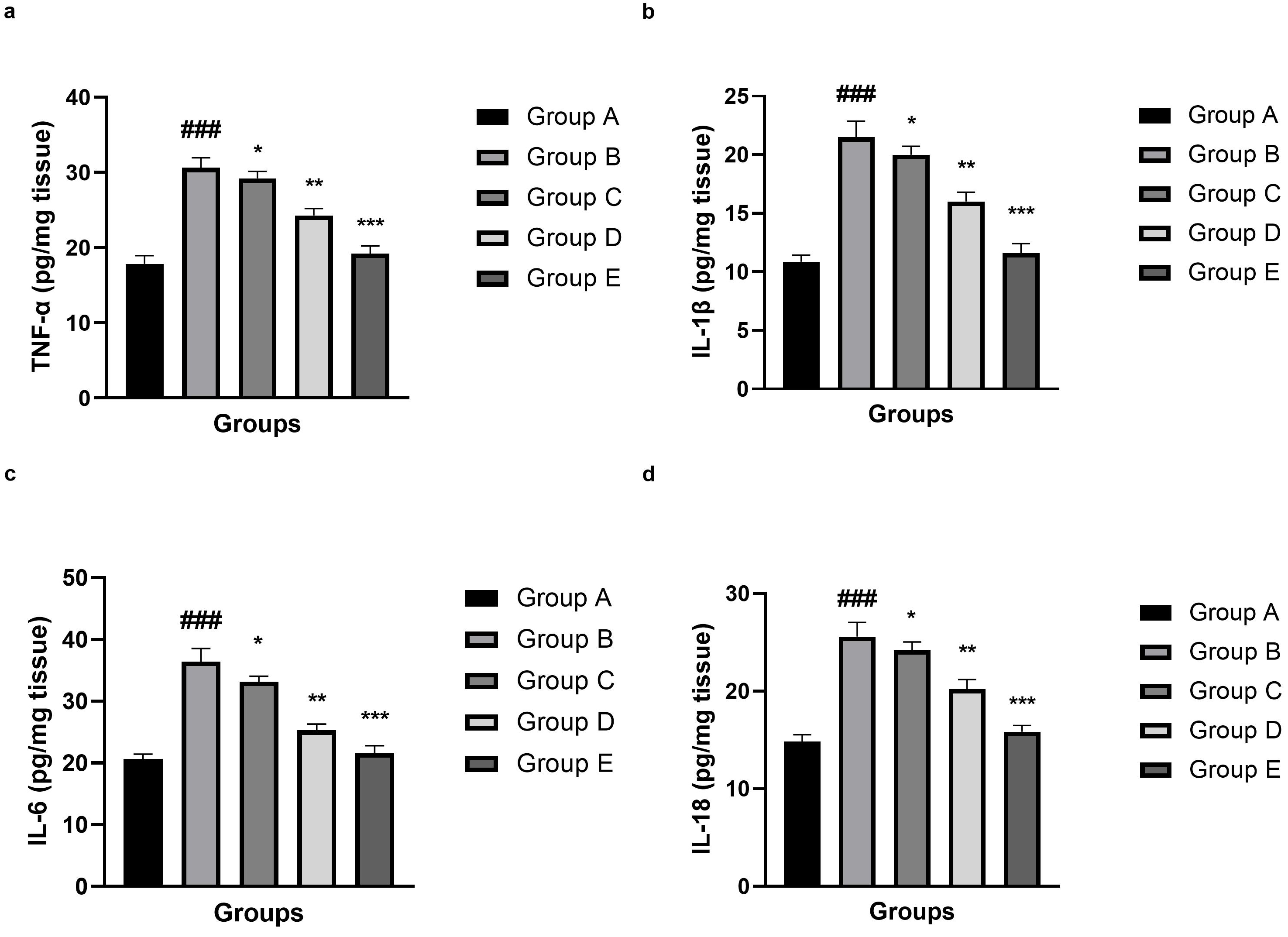 Fig. 7.
Fig. 7.
