



1 Institute of Integrated Traditional Chinese and Western Medicine, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, 430030 Wuhan, Hubei, China
2 Department of Dermatology, Wuhan No. 1 Hospital, Tongji Medical College, Huazhong University of Science and Technology, 430033 Wuhan, Hubei, China
3 Hubei Province & Key Laboratory of Skin Infection and Immunity, 430071 Wuhan, Hubei, China
4 Department of Integrated Traditional Chinese and Western Medicine, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, 430030 Wuhan, Hubei, China
Abstract
The female reproductive tract consists of the vagina, cervix, uterus, and fallopian tubes. In particular, the lower region of the reproductive tract, which contains the vagina and cervix, is often attacked by various pathogenic microorganisms such as bacteria, fungi, and viruses. The immune response of the female lower genital tract is the first line of defense against pathogenic microorganisms. The toll-like receptors (TLRs), a critical pattern recognition receptor, are essential for fighting infections in the female lower genital tract. Here we give an overview of the current research on TLR expression in the female lower genital tract and review the role of TLRs and their signaling pathways in the identification of numerous pathogens in female lower genital tract infections. Our review will contribute to a deeper understanding of the connection between TLRs and the pathological mechanisms of female lower reproductive tract infections, serving as a reference for both fundamental research and preventative strategies for these diseases.
Keywords
- toll-like receptor
- infectious diseases
- female genital tract
- innate immunity
Toll-like receptors (TLRs) are protein molecules involved in non-specific immunity, and are a type of pattern-recognition receptor (PRR) expressed by innate immune cells. TLRs recognize specific structural molecules derived from pathogens essential for maintaining pathogen survival and are described as pathogen-associated molecular patterns (PAMPs) [1]. TLRs recognize the corresponding ligands when microorganisms invade the body’s barriers and activate non-specific immunity. Furthermore, they can initiate the body’s defense system to respond accordingly and modulate specific immunity to protect the host from pathogenic threats [2].
TLRs are expressed in a variety of cells, including macrophage dendritic cells (DCs), neutrophilic-granulocyte (NE), natural killer cells (NK), and fibroblasts. TLRs can bind many ligands, including peptidoglycans, lipoproteins, yeast glycans, flagellin, microbial DNA/RNA, and other pathogen-derived ligands (Fig. 1). To date, 10 TLR family members have been identified in humans. Depending on their sites of expression, TLR can be classified into two major groups: those expressed within endosomal compartments, including TLR3/7/8/9, which is mainly used to recognize viral and bacterial DNA [3]; and those primarily distributed on the plasma membrane, mainly recognizing other PAMPs (Fig. 2). TLR1, TLR2, and TLR6 can recognize bacterial lipoproteins, TLR4 recognizes lipopolysaccharide (LPS), and TLR5 recognizes flagellin binding [4]. In addition, TLR10 is also distributed inside the cell membrane, but its corresponding ligand has yet to be discovered.
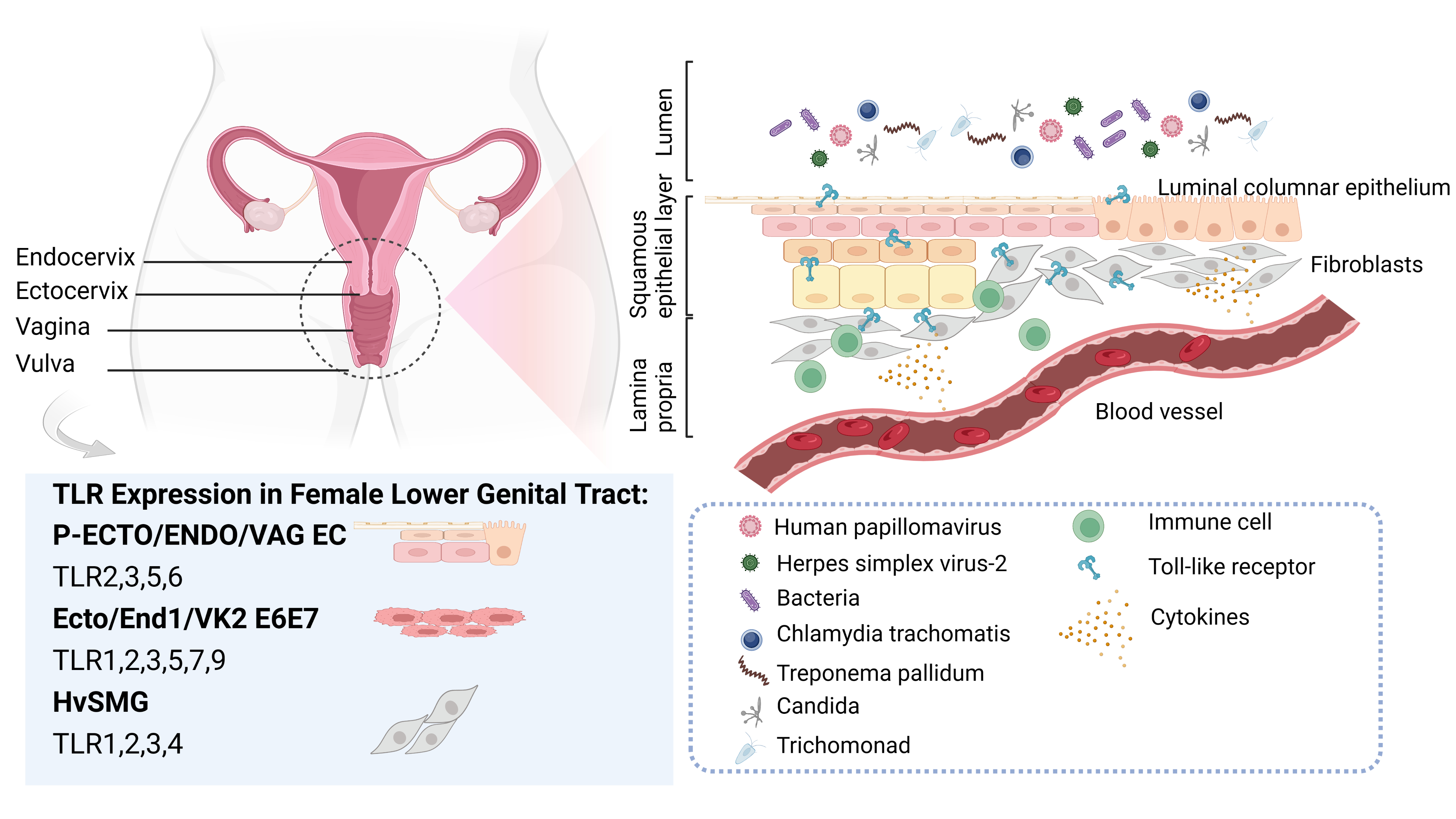 Fig. 1.
