

 , Dongqing Mi 2,3,†, Yan Jiang 1,2, Caiping Geng 1,2, Jingchuan Yuan 1,2, Tingting Zhao 1,2, Han Zhou 1,2, Xuhui Zhang 1,2, Yi Hu 4,*
, Dongqing Mi 2,3,†, Yan Jiang 1,2, Caiping Geng 1,2, Jingchuan Yuan 1,2, Tingting Zhao 1,2, Han Zhou 1,2, Xuhui Zhang 1,2, Yi Hu 4,* , Xiaohua Wu 1,2,*
, Xiaohua Wu 1,2,*1 Center for Reproductive Medicine, The Fourth Hospital of Shijiazhuang, Assisted Reproductive Technology Innovation Center of Shijiazhuang City, 050010 Shijiazhuang, Hebei, China
2 Key Laboratory of Maternal and Fetal Medicine of Hebei Province, 050011 Shijiazhuang, Hebei, China
3 Clinical Laboratory, The Fourth Hospital of Shijiazhuang, 050010 Shijiazhuang, Hebei, China
4 Biomedical Innovation Center, Beijing Shijitan Hospital, Capital Medical University, 100038 Beijing, China
†These authors contributed equally.
Abstract
Endometriosis is a complex, multifactorial disease characterized by the growth of endometrial-derived cells outside the uterus. Abnormal proliferation of ectopic lesions and chronic inflammation contribute to the onset and progression of the condition. This study aimed to identify dysregulated genes involved in the pathogenesis of endometriosis.
We performed differential expression analysis and Weighted Gene Co-expression Network Analysis (WGCNA) using gene expression datasets (GSE11691 and GSE23339) from the Gene Expression Omnibus (GEO). We then analyzed significant differentially expressed genes (DEGs) using Gene Set Enrichment Analysis (GSEA) to predict their functional roles. The expression of key DEGs was validated by immunohistochemistry (IHC) in ectopic and eutopic endometrial tissues from 14 patients with endometriosis. Primary endometrial cells were isolated from normal endometrial tissue. A candidate gene was overexpressed in these cells, and its effect on cellular metabolism was assessed by measurement of the oxygen consumption rate (OCR).
Our data identified 48 key dysregulated genes associated with endometriosis. These genes showed functional enrichment in processes such as complement activation and cell adhesion, both of which are implicated in disease pathogenesis. Notably, fatty acid binding protein 4 (FABP4), linked to mitochondrial dysfunction in endometrial stromal cells (ESCs), was one of the most significantly upregulated genes in ectopic endometrium. Overexpression of FABP4 enhanced cell growth and increased the OCR in primary endometrial cells.
This study elucidates the functional role of FABP4 dysregulation in endometriosis and identifies it as a potential therapeutic target.
Keywords
- endometriosis
- fatty acid binding protein 4
- mitochondria
Endometriosis is a multifaceted gynecological disorder characterized by the presence of endometrial-like tissue outside the uterine cavity. It affects approximately 10–15% of women of reproductive age and 20–50% of women who experience infertility [1]. A study has reported associations between endometriosis, infertility, and ovarian cancer [2]. Common symptoms include dysmenorrhea, dyspareunia, chronic pelvic pain, and irregular uterine bleeding. Although surgical intervention remains the primary treatment for removal of ectopic lesions, approximately 40% of patients experience recurrence [3]. Abnormal growth of endometrial tissue and chronic inflammation contribute to the onset and progression of endometriosis; however, the precise mechanisms remain poorly understood [4].