Effect of umbelliferone on the inflammatory cytokine parameters
against SAH in mice. (a) TNF-
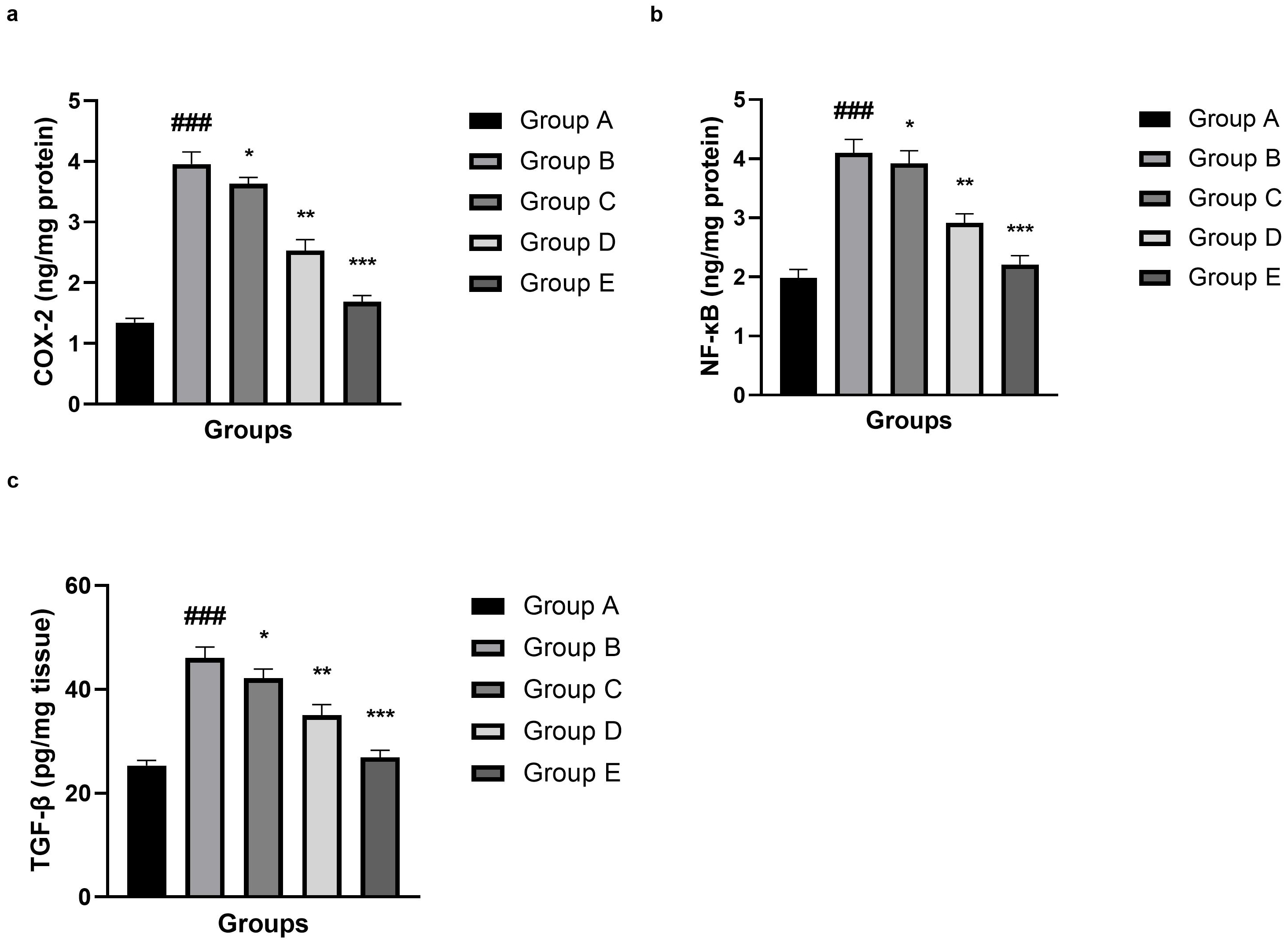 Fig. 8.
Fig. 8.