Fig. 1.Toll-like receptors (TLRs) and common pathogens in the female lower genital tract. The vulva, vagina, and cervix comprises the lower part of the female genital canal. The mucosa of the cervical canal is covered by a single layer of highly columnar epithelium, whereas a complex layer of squamous epithelium covers the vaginal wall. The complex squamous epithelium meets the single columnar epithelium at the ectocervix. The lower region of the female genital tract is often subjected to various pathogenic microorganisms, such as human papillomavirus, herpes simplex virus-2, bacteria, chlamydia trachomatis, treponema pallidum, candida, and trichomonad. Confronted with the invasion of these pathogens, each part plays a unique role in the local immune response mechanism, and the distribution, expression, and response of TLRs within it also display subtle differences. Among the commonly employed in vitro research models for female lower reproductive tract infections, TLRs 2/3/5/6 are highly expressed in primary epithelial cell cultures derived from ectocervix (P-ECTO EC), primary epithelial cell cultures derived from endocervix (P-ENDO EC), and primary epithelial cell cultures derived from vaginal tissue (P-VAG EC). The TLR1/2/3/5/6/7/9 are expressed by immortalized cervical epithelial cell line (End1/E6E7), immortalized ectocervical cell line (Ect1/E6E7), and immortalized vaginal cell line (Vk2/E6E7). Human vagina smooth muscle cells (HvSMCs) express all TLRs, with TLR1/2/3/4 being the most abundant.
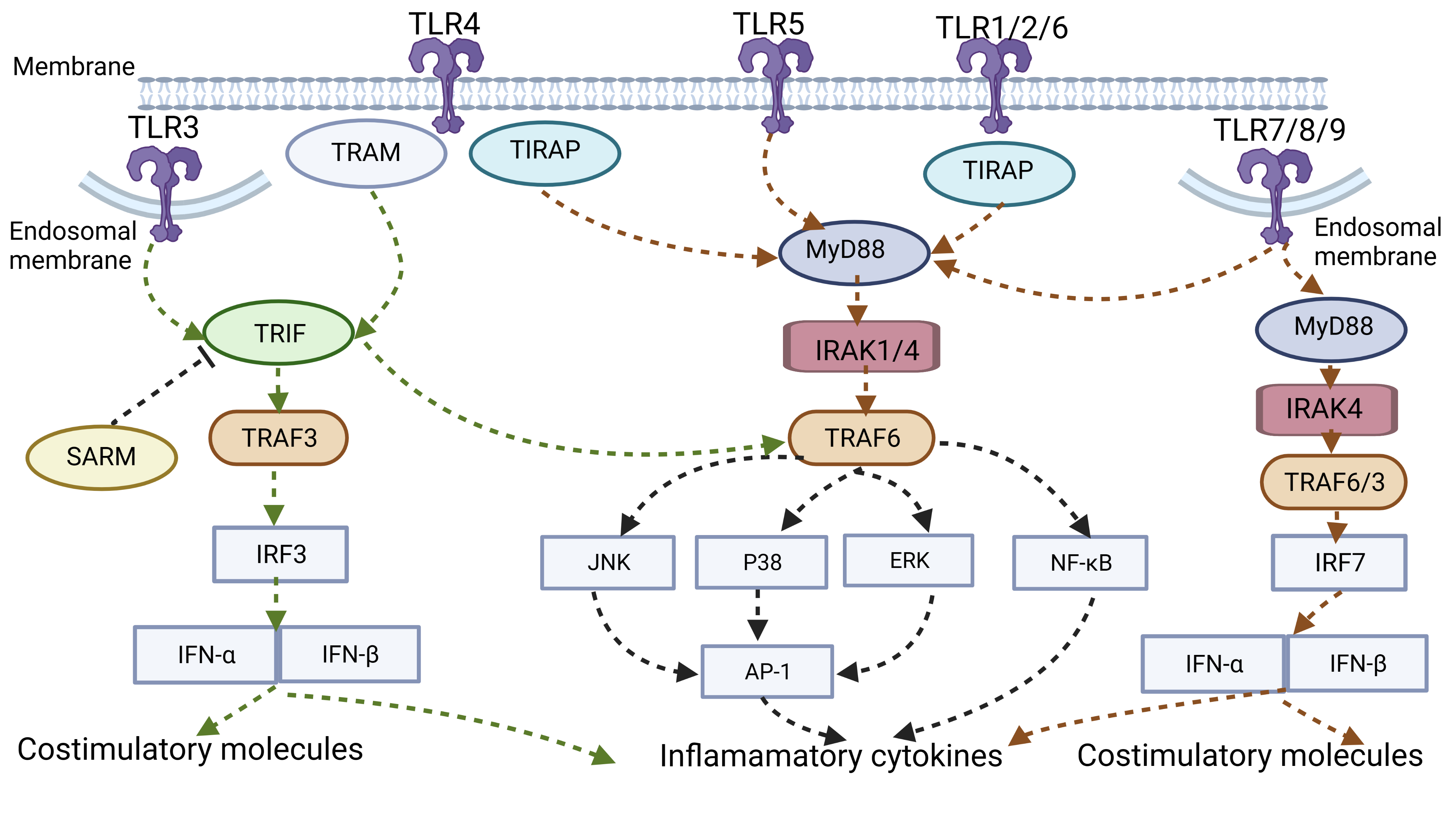 Fig. 2.