Fatty acid-binding protein 4 (FABP4) is a member of the FABP family, a group of highly conserved cytosolic proteins that bind various hydrophobic ligands, including long-chain fatty acids, eicosanoids, leukotrienes, and prostaglandins. FABP4 plays a critical role in numerous cellular processes, such as fatty acids uptake, intracellular transport, and gene expression regulation [5, 6]. Tian et al. [7] reported that FABP4 was undetectable in the mouse uterus from days 1 to 4 of pregnancy, appeared in the primary decidual zone on days 5 and 6, and was significantly upregulated on days 7 and 8. They further identified FABP4 as crucial for decidualization in mice. In humans, FABP4 mRNA levels were higher in the endometrium on day 7 after the luteinizing hormone (LH+7) surge than in the early secretory phase (LH+2) [8]. Furthermore, estrogen alone and in combination with progesterone upregulates FABP4 expression in mice [7]. Zhu et al. [8] reported that FABP4 is consistently expressed in endometrial epithelial cells during both the proliferative and secretory phases, whereas stromal cells express FABP4 only during the secretory phase. They also confirmed that FABP4 is a key regulator of proliferation, migration, and invasion in endometrial epithelial cells, and that its downregulation reduces endometrial receptivity [8, 9]. However, the role of FABP4 in endometriosis remains unclear.
Mitochondria are considered the “powerhouses of the cell” due to their efficient ATP-generating capacity. Recent studies indicated that altered mitochondrial function may contribute to the development of endometriosis [10, 11]. For instance, melatonin has been shown to inhibit the development of endometriosis by disrupting mitochondrial function and reducing oxidative stress [12, 13, 14]. Chen et al. [10] examined the morphology and oxygen consumption rates (OCR) of isolated primary endometrial stromal cells (ESCs). They observed that ectopic ESCs exhibited elongated mitochondria and a 95% increase in basal OCR compared with eutopic ESCs and a 51% increase compared with controls [10]. They also found that genes that control mitochondrial fission and fusion were dysregulated in ectopic ESCs, which contributed to alterations in mitochondrial morphology [15]. Nevertheless, the mechanisms underlying mitochondrial dysfunction in endometriosis remain unclear.
In this study, we analyzed two gene expression profiling datasets (GSE11691 and GSE23339) from the Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo) with Weighted Gene Co-expression Network Analysis (WGCNA). We identified 48 genes that were dysregulated in patients with endometriosis. We analyzed the potential function of FABP4 with Gene Set Enrichment Analysis (GSEA) and examined it in ESCs.
This study was conducted at The Fourth Hospital of Shijiazhuang. Two microarray datasets were analyzed to identify differentially expressed genes (DEGs) in endometrial tissue from patients with endometriosis. We included 14 patients with a surgically and histologically confirmed diagnosis of endometriosis who underwent laparoscopic excision during the study period for further validation. Ectopic endometrial samples were collected for mRNA and protein analysis. Eutopic endometrial tissue from the same patient served as the control. Additionally, endometrial samples from 5 healthy participants were obtained to assess target gene expression in normal endometrium. All clinical data were retrieved from the patients’ electronic medical records. Primary ESCs were isolated to investigate the biological function of the candidate gene.
Gene expression datasets were retrieved from the GEO portal and filtered based
on data characteristics, experimental design, and sample size. Two microarray
datasets (GSE11691 and GSE23339) were selected for DEGs analysis. Transcriptome
analysis was performed using R (version 4.4.1) in RStudio (Desktop version,
2024.04.2+764; Boston, MA, USA). Background adjustment was performed using the
gcRMA software package [16]. Data quality was assessed with the
array QualityMetrics software package (v3.65.0, Bioconductor Project, Boston, MA,
USA) [17], and samples of poor quality were excluded from subsequent analyses.
Multiple probes corresponding to the same gene were consolidated into a single
value by summarizing the median expression levels. DEGs were identified using the
limma software package (version 3.52.4, Walter and Eliza Hall Institute of Medical Research, Royal Parade, PV, Australia), with thresholds set at an adjusted
p-value
WGCNA was applied to the GSE11691 and GSE23339 datasets to identify critical
module genes. Pearson correlation coefficients were calculated to assess gene to
gene relationships. A scale-free network was constructed, and an appropriate soft
threshold was selected for network formation. The adjacency matrix was then
transformed into a topological overlap matrix, and hierarchical clustering was
used to generate a clustering tree. Dynamic tree cutting defined the
co-expression modules. Modules with a correlation coefficient
Intersection genes underwent GO enrichment analysis, covering cellular components (CCs), molecular functions (MFs), and biological pathways (BPs), as well as KEGG pathway analysis. The clusterProfiler R package (v4.12.6) was used for statistical analysis.