Effect of umbelliferone on the inflammatory parameters against
SAH in mice. (a) COX-2, (b) NF-
The result revealed an obvious upregulation of mitochondrial-dependent apoptosis
after SAH onset. As illustrated in Fig. 9a–d, pro-apoptotic parameters were
significantly (p
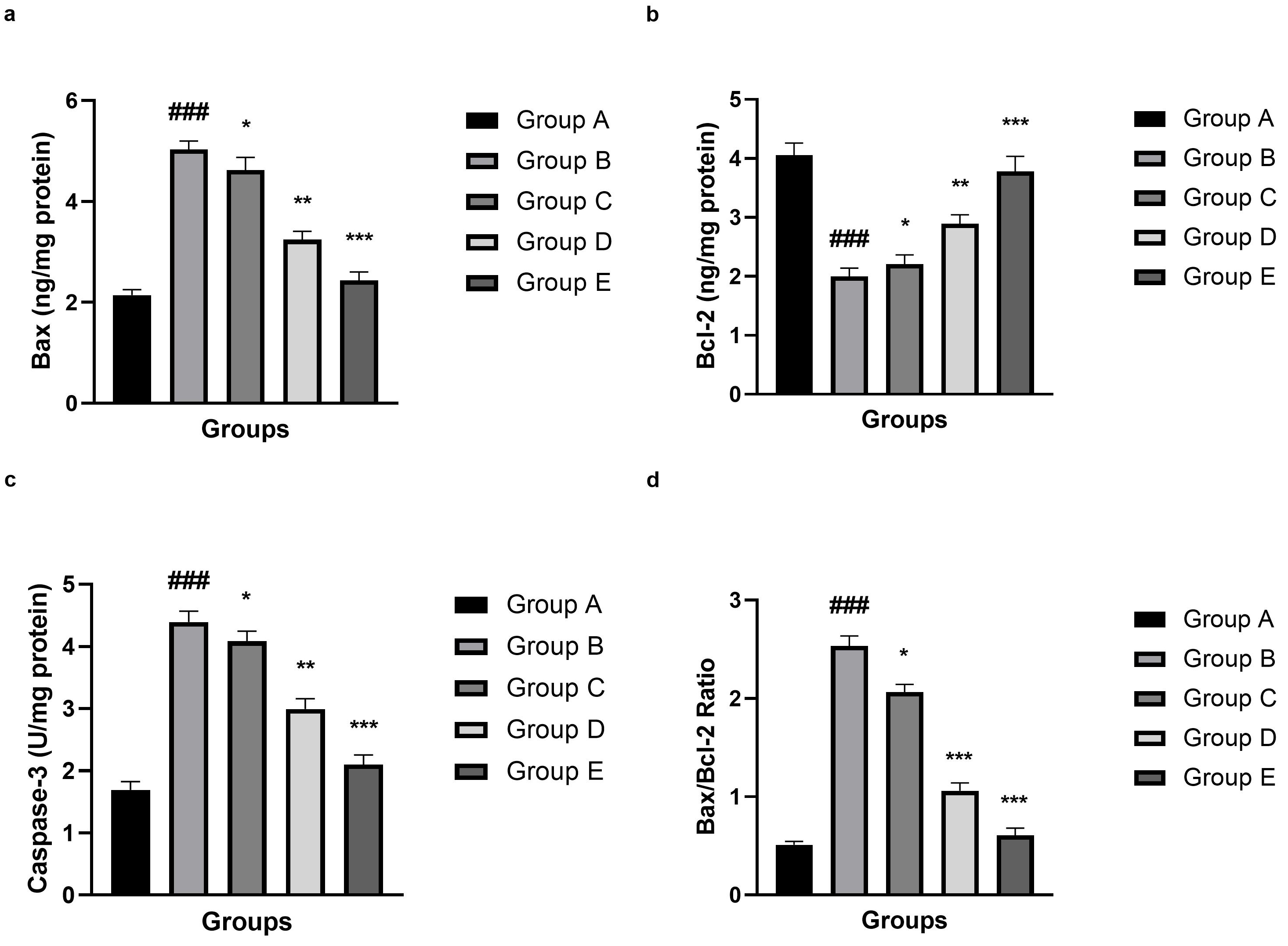 Fig. 9.