Fig. 2.TLR signal pathway. TLR3/7/8/9 is expressed within endosomal
compartments, whereas others are distributed on the plasma membrane. TLRs
interact with intracellular bridging proteins (MyD88/TIRAP/TRIF/TRAM) to recruit
various kinases (IRAK4/IRAK1/IRAK2) and ubiquitin ligases (TRAF6/TRAF3), thereby
activating downstream NF-
TLR recognizes PAMP and interacts with other intracellular bridging proteins
with the same TIR domain (Toll/IL-1R) to recruit various kinases and ubiquitin
ligases, thereby activating downstream NF-
Myeloid differentiation factor 88 (MyD88), TIR domain-containing adaptor protein
(TIRAP), TIR domain-containing adapter-inducing interferon (TRIF), TRIF-related
adaptor molecule (TRAM), and sterile alpha and armadillo-motif-containing protein
(SARM) are the primary transduction bridging molecules (Fig. 2). SARM is a
protein that has been shown to suppress TRIF-dependent activity in human cell
lines [6]. Among them, MyD88 is a universal adapter for inflammatory pathway
activation shared by all TLRs except TLR3. Under the action of MyD88, TLRs
attract and activate different kinases such as IRAK4, IRAK1, IRAK2, and ubiquitin
ligase TRAF6 and subsequently assist in the activation of MAPK (ERK, JNK, p38)
and transcription factor NF-
The female genitalia comprises the vulva, vagina, uterus, fallopian tubes, and ovaries (Fig. 1). Due to its anatomy, the female genital tract, especially the lower region of the genital tract containing the vulva, vagina, and cervix, is often subjected to various pathogenic microorganisms. The natural immunity of the mucosa of the genital tract is essential to fight against infections in the female lower genital tract [7], and TLRs play an important role as the first barrier against infectious diseases [8]. According to the recent studies, the associations between TLRs and common female lower genital tract infections were enumerated in Table 1.
| Disease | PAMP | TLR | Function | Reference |
| Human papillomavirus infections | CpG | TLR9 | Recognize HPV16E6 code DNA sequence | [16] |
| VLPs | TLR4 | HPV16 L1 VLPs activate B cell-mediated humoral immunity via TLR4 and MyD88-dependent signaling | [17] | |
| HPV6b/11E7 protein | TLR7/9 | Promoted by HPV6b/11E7 protein | [18] | |
| Increase JNK/p38MAP kinase signaling pathway | ||||
| HMGB1 | TLR4 | Bind to HMGB1 to activate downstream responses | [19] | |
| Overexpressed by HPV11 | ||||
| Promote NF- | ||||
| TLR9 | Depressed by HPV11 | [20] | ||
| Involve in reversed activation of NF- |
||||
| TLR4/5/9 | Overexpressed in the skin of HR-HPV patients | [21, 22] | ||
| TLR7 | Overexpressed in skin tissue of HR-HPV patients | |||
| TLR4/9 | Consistent with pathologic histology-grading | [23, 25] | ||
| TLR2/3/7/8/9 | Associated with the clearance of HPV16 | [26, 27] | ||
| TLR4 | Decreased during cervical tumor development | [28] | ||
| Associated with the expression of p16INK4a | ||||
| TLR1 | Elevated in squamous cervical cancer tissue | [30] | ||
| TLR3/4/5 | Downregulated in squamous cervical cancer tissue | |||
| TLR3/4/7/8 | Overexpressed in squamous cervical cancer tissue | [31, 32] | ||
| TLR2/4/9 | Increases the risk of cervical cancer | [33] | ||
| Genital herpes | Viral glycoproteins | TLR4 | First expressed in VK2/E6E7 cells and infected by HSV-2 | [33] |
| Viral nucleic acids | TLR3/7/9 | Expressed in a time-dependent manner in VK2/E6E7 cells infected by HSV-2 | ||
| TLR4 | Mediated by ICP0 | [37] | ||
| Associated with host cell AP-1 activation | ||||
| TLR4 | Activates the NF- |
[38] | ||
| Induces the expression and secretion of downstream pro-inflammatory factors IL-6 and IFN- | ||||
| TLR4 | Responds to HSV-2 infection via the TLR4-Mal/MyD88-IRAK1-NF- |
[39] | ||
| TLR4 | TLR4-TRIF/TRAM response to HSV-2 infection in cervical epithelial cells | [40] | ||
| TLR9 | Overexpression by binding with SP1 | [41] | ||
| Exhibits a dose-dependent and time-dependent HSV-2 infection | ||||
| TLR9 | Mediate the early IFN response | [43] | ||
| TLR2/3 | Reduced by VHS to facilitate immune evasion | [44] | ||
| TLR2 | Gene polymorphisms associated with the severity of HSV-2 infection | [45] | ||
| TLR3 | Gene polymorphisms associated with the susceptibility to HSV-2 | [46] | ||
| TLR3/5/6/9 | Ligand treatment inhibited HSV-2 replication | [47, 48] | ||
| Bacterial vaginosis | LPS | TLR4 | Recognize gram-negative LPS | [1] |
| Lipopeptide | TLR6 | Recognize lipopeptide of Mycobacterium and other bacteria | ||
| Flagellin | TLR5 | Recognize flagellin of flagellated bacteria | ||
| CpG DNA | TLR9 | Recognize non-methylated CpG DNA of various bacteria | ||
| SCFAs | TLR2/7 | Increase the release of IL-8, IL-6, IL-1 |
[52] | |
| TLR2/4 | Upregulated in vitro by stimulation of vaginal lavage solution from BV patients | [53] | ||
| TLR2 | Induces the production of IL-6, IL-8, and | |||
| TLR5 | Stimulated by Mobiluncus mulieris and BVAB1 and induce NF- |
[49] | ||
| TLR2/7 | Increases the incidence of BV in African women by gene polymorphisms | [54] | ||
| TLR1/2/9 | Variants associated with BV in HIV-1 positive African Americans | [50] | ||
| TLR4 | Missense mutations cause a 10-fold increase in Gardnerella vaginalis levels | [55] | ||
| TLR9 | Genetic variants strongly connected with BV diagnosis | |||