To identify biological pathways, MFs, and CCs significantly associated with genes whose expression correlated with FABP4, GSEA was performed. First, expression profiles for FABP4 and all other genes were extracted across samples. Spearman correlation coefficients between FABP4 and each gene were then computed. Genes were ranked based on their correlation coefficients, from the highest positive to the lowest negative correlation, and the ranked gene list was subsequently subjected to GSEA.
Endometriotic tissues were collected from 14 Chinese-Han women diagnosed with
endometriosis at The Fourth Hospital of Shijiazhuang (mean age: 30.2 years;
range: 28–34 years; follicular phase, n = 5; luteal phase, n = 9) between
October 2022 and May 2025. The inclusion criteria were as follows: (1) a history
of infertility for more than one year; (2) age between 20 and 38 years; (3) a
basal serum follicle-stimulating hormone (FSH) level
5 healthy women (mean age: 27.4 years; range 26–30 years; follicular phase, n = 2; luteal phase, n = 3) without endometriosis were recruited in this study, and endometrial samples were obtained using a pipelle device.
Written informed consent was obtained from all participants prior to enrollment in this study. All procedures adhered to the ethical standards of the institutional and national research committees, as well as the Helsinki Declaration and its subsequent amendments. The Bioethics Committee of The Fourth Hospital of Shijiazhuang approved this study (Approval number: 20220039).
Medical records were obtained from the Shijiazhuang Obstetrics and Gynecology Hospital. Each tissue sample was divided for total RNA extraction, protein extraction, and cell isolation.
Paraffin-embedded tissue sections (4 µm thick) were deparaffinized through
sequential xylene immersion (2
Sections were blocked with 5% normal goat serum (cat.no.C0265, Beyotime Biotechnology, Shanghai, China) in PBS with 0.3% Triton X-100 (cat.no. X100-100ML, Sigma-Aldrich, St. Louis, MO, USA) for 1 h at RT, then incubated overnight at 4 °C with primary antibody (rabbit anti-FABP4 polyclonal antibody, 1:200 dilution, cat.no. ab13979, Abcam, Waltham, MA, USA). After three 10 min washes with TBST (Tris-buffered saline with 0.1% Tween 20), sections were incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary antibody (1:500 dilution, cat.no. #7074, Cell Signaling Technology, Inc., Danvers, MA, USA) for 2 h at RT.
Following three additional TBST washes (5 min each), immunoreactivity was visualized using a DAB substrate kit (cat.no. #8059, Cell Signaling Technology, Inc. Danvers, MA, USA) with a development time of 3–5 min. Sections were counterstained with Mayer’s hematoxylin for 1 min, dehydrated through graded alcohol series, cleared in xylene, and mounted with mounting medium.
Images were captured using microscopy. Two pathologists independently evaluated staining intensity (0: negative; 1: weak; 2: moderate; 3: strong) and the percentage of positively stained cells (0–100%). An H-score (range: 0–300) was calculated by multiplying the staining intensity by the percentage of positive cells.
ESCs were isolated from endometrial tissue with established methods [18, 19].
Briefly, tissue samples were minced and then digested in DMEM/F12 with type I
collagenase (2.5 mg/mL, cat.no. C1-22-1G, Sigma-Aldrich, St. Louis, MO, USA) and
DNase I (15 U/mL, cat.no. D7291, Sigma-Aldrich, St. Louis, MO, USA) at 37 °C for 1
h. Debris was removed by filtration through a 40 µm nylon cell strainers.
Cells were collected by centrifugation at 400
The full length of 396 bp FABP4 coding sequence was cloned into the pcDNA3.1 plasmid between BamHI and XbaI sites to generate the FABP4 overexpression vector. The plasmid was transfected into primary ESCs by electroporation, and the expression of FABP4 was examined by immunoblotting 48 h after transfection.
Proteins were extracted from cell samples using Radioimmunoprecipitation Assay
(RIPA) buffer (cat.no.J63306-AP, Thermo Fisher Scientific, Waltham, MA, USA), separated on 4–12% Bis-Tris polyacrylamide gels (cat.no.