Fig. 9.
Effect of umbelliferone on the apoptosis parameters against SAH
in mice. (a) Bax, (b) Bcl-2, (c) caspase-3 and (d) Bax/Bcl-2 ratio. Data are
presented as the mean
Gene expression experiments revealed significant changes in inflammatory,
matrix-degrading and blood–brain barrier-related genes during SAH. As shown in
Fig. 10, there was a significant increase of mRNA levels of MMP-2 (Fig. 10a),
MMP-9 (Fig. 10b), BSG2 (Fig. 10c), TNF-
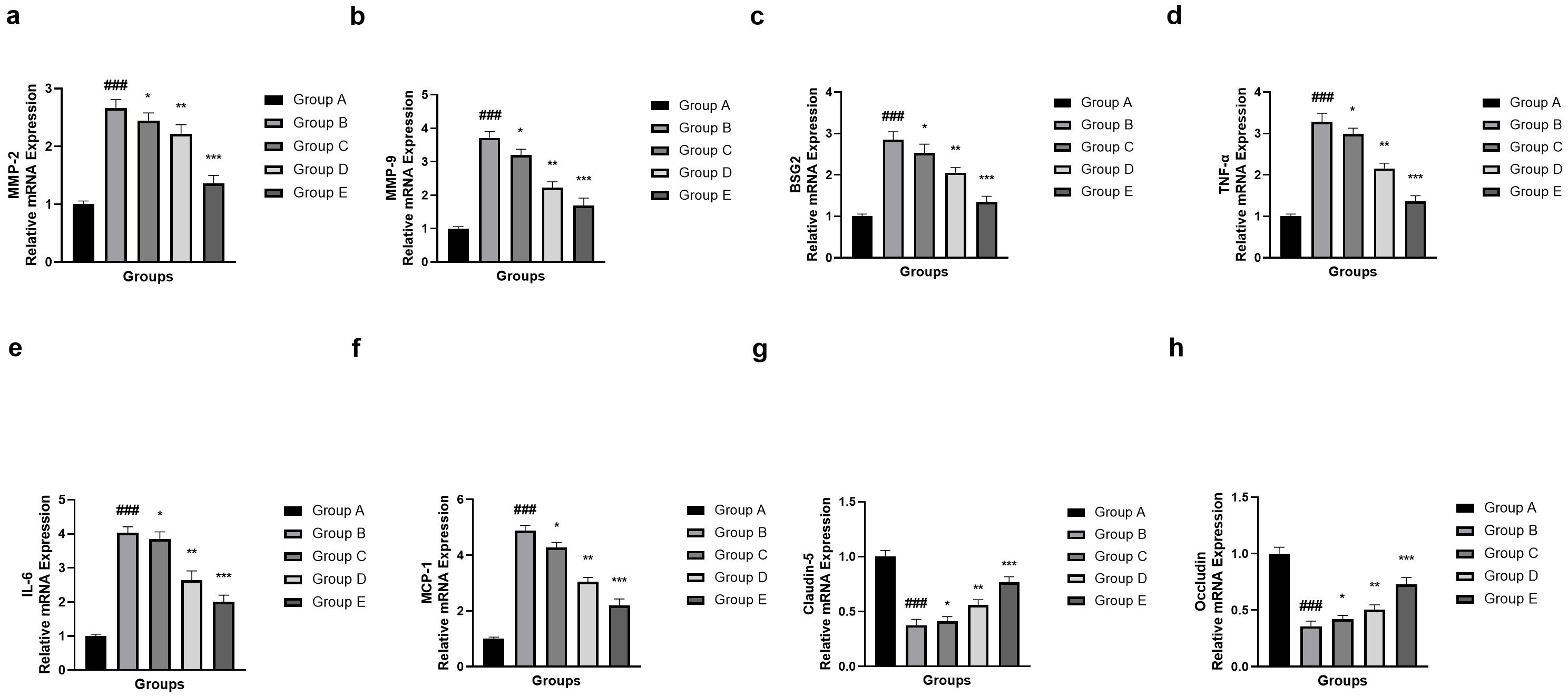 Fig. 10.