| Vulvovaginal candidiasis | Mannans | TLR4 | Recognize mannans | [57] |
| Phospholipid mannans | TLR2 | Recognize phospholipid mannans | ||
| Prevent colonization and endogenous invasion of Candida albicans | ||||
| Single-stranded RNA | TLR7 | Recognize single-stranded RNA of Candida albicans | ||
| TLR2/4 | Increase in vaginal endothelial cells obtained from vaginal swabs of VVC patients | [58] | ||
| Activate c-fos and p38 signaling | ||||
| TLR2 | Increases the fungal load in the vagina | [56] | ||
| TLR2/6 | Expressed in Candida albicans infected VK2 cells | [59] | ||
| Activate IL6 and TNF | ||||
| TLR2/4 | Involved in the release of PGE2 | [60] | ||
| Trichomonas vaginitis | LPS | MyD88 non-dependent pathway of TLR | Induces IL-8, MIP-3 |
[61] |
| TLR2/4/9 | Upregulated through the p38 MAPK signaling pathway and regulate the release IL-8 TNF-a | [62] | ||
| Activate Bcl-xl and NF- | ||||
| TLR4 | Activate TLR4, MAPKs, and NF- |
[63] | ||
| TLR4 | Gene polymorphisms are linked with a high risk of TV-induced cervicitis | [63] | ||
| Syphilis | Tp92 | TLR2 | Signals the downstream MyD88 pathway activating NF- |
[69] |
| Tp0136 | TLR4 | Mediates the activation of ERK, JNK, PI3K, and NF- |
[70] | |
| Flagellin | TLR5 | Produces pro-inflammatory cytokines such as IL-6 and IL-8 | [68] | |
| FlaB1, FlaB2 and FlaB3 | TLR5 | Induces IL-6, IL-8 production in THP-1 cells activating ERK, p38 and NF- | ||
| FlaA2 | TLR2 | Stimulate IL-6, IL-8 expression in THP-1 cells through p38, NF- |
[71] | |
| TLR1/2/6 | Associated with an increased risk of neurosyphilis | [72] | ||
| TLR2 | Loci in the TLR2 TIR structural domain helped prevent syphilis infection | [73] | ||
| Genital chlamydial infections | MOMP | TLR2 | Induces TLR2-dependent IL-8 secretion | [77] |
| Increases NF- | ||||
| Induces IL-8 and IL-6 secretion in End/E6E7 cells | ||||
| cHSP60 | TLR2/4 | Activates NF- |
[76] | |
| Triggers the release of IL-1 and IL-18 in epithelial cells | ||||
| Mip | TLR1/2/6 | Involved in diminishing pro-inflammatory activity | [78] | |
| GlgA | TLR2/4 | Mediates the MAPK/NF- |
[79] | |
| Increases IL-8, IL-1 | ||||
| TLR2 | Involved in the production of TNF- |
[80] | ||
| LPS | TLR4 | Involved in LPS’s endotoxic activity | [81] | |
| TLR2/4 | Increases GM-CSF, TNF, IL-1 |
[82] | ||
| TLR2 | Involved in intracellular signaling | [81] | ||
| TLR4 | Activates PKR | [81] | ||
| TLR3 | Deficiency results in increased LPS within the chlamydial inclusion body | [75] | ||
| TLR2/9 | Haplotype genes prevent the exacerbation of clinical infection | [85] | ||
| TLR4 | Increased odds of Chlamydia trachomatis infection in women with pelvic inflammatory disease |
PAMP, pathogen-associated molecular pattern; TLR, Toll-like receptor; VLPs,
virus-like particles; HPV, human papillomavirus; HMGB1, high mobility group
protein B1; HR-HPV, high-risk human papillomavirus; HSV-2, herpes simplex virus
type 2; SP1, specificity protein 1; LPS, lipopolysaccharide; IFN, interferon;
VHS, virion host shutoff protein; SCFAs, short-chain fatty acids; BV, bacterial
vaginosis; BVAB, bacterial vaginitis-associated bacteria; VVC, vulvovaginal
candidiasis; PGE2, prostaglandin E2; MIP-3
Various hormones and commensal flora influence the female lower genital tract where a complex structure and precise function characterize it. Confronted with the invasion of pathogenic microorganisms, each part plays a unique role in the local immune response, and the distribution, expression, and response of TLRs within it also display subtle differences (Fig. 1).
To clarify whether TLRs are differentially expressed between parts of the female lower genital tract, researchers isolated primary cells from various parts of the female lower genital tract. They found that TLR1, TLR 3, and TLR 6-9 were similarly expressed in primary epithelial cell cultures derived from ectocervix (P-ECTO EC), primary epithelial cell cultures derived from endocervix (P-ENDO EC), and primary epithelial cell cultures derived from vaginal tissue (P-VAG EC). TLR2, TLR3, TLR5 and TLR6 were highly expressed compared with TLR1, TLR4 and TLR7-9 which were less expressed. TLR2, TLR4, and TLR5 expression levels in P-ECTO EC were significantly different from those in the other two tissues, with P-VAG EC expressing relatively higher TLR4 RNA [9]. Compared to epithelial cells, human vagina smooth muscle cells (HvSMCs) expressed all TLRs, with TLR1, TLR2, TLR3, and TLR4 being the most abundant [10]. Herbst-Kralovetz et al. [9] reported that immortalized cervical epithelial cell line (End1/E6E7), immortalized ectocervical cell line (Ect1/E6E7), and immortalized vaginal cell line (Vk2/E6E7) share the same TLRs expression pattern. The TLR1/2/3/5/6/7/9 are expressed by all of them, although TLR4 and TLR8 are only faintly expressed. Pioli et al. [11] obtained tissue lysates from various parts of the female genital tract, and found that all parts of the human female lower genital tract expressed TLR1-6 and the myeloid differentiation factor MyD88, but TLR4 RNA was found to be present only in the fallopian tubes instead of the female lower genital tract. Another study also confirmed that both vaginal epithelial cells and ectocervix cells were negative for anti-TLR4 antibodies [12].