NP0321BOX, Thermo Fisher Scientific, Waltham, MA, USA), and transferred to
polyvinylidene difluoride (PVDF) membranes (cat.no. IPVH00010, Millipore,
Burlington, MA, USA). Membranes were blocked with 5% fat-free milk and incubated
overnight at 4 °C with a primary antibody against FABP4. After washing with TBST,
membranes were incubated with an HRP-conjugated secondary antibody diluted in
TBST containing 5% fat-free milk (cat.no.P0216-300g, Beyotime Biotechnology, Shanghai, China). Protein bands were visualized using a
SuperSignal West Femto Maximum Sensitivity Substrate kit (cat.no. 34094, Thermo
Fisher Scientific, Waltham, MA, USA), with
Antibody information:
Rabbit anti-FABP4 polyclonal antibody (1:1000 dilution, cat.no. ab13979, Abcam, Waltham, MA, USA).
HRP-conjugated goat anti-rabbit secondary antibody (1:3000 dilution, cat.no. #7074, Cell Signaling Technology, Danvers, MA, USA).
HRP-conjugated mouse anti-
Cell viability was assessed using the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. A total
of 2
Cells were harvested by gentle trypsinization, washed once with ice-cold PBS (pH
7.4), and pelleted by centrifugation at 400
Cells were incubated with 5 µL of Annexin V-FITC (Cat. No. 640906, BioLegend, San Diego, CA, USA) for 15 min at RT in the dark. Prior to analysis, 10 µL of propidium iodide (PI) solution (50 µg/mL) was added to discriminate late apoptotic or necrotic cells. Samples were acquired within 1 h of staining using a BD FACSCanto II flow cytometer (BD biosciences, Franklin Lakes, NJ, USA), with a minimum of 20,000 events recorded per sample. Fluorescence was detected using: FITC (Annexin V): 488 nm excitation, 530/30 nm emission filter. PI: 488 nm excitation, 585/42 nm emission filter. Results were analyzed using FlowJo software (v10.4.1, Tree Star, Inc. Ashland, OR, USA).
Mitochondrial function was evaluated using a Seahorse XF24 Analyzer (Agilent
Technologies, Santa Clara, CA, USA) and the Seahorse XF Cell Mito Stress Test
Assay (cat.no. 103016-100, Agilent Technologies, Santa Clara, CA, USA). Briefly,
1
The cells were fixed using 4% formaldehyde at RT for 10 minutes, followed by permeabilization on ice for an additional 10 minutes with PBS containing 0.5% Triton-X100. After a 30 min incubation with blocking buffer composed of PBS, 0.1% Tween 20, and 1% BSA, cells were incubated overnight at 4 °C with diluted antibodies: Alexa Fluor 488 labeled VIM antibody (1:300 dilution, cat.no. ab154207, Abcam, Waltham, MA, USA), Alexa Fluor 488 labeled mouse anti-cytokeratin monoclonal antibody (1:200 dilution, cat.no. sc-57004 AF488, Santa Cruz Biotechnology, Dallas, TX, USA), or FITC labeled mouse anti-CD45 monoclonal antibody (1:200 dilution, cat.no. sc-53201 FITC, Santa Cruz Biotechnology, Dallas, TX, USA). After three washes, slides were mounted with an antifade mounting medium containing DAPI (cat.no. H-1200-10, Vector Laboratories, Inc., Newark, CA, USA), and images were captured using fluorescence microscope (ECLIPSE Ti2, Nikon, Melville, NY, USA).