Fig. 10.
Effect of umbelliferone on the mRNA expression (MMP-2, MMP-9,
BSG2, TNF-
This analysis revealed that the mRNA expression of PI3K (Fig. 11a), Akt (Fig. 11b), TLR2 (Fig. 11c) and TLR4 (Fig. 11d) varied significantly (p
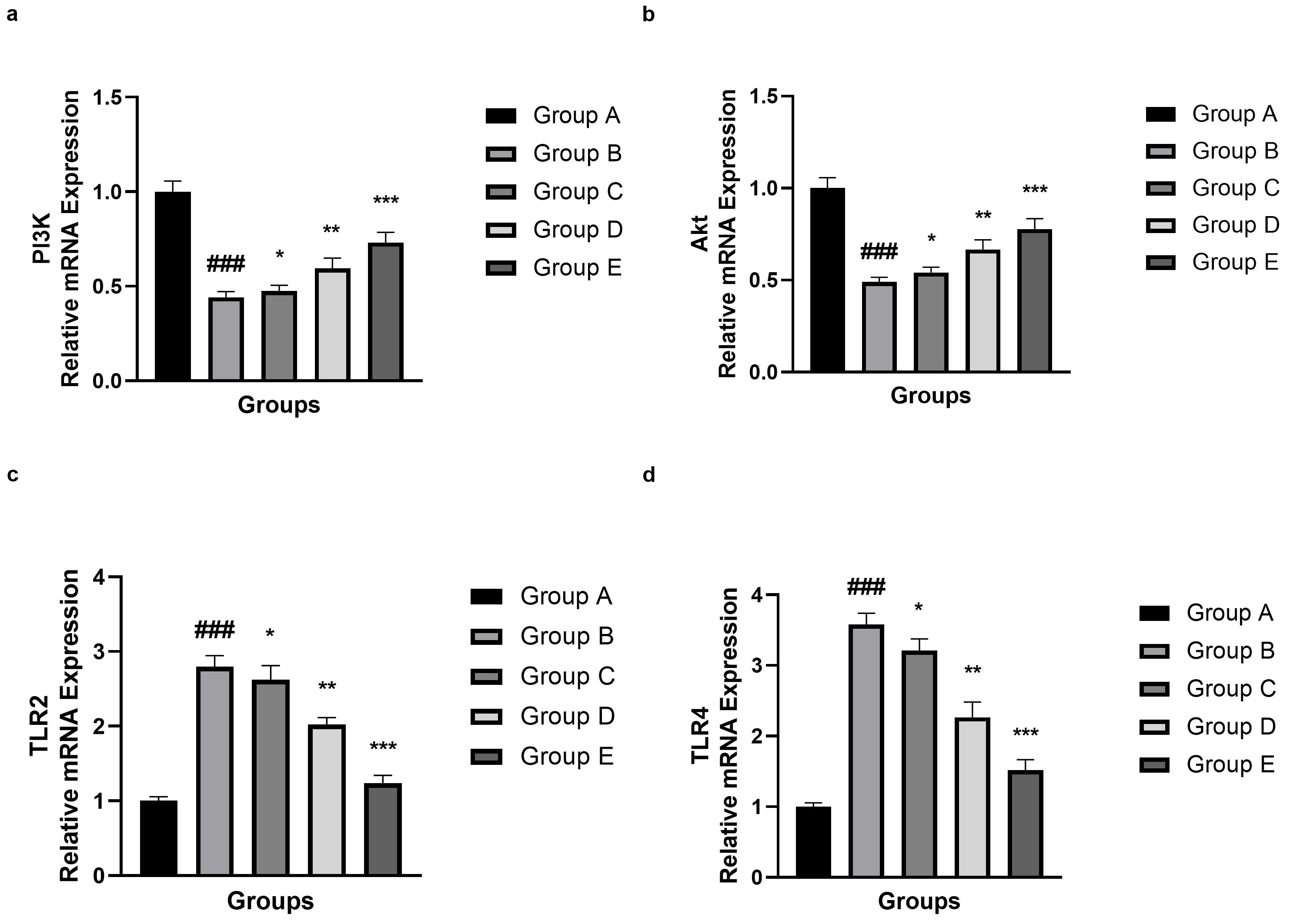 Fig. 11.