In addition to the distribution of TLRs in standard samples from the female
lower genital tract, the response profile of TLRs varies from site to site. By
treating primary cells obtained from normal female reproductive tract tissue
using various TLR activators and comparing cytokine expression profiles in each
segment of the female reproductive tract, it was found that pro-inflammatory
markers were increased in each segment when stimulated by TLR3, TLR4, TLR7, TLR8,
and TLR9 ligands. Specifically, cervical cells responded more strongly to TLRs
agonists, significantly increasing various cytokines. The levels of IL-2R,
CCL3/4/5/9 in the endocervical lining were increased upon TLR3 ligand activation,
and IL-8, IL-2R, IFN-
HPV is a double-stranded DNA virus without an envelope with epithelial tropism, often invading the human oral cavity, skin, and perianal and genital tract. According to their carcinogenic potential, they are further classified as low-risk human papillomavirus (LR-HPV) and high-risk human papillomavirus (HR-HPV) [14].
The coding region of HPV is divided into the long control region (LCR), early gene region (E), and late gene region (L). Early genes (E1–E7) encode proteins associated with the regulation of viral replication, regulating the life cycle of viruses, DNA replication, DNA transcription, and viral protein translation. Among them, E6 and E7 are oncogenes encoding oncoproteins that play an essential role in interfering with signaling pathways to induce cell immortalization [15]. Late genes (L1 and L2) are responsible for encoding capsid proteins involved in cell surface adsorption, viral particle packaging, and virus entry into the cytoplasm. CpG sequences are present in the HPV genome. Furthermore, the HPV16E6 coding DNA sequence is recognized by TLR9 [16]. Research has found that HPV16 L1 virus-like particles (VLPs) directly activate B cell-mediated humoral immunity via TLR4 and MyD88-dependent signaling [17]. Han R et al. [18] explained that the HPV6b/11E7 protein could promote TLR7 and TLR9 expression. When plasmacytoid dendritic cells (PDCs) were treated with E7 protein, the expression of key factors of the TLR signaling pathway and the JNK/p38MAP kinase signaling pathway were increased. Weng H et al. [19] reported that novel pro-inflammatory cytokine high mobility group protein B1 (HMGB1) is significantly increased in pointed condyloma samples, which also binds to TLR4 to activate downstream responses. Therefore, these immunogenic proteins are all potential PAMPs for HPV recognition by TLRs.
LR-HPV infection often causes benign lesions like condyloma to acuminate (CA).
Moreover, the common genotypes are HPV6 and HPV11. TLR9 expression in the skin
tissue of CA patients was lower than in normal skin tissue, and TLR4 levels were
significantly higher than in normal tissue [20]. HPV11-transfected HACAT cells
had reduced TLR9 expression and reversed activation of NF-
HR-HPV infection often triggers invasive cervical cancer (ICC); most commonly HPV16 and HPV18. In clinical studies [21, 22], the expression of TLR4, TLR5, and TLR9 is increased, and the expression of TLR7 is decreased in the skin tissue of HR-HPV patients. Besides, the expression levels of TLR4 and TLR9 showed a trend consistent with pathologic histology grading. TLR4 expression was higher in ICC tissue than in cervical intraepithelial neoplasia (CIN) samples, whereas it was lower in normal cervical tissue [23]. Similarly, during the progression of CIN, TLR9 expression increased sequentially [24, 25]. There are also slight differences in TLR expression by HPV type. For example, the TLR4 expression was higher in SiHa (HPV16+) cells than in HeLa (HPV18+) cells and lower in C33A (HPV-) cells than in HeLa (HPV18+) cells [23].
Most HPV infections are transient and will be cleared by the immune system over time. The relationship between HPV clearance and the expression of TLRs is intimate. Daud et al. [26] found that clearance of HPV16 was associated with increased expression of TLR2, TLR3, TLR7, TLR8, and TLR9 during clinical follow-up. TLR2 and TLR7 levels were significantly higher in women with the regression of CIN2; whereas, TLR2, TLR7, and TLR8 were significantly increased in women with HPV conversion [27]. This suggests that TLRs play an essential role in viral clearance. Furthermore, the signal transduction and secretion of a series of cytokines triggered by the upregulation of TLRs exert antiviral effects.
While HPV can disrupt innate immunity by evading TLR identification, it can also cause disease progression and, eventually, the emergence of aggressive malignancies. In general, activation of TLRs on immune cells can exert anti-tumor effects. TLR expression on the surface of tumor cells impedes immune cell infiltration and creates an immunosuppressive microenvironment for promoting tumorigenesis and progression.
Yu L et al. [28] found that TLR4 expression gradually decreases during cervical tumor development and that this appears to be associated with the expression of p16INK4a, a key marker of integration into host cells. Myeloid differentiation factor88 (MyD88), responsible for TLR pathway activation, was also downregulated in cervical cancer cell lines, but SARM1 expression levels were upregulated. SARM1 acted as a competitor for TLR binding and caused signaling disruption, inhibiting the innate immune response triggered by TLR [29]. Therefore, inactivation of the TLR pathway and SARM1-specific activation may be involved in HPV immune escape. Aggarwal et al. [30] discovered that TLR1 was elevated in squamous cervical cancer tissue whereas TLR3, 4, and 5 were downregulated. However, the opposite conclusion was reached by Li J et al. [31] and Xiao J et al. [32], namely that TLR3, TLR4, TLR7, and TLR8 showed an upward trend in samples from patients with squamous cell carcinoma of the cervix than in samples from normal subjects. In view of this, there is a dynamic process of TLR expression in cervical tumors. On the one hand, the organism exerts anti-tumor effects by upregulating TLRs-mediated immune responses.
On the other hand, tumors avoid TLR identification by interfering with innate immunity and downregulating TLR transcription to increase virus persistence. In addition, the development of cervical tumors is also influenced by genetic factors. TLR gene polymorphism has a close connection, and the polymorphisms in TLR2, TLR4, and TLR9 genes can increase the risk of cervical cancer [33].