Data was analyzed using R (version 4.4.1). Statistical significance between the
two groups was evaluated by using two-tailed Student’s t-test, and
results with a p-value
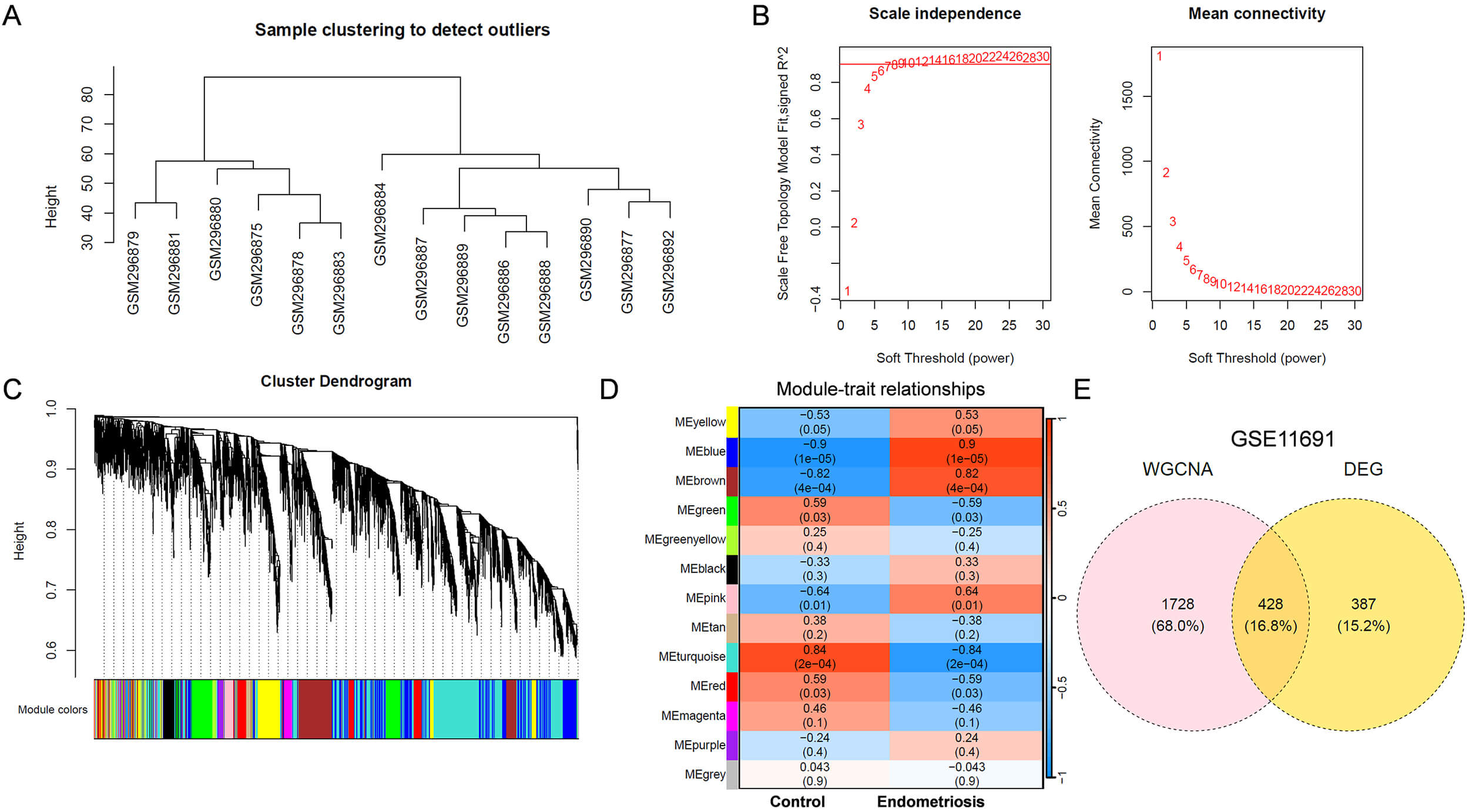
To identify critical genes involved in the initiation and progression of
endometriosis, WGCNA was performed on the GSE11691 dataset. A sample clustering
tree was constructed (Fig. 1A), and no significant outliers were identified. A
soft-thresholding power of 6 was selected, as shown in Fig. 1B. Genes were
clustered into 13 modules (Fig. 1C), and 2156 genes in 4 key modules (blue,
brown, pink and yellow) were positively correlated (r