Fig. 11.
Effect of umbelliferone on the mRNA expression (PI3K, Akt,
TLR2 and TLR) against SAH in mice. (a) PI3K, (b) Akt, (c) TLR2 and (d)
TLR4. Data are presented as the mean
Neuroinflammation is a key factor in secondary brain injury and poor outcomes in patients with SAH. Natural products with antioxidant and anti-inflammatory properties have gained attention as potential treatments for these damaging processes. Umbelliferone may reduce oxidative stress and the inflammatory cascade triggered by SAH and could help protect neurological tissue from damage [1, 2]. However, the exact mechanism through which umbelliferone influences SAH-induced neuroinflammation has not yet been fully determined. It may also be valuable to investigate whether umbelliferone’s effects are mediated by main molecular targets involved in post-SAH inflammatory responses, such as microglial activation, cytokine release, and BBB permeability [2, 3] Umbelliferone is a free radical scavenger, possessing strong antioxidant activities which are ascribed to its ability to inhibit xanthine oxidases and metal chelating properties. These features indicate that umbelliferone may inhibit oxidative stress and the inflammatory cascade induced by SAH, and may help protect neurological tissue from damage. However, the exact mechanism by which umbelliferone exerts its effects on SAH-induced neuroinflammation has yet to be fully elucidated. It might be also interesting to validate whether the umbelliferone action is mediated via main molecular targets associated with post-SAH inflammatory responses, such as microglial activation, cytokine release and blood-brain barrier (BBB) permeability [1, 3, 6]. In the present study, we aim to clarify these mechanisms by discussing current evidence on umbelliferone’s effects on inflammatory mediators and oxidative stress markers in SAH, as well as laying a solid groundwork for future preclinical and clinical studies aimed at improving outcomes for patients with this morbid disease [1, 3].
Antioxidants such as SOD, CAT, GSH, and GPx form a coordinated defense system
against reactive oxygen species (ROS), detoxifying them and thereby protecting
against oxidative damage. Redox balance is impaired by a decrease in antioxidant
activity or levels, predisposing tissues to oxidative damage and disease
progression [22]. Inflammation, which is closely associated with oxidative
stress, is a complex network of signaling molecules and transcription factors
that controls immune responses. Pro-inflammatory cytokines that induce leukocyte
recruitment to the site of injury produce more ROS and initiate apoptotic
signaling pathways which can further destroy tissue [1, 4, 23]. Conversely,
anti-inflammatory cytokines such as IL-10 check this response by repressing
inflammation signaling and promoting tissue healing. Inflammatory mediators also
participate in inflammation spreading and pain sensation, with NF-
The pro-inflammatory response significantly contributes to the pathophysiology
of EBI after experimental SAH and transcription factor NF-
MMP-9-mediated disruption of the BBB enhances the entry of plasma proteins and immune cells into the brain parenchyma, exacerbating neuroinflammation and leading to vasogenic edema [28, 29]. This pathological elevation of vascular permeability exacerbates brain edema, and most importantly, forms the basis for secondary neuronal injuries due to oxidative stress and excitotoxicity. The fact that brain pericytes are a major source of MMP-9 upon thrombin stimulation highlights the complicated crosstalk between the coagulation cascade and NVCU members in the acute phase of an insult to the brain. Pharmacologically, drugs against thrombin can be explored as an effective strategy in reducing MMP-9 induced BBB disruption and edema. Blocking thrombin activity early after injury may downregulate MMP-9 expression in pericytes, thus preventing BBB damage and the consequent entry of detrimental inflammatory mediators. This treatment not only prevents the development of secondary injury cascades (e.g., microglial activation and neuronal apoptosis) but also promotes functional recovery through stabilization of the neurovascular microenvironment [28, 30, 31, 32]. Elucidation of relationships among thrombin signaling, pericyte activation, and MMP-9 release would provide additional approaches to treat edema associated with a large number of devastating neuropathological conditions, including TBI, stroke, and other ischemic insults. Gaining control over the formation of edema would likely be therapeutic [28, 30].