HSV is a double-stranded DNA virus that primarily infects the skin mucosa of the genital tract and can also be latent in the spinal ganglia for a lifetime [34]. Herpes simplex virus 2 (HSV-2) mainly causes genital herpes, with an estimated 491.6 million infections worldwide. It is considered one of the most common sexually transmitted diseases [35]. Since HSV-2 often disrupts the mucosal barrier of the genital tract, HSV-2 infection is also an essential synergistic factor for HIV and HPV infection. Natural immune responses are critical in maintaining the integrity of the genital tract mucosa, and the TLR also plays an important role in responding to HSV-2 infection.
First, HSV-2 relies on glycoproteins for adsorption and penetration into the host cell, followed by microtubules with kinesin-mediated migration of the viral capsid to the nucleus and release of chromatin. The capsid protein then forms and wraps the viral DNA to leave the nucleus. Finally, it is processed in the cytoplasm to form progeny viruses and released outside the cell. TLR mediates the immune response during HSV-2 infection, and TLR expression shows a temporal correlation with infection. A study by Duan Q et al. [36] reported that TLR4, located on the cell surface and recognizes viral glycoproteins, was first expressed after HSV-2 infection of VK2/E6E7 cells. TLR3, TLR7, and TLR9, which are located in the cell and recognize viral nucleic acids, were expressed in a time-dependent manner.
TLR4 is a pattern recognition receptor for LPS, a product of gram-negative bacteria. Recent reports have revealed that TLR4 can also recognize viruses and related proteins as a signal to trigger downstream-related responses [4], among which HSV-2 can upregulate TLR4 expression. Medullary differentiation protein 2 (MD2) is an auxiliary protein of TLR4-mediated endotoxin/LPS signaling that helps TLR4 recognize pathogen-associated molecular patterns. In HSV-2-infected human cervical epithelial cells (HCE), MD2 overexpression was found to enhance HSV-2-mediated activator protein 1 (AP-1) activation, and thus the TLR4/MD2 complex plays a role in HSV-2 infection similar to that of TLR4 recognition of LPS. In addition, ICP0, the immediate early protein of HSV-2, induces a significant increase in AP-1-driven transcription and TLR4 promoter activation in HEC-1 cells. Furthermore, the accumulation of ICP0 allows host cell AP-1 activation, which in turn mediates TLR4 activation [37].
HSV-2 infection of HCE induces the expression of TLR4, which activates the
NF-
TLR9 recognizes bacterial and viral DNA and CpG oligonucleotide (CpG-ODN). Although TLR9 can be activated by the DNA sequence of CpG-ODN and the HSV-2 genome is rich in CpG, HSV-2 infection induces TLR9 activation in a viral replication-dependent rather than a CpG-dependent manner. Infection of human cervical epithelial cell line ME-180 with HSV-2 induces TLR9 activation through the binding of the JNK signaling pathway promoting transcription factor specificity protein 1 (SP1) to the TLR9 promoter, which in turn enhances TLR9 expression, and TLR9 activation exhibits a dose-dependent and time-dependent HSV-2 infection [41]. Malmgaard L et al. [42] also confirmed that although TLR9 is one of the TLRs activated by HSV-2, the production of interferon signals occurs mainly through TLR non-dependent pathways rather than those mediated by the TLR9 signaling pathway. In particular, TLR9 and plasmacytoid dendritic cells mediate the early IFN response, but later responses come from different cell types and are independent of TLR9 induction [43]. This suggests that HSV-2 has evolved a mechanism to antagonize the TLR9 signaling pathway.
HSV-2, which causes persistent infection, has evolved many strategies to evade the host’s intrinsic antiviral response. The virion host shutoff protein (VHS) of HSV-2 inhibits TLR2 and TLR3 expression in VK2 cells to facilitate immune evasion during HSV-2 infection [44]. In addition, TLR2 gene polymorphisms were associated with the severity of HSV-2 infection [45], and polymorphisms in the TLR3 gene were also a factor in susceptibility to HSV-2 [46].
By exploiting the vital role of the TLR family in recognizing pathogenic microorganisms to activate natural immunity, agonists targeting the activation of these pattern recognition receptors may be effective in preventing primary HSV-2 infection in vaginal cells. In the research, ligand treatment of TLR3, TLR5, and TLR9 significantly inhibited HSV-2 replication [47]. FSL-1, the TLR6 agonist, exerted antiviral effects on mouse and human vaginal endothelial cells [48].
Bacterial vaginosis is a condition brought on by dysbiosis of the vaginal microbiota, which results in an increase in anaerobium and facultative anaerobe and a decrease in lactobacilli that produce hydrogen peroxide [49]. Typical causative agents include Gardnerella vaginalis, Mobiluncus mulieris, Mycobacterium vaginalis, and bacterial vaginitis-associated bacteria 1-3 (BVAB-1, BVAB-2, and BVAB-3) [50]. BV can increase the risk of infection by other common pathogens of the female reproductive tract, leading to pelvic inflammatory disease, premature birth, and associated diseases such as postpartum endometritis [51].
TLR-recognized compounds in the BV-associated microbiome include peptidoglycans
and LPSs [50]. In response to the body’s natural immune response, these ligands
bind to the relevant receptors to produce various products, including the
corresponding cytokines and chemokines. TLR4 can recognize gram-negative LPS.
Peptidoglycan of Mycobacterium can be recognized by TLR2, and lipopeptide of
Mycobacterium can be recognized by TLR6. Flagellin of flagellated bacteria can be
recognized by TLR5, common to various bacteria, and bacterial lipopeptide and
non-methylated CpG DNA can be recognized by TLR2 and TLR9, respectively [1]. In
addition, anaerobic bacteria synthesize short-chain fatty acids (SCFAs) from
incompletely digested carbohydrates, consisting mainly of acetic acid, butyric
acid, and propionic acid, which have been shown to modulate immune responses
broadly. In the lower genital mucosa of women with BV, there are high
concentrations of SCFAs. SCFAs significantly increase the release of IL-8 and
TNF
It has been demonstrated that vaginal lavage solution from BV patients could
induce TLR4 and TLR2 expression in vitro and induce IL-8 production from
NF-
In genomic studies, TLR polymorphisms are also strongly associated with the dysregulation of vaginal microecology. Gene polymorphisms in TLR2 and TLR7 were proven to cause a significant increase in the incidence of BV in African women [54]. BV is associated with TLR1, TLR9, and TLR2 variants in a non-pregnancy cohort study of 159 HIV-1-positive African Americans [50]. In a study examining the impact of genetic variants in the TLR gene on the different host immune responses that occur in HIV-1-infected adolescents facing BV, missense mutations in TLR4 were found to cause a 10-fold increase in Gardnerella vaginalis levels, and genetic variants in the TLR9 gene were also discovered to be strongly connected with BV diagnosis [55].