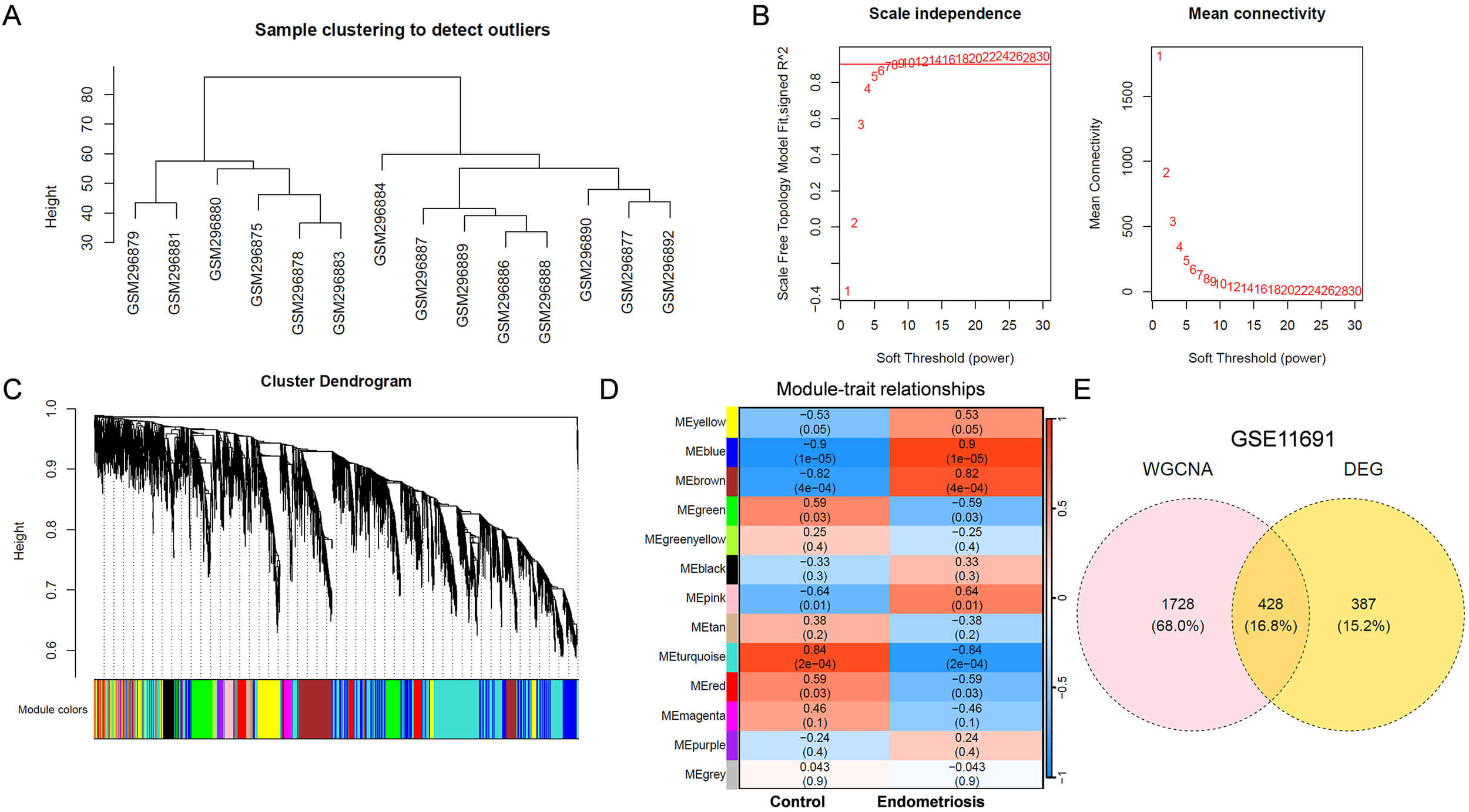 Fig. 1.
Fig. 1.
Identification of key DEGs associated with endometriosis using GSE11691 dataset. (A) Sample clustering tree. (B) The relationship between scale-free topology model fit or mean connectivity and soft-threshold power. The soft-threshold power was 6. (C) Cluster dendrogram of genes. Different colors represented different modules. (D) Module-trait heatmap showing Pearson correlation coefficients between modules and endometriosis. (E) Venn diagram showing DEGs related to endometriosis phenotype. DEGs, differentially expressed genes; WGCNA, Weighted Gene Co-expression Network Analysis.