Activation of the PI3K/Akt signaling pathway is associated with increased neuronal survival by blocking apoptotic cascade via effectors including phosphorylation of Bad and caspase-9, which are known to counteract programmed cell death [33, 34]. Furthermore, PI3K/Akt signaling also regulates oxidative stress by up-regulating oxidant scavenger molecules and inhibits pro-inflammatory mediators, promoting overall cerebrovascular hemostasis and blood–brain barrier preservation following SAH [34, 35]. Blockade of this pathway enhances EBI by promoting neuronal apoptosis, inflammation, and vascular permeability that contribute to the development of SAH-related neurological deficits. The neuroprotective effects of umbelliferone are associated with the ability to modulate the PI3K/Akt pathway and to leverage its antioxidant and anti-inflammatory properties to prevent damage caused by SAH. Through the mediation of PI3K/Akt activity to restore, umbelliferone eliminates neuronal apoptosis and oxidative damage, providing structural integration as well as functional maintenance for blood–brain barrier integrity, which is essential to protect secondary injurious cascades [33, 35, 36]. These multi-target effects emphasize umbelliferone’s therapeutic potential, not only by targeting molecular signaling pathways but also by ameliorating complex pathophysiological changes following SAH.
Importantly, although current work focuses on the role of TLR2, the contribution
of TLR4 as a co-stimulatory receptor for T cell activation has been less
investigated. In view of the complex immune environment in the brain, in which
cytokines such as IFN-
Several studies found similar neuroprotective, antioxidant and anti-inflammatory
effects for coumarin derivatives and related phenolic compounds as we have
obtained with umbelliferone. For instance, numerous simple coumarins (e.g.,
esculetin and scopoletin) as well as more complex derivatives (e.g., osthole and
daphnetin) have been shown to abrogate oxidative stress, decrease the production
of pro-inflammatory cytokines, and restrain apoptotic signaling in models of
cerebral ischemia, traumatic brain injury and other acute CNS insults [41]. These
compounds frequently modulate shared injury pathways specifically ROS scavenging,
inhibition of NF-