VVC is an inflammatory vaginal disease primarily due to Candida albicans infection, with 75% of women of childbearing age becoming infected at least once during their lifetime and 6% to 8% of women suffering from recurrent vulvovaginal candidiasis (RVVC) [56]. Candida albicans is a saprophytic parasite. As an opportunistic pathogen, its pathogenicity depends on the vaginal microecological environment, the host’s immune response, and the virulence of the fungus. Host-microbe interactions during the natural immune response play an essential role in the pathogenesis of VVC.
The cell wall of Candida albicans contains chitin, dextran, and mannans, as well as intracellular DNA and RNA molecules that are pathogen-associated molecular patterns that can be recognized by the host immune system and activate an immune response. TLR4 recognizes mannans, whereas TLR2 recognizes phospholipid mannans, prevents colonization and endogenous invasion of Candida albicans, and TLR7 recognizes single-stranded RNA of Candida albicans [57].
A clinical study has shown that TLR2 and TLR4 expression was significantly increased in vaginal endothelial cells obtained from vaginal swabs of VVC patients compared with samples obtained from normal subjects. In addition, this was accompanied by activation of c-fos and p38 signaling, and phosphorylation of p-38 occurred in close association with the presence of pseudohyphal/mycelia [58].
Animal studies have shown that the lack of TLR2-mediated signaling increases the fungal load in the vagina. Usually, the fungus adheres to keratinized epithelial cells. However, in TLR2-deficient animals, the epithelial barrier can be easily crossed by Candida albicans hyphae, which invade the deep mucosa and produce severe tissue damage [56]. This confirms that TLR2 is an important factor in controlling the progression of VVC disease.
In vitro studies revealed that Candida albicans could induce
TLR2 and TLR6 expression in VK2 cells and activate related genes and cytokines to
produce inflammatory signals such as IL6 and TNF
TV is an inflammation of the vagina caused by the production and reproduction of Trichomonas vaginalis. It is one of the most common infectious diseases, often accompanied by severe vulvar itching, increased discharge, and urinary tract infections. Numerous diseases are closely associated with TV, such as premature birth, infertility, and HIV infection [61]. Trichomonas vaginalis is a flagellated, extracellular parasitic protozoan often found in the female vagina, producing extracellular virulence factors by adhering to female vaginal epithelial cells and destroying the genital tract barrier. Therefore, local natural immunity of the vagina is crucial for Trichomonas vaginalis defense [62].
For parasitic protozoa, adherence to epithelial cells is a key link to infection
and parasitism of the host. The surface of trichomonad contains a variety of
complex carbohydrates, such as glycolipids, glycoproteins, and
glycophosphatidylinositol, which play a significant role in host cell invasion
and evasion of the host immune response. Lipophosphoglycan (LPG) is the primary
cell surface glycoconjugate of Trichomonas vaginalis, assisting in
adherence to epithelial cells and initiating the secretion of cytokines. IL-8 and
macrophage inflammatory protein-3
As indicated by Chang [62], Trichomonas vaginalis infection of cervical
cancer cells HeLa cells upregulated the expression of TLR2, TLR4, and TLR9
through the p38 MAPK signaling pathway, thereby regulating the release of the p38
MAPK-dependent prototype chemokine IL-8, as well as tumor necrosis
factor-
Notably, TLR4 is vital in the inflammatory response triggered by
Trichomonas vaginalis. In the study of Zariffard [63], vaginal
irrigation fluid from TV patients stimulated cytokine production in mouse
splenocytes through TLR4 response. Trichomonas vaginalis activates TLR4,
MAPKs, and NF-
Treponema pallidum (TP) is the causative agent of the sexually transmitted disease syphilis, which infects about 36 million people worldwide, with 12 million new cases per year [66]. TP, a pathogenic microorganism that infects only humans, is spiral shaped like a spring and is highly motile. TP generally invades the body through broken skin mucosa, spreads through lymphatic vessels and the bloodstream, penetrates systemic tissue and breaks the vascular barrier [67]. This induces the host to establish a rapid immune response, and produces an inflammatory response [68]. Symptoms subside into latency, and TP enters the blood and lymphatic circulation from the site of infection, spreading to organs and skin mucosa throughout the body and causing widespread lesions. TP possesses a mechanism to evade the natural immune response, and persistent infection is one of the characteristics of syphilis [69]. TP has a complex structure comprised of an exterior membrane, a full-length spiral flagellum, or axial filament, and a cylindrical bacterium from the outer to the inner part.
Outer membrane proteins bind to the fibronectin of the host’s extracellular
matrix through adhesion. They are involved in the colonization of host tissues by
TP and the disruption of tissue barriers to facilitate the spread of the organism
in vivo. As antigens are directly exposed to the external environment,
they are recognized rapidly by the innate immune response system, activating a
series of immunopathological effects and immune escape mechanisms. Tp92, the only
TP membrane protein with structural features similar to other gram-negative
bacterial outer membrane proteins, can mediate cell death by being recognized by
CD14 and or TLR2 on the surface of human monocytes. When Tp92 is recognized by
CD14 and TLR2, the downstream MyD88 pathway is signaled, which activates
NF-
Flagellar axoneme proteins are mainly composed of two outer proteins (FlaA1 and
FlaA2) and three core proteins (FlaB1, FlaB2, and FlaB3) [66], which are of great
importance in the induction of immune responses. Flagellin is the only ligand for
TLR5 in many bacteria and binds to TLR5 at the cell surface to stimulate
downstream signaling through a MyD88-dependent pathway. TP flagellin stimulates
monocytes to produce pro-inflammatory cytokines such as IL-6 and IL-8 in a
TLR5-dependent manner. Three TP flagellar core proteins (FlaB1, FlaB2, and FlaB3)
induce IL-6 and IL-8 production in human acute monocytic leukemia cells THP-1 by
activating ERK, p38, and NF-
The flagellin molecule contains two to four structural domains, D0–D3. Among them, the D0 and D1 structural domains (N/C regions) are highly conserved among organisms, which are essential for the immunostimulatory activity of flagellin. The ND1 structural domain of TP flagellin was found to be one of the sites on flagellin in TP-mediated TLR5 activation [66].