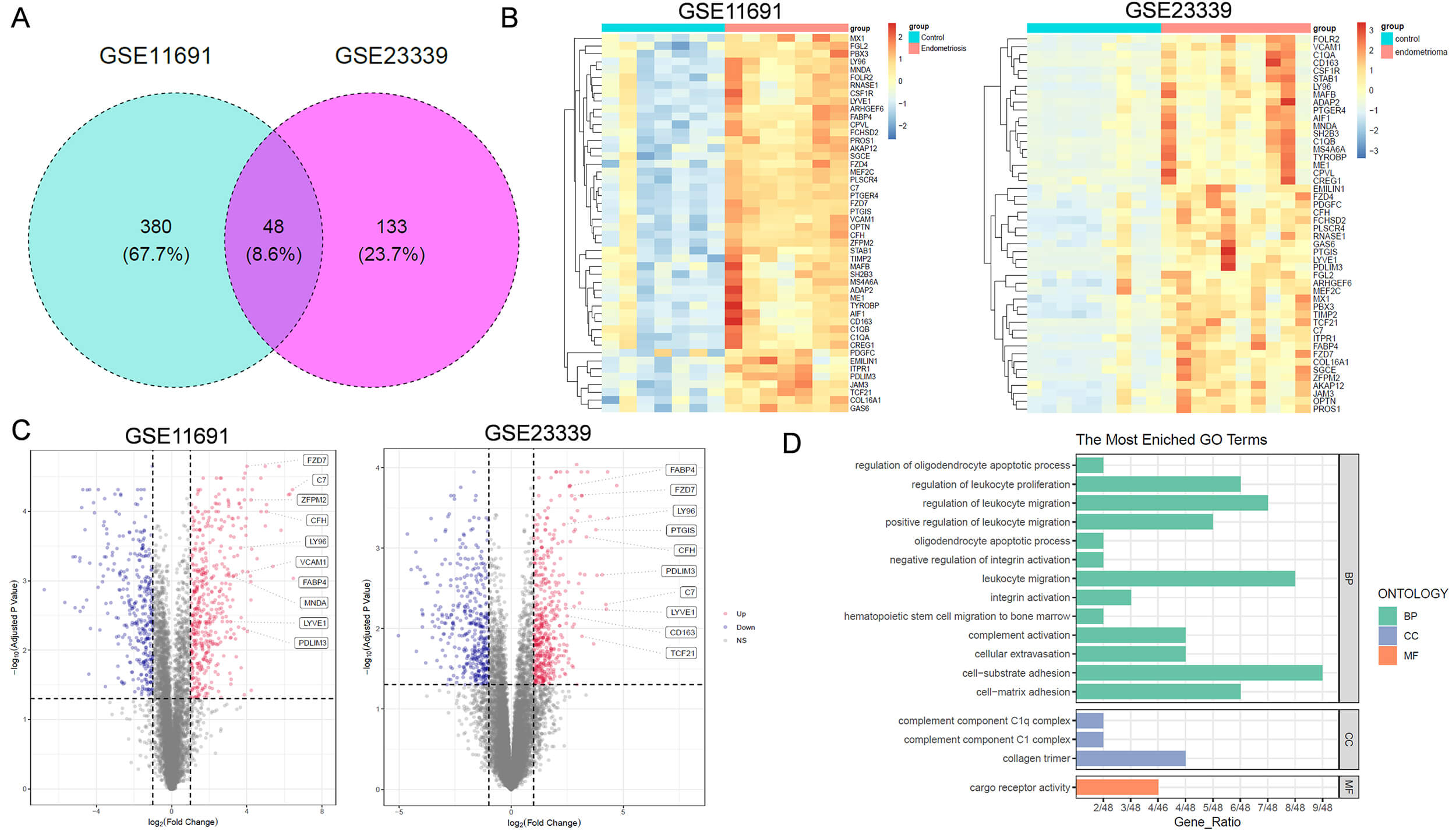