This study is not without its limitations, which the reader should keep in mind
when interpreting the results. The experiments were performed in a single mouse
model of SAH, and therefore, the findings may not be directly translated to human
disease due to species differences in pathophysiology or drug metabolism. Also,
the experimental study was confined to short-term biochemical and molecular
results and no long-term (neurological, behavioral or cognitive) responses were
measured; hence, it is not known whether the demonstrated molecular/cell
improvements lead to sustained functional recovery. Although changes in the
TLR2/TLR4–NF-
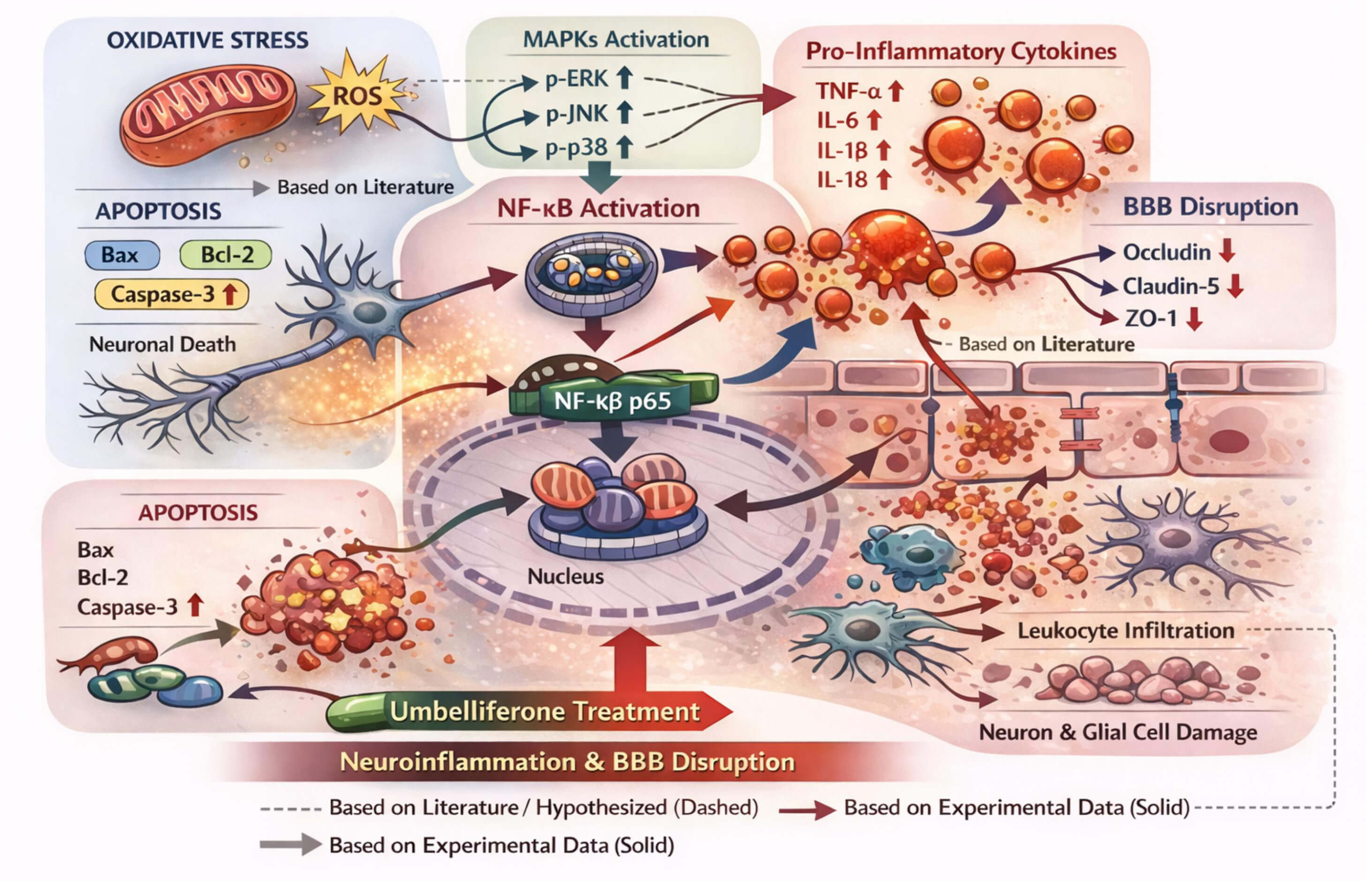 Fig. 12.
Fig. 12.
The mechanism diagram shows that SAH causes excessive oxidative
stress, leading to the overproduction of reactive oxygen species (ROS), which
activate the MAPK signaling pathway (p-ERK, p-JNK, and p-p38). MAPK activation
activates NF-
The current study demonstrated that umbelliferone has a strong neuroprotective
effect on SAH-induced EBI via multiple signaling pathways. The protective effects
of umbelliferone against oxidative stress and neuroinflammation were successfully
ameliorated, as indicated by the restoration of antioxidant status and inhibition
of pro-inflammatory cytokines. At the molecular level, these benefits were
associated with inactivation of the TLR2/TLR4–NF-
The raw data will be available on the request to the corresponding author.
HZ and XL performed the experimental study. YZ designed the experimental study. HZ draft the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The experimental protocols were reviewed and approved by the Animal Ethics Committee of Baotou Central Hospital, China and all procedures were performed under the approved ethical standards. The ethics approval number is [Approval No. 12783948]. All animal testing follows the 3R principle and the ARRIVE guidelines.
Not applicable.
This research received no external funding.
The authors declare no conflicts of interest.
During the preparation of this work, the author used the ChatGPT 4 tool to create Figures 1 and 12. After using this tool, the authors reviewed the content as needed and took full responsibility for the content of the publication.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.