In genetic polymorphism studies, TLR1, TLR2, and TLR6 were found to be associated with an increased risk of neurosyphilis [72], whereas loci in the TLR2 TIR structural domain helped prevent syphilis infection [73].
Chlamydia trachomatis infection is a common sexually transmitted infection (STI) and is asymptomatic in 60-80% of cases. Chlamydia trachomatis (CT) is a specialized intracellular gram-negative bacterium that can cause various diseases. There are nineteen serotypes of CT. Infections of serotypes A-C cause ocular disease and are currently the leading cause of visual impairment due to infection, whereas serotypes D–K infections cause genitourinary tract disease. Aggressive serotypes L1–L3 cause genital lesions, leading to lymphogranuloma venereum (LGV). Repeated or persistent CT infections of the reproductive tract can cause urethritis, pelvic inflammatory disease, infertility, and neonatal infections [74]. Repeated or persistent CT infections of the genital tract can cause urethritis, pelvic inflammatory disease, infertility, and neonatal infections. Early-stage infections are easily cured. However, because 75% to 90% of women with chlamydia are asymptomatic in clinical disease, opportunities for therapeutic intervention are often missed. Asymptomatic infections are a significant contributor to sexual transmission [75]. CT is an intracellular pathogen with a bidirectional developmental cycle. In the extracellular form of infection, it is manifested as a metabolically inert elementary body (EB). After infecting the host cell to form inclusion bodies, it is transformed into the replication-competent reticulate body (RB), which replicates through bifurcation.
Furthermore, RB recombines into infectious EB and then moves extracellularly [76]. The cell wall and outer membrane components of CT can be used as a PAMP for TLR recognition.
Major outer membrane protein (MOMP) is the most abundant protein in the cell
membrane of chlamydia. MOMP induces TLR2-dependent IL-8 secretion and increases
NF-
Heat shock proteins (HSPs) are present in the outer membrane complexes of
protozoan EB and reticulon RB, which are expressed throughout the life cycle of
Chlamydia trachomatis [76]. According to Jha [76], cHSP60 stimulation
enhanced TLR2 and TLR4 mRNA expression, activated NF-
Macrophage infectivity potentiator (Mip), a classical bacterial lipoprotein, is
present on the surface of chlamydia and induces the secretion of TNF-
Chlamydial secretory protein GlgA, an enzyme related to glycogen metabolism, is
found in chlamydial inclusion bodies. Sun et al. [79] proved that GlgA
contributes to the secretion of inflammatory factors IL-8, IL-1
Gram-negative bacterial LPS is a well-known inflammatory trigger, however, chlamydia LPS has low endotoxic action compared with other bacteria and depends on membrane CD14 for TLR4 activation [81].
TLR2 and TLR4 have a significant role in the process of CT infection. Yilma et al. [82] found enhanced expression of TLR2 and TLR4 on Chlamydia trachomatis-infected macrophages, producing a broad spectrum of inflammatory cytokines and chemokines. O’Connell et al. [81] identified that TLR2 and MyD88 were specifically recruited to the inclusion body membrane during productive infection with chlamydia and that TLR2 was actively involved in intracellular signaling. Webster et al. [83] discovered that Chlamydia trachomatis infection also resulted in TLR4-dependent and MyD88 signaling-dependent protein kinase RNA activation (PKR) was also efficiently activated.
In addition, TLR3 deficiency leads to enhanced chlamydial replication and increases the risk of reproductive tract morbidity during infection. In a study, Xu et al. [75] used the immortalized human oviductal epithelial cell line (HOE) OE-E6/E7 to assess the role of TLR3 in the immune response to L2 CT infection and found that TLR3 deficiency resulted in increased levels of LPS within the chlamydial inclusion body.
There is a robust relationship between single nucleotide polymorphisms and susceptibility and severity of CT infection. Among them, Verweij et al. [84] showed that specific TLR2 and TLR9 haplotype genes prevented the exacerbation of clinical infection; TLR4 genotype polymorphisms were associated with increased odds of Chlamydia trachomatis infection in women with pelvic inflammatory disease in a clinical study by Taylor et al. [85].
TLR, the first line of immune defense in the female lower genital tract, is
abundantly expressed in the lower genital tract. TLR recognizes pathogens and
induces activation of the human immune response in common infectious diseases of
the female lower genital tract and is intimately involved in the development and
regression of disease. Current studies have focused on TLR recognition of various
pathogens and immune response activation. The specific role of TLR in multiple
aspects of disease evolution requires in-depth investigation. The wide
distribution of TLR in the female lower genital tract reflects the immune
tolerance to commensal flora and resistance to invasion by various pathogenic
microorganisms. Despite differences in the intervention of different pathogens on
the immune system, the TLR family, which recognizes a wide range of
pathogen-associated molecular patterns, is often involved in the same signaling
in defense against various diseases. TLR-mediated activation of MyD88-dependent
pathways or MyD88-independent pathways such as NF-
XZ–conceptualization, literature collection, and writing-original draft; ZC–conceptualization, editing, and giving final approval of the version; WJ, XJ, and WW–data acquisition, analysis, review and editing; QS, TL, and CH–literature analysis, picture-processing, review and editing. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We are grateful for the drawing materials provided by BioRender.
This research was supported by grants from the National Natural Science Foundation of China (No. 81874483).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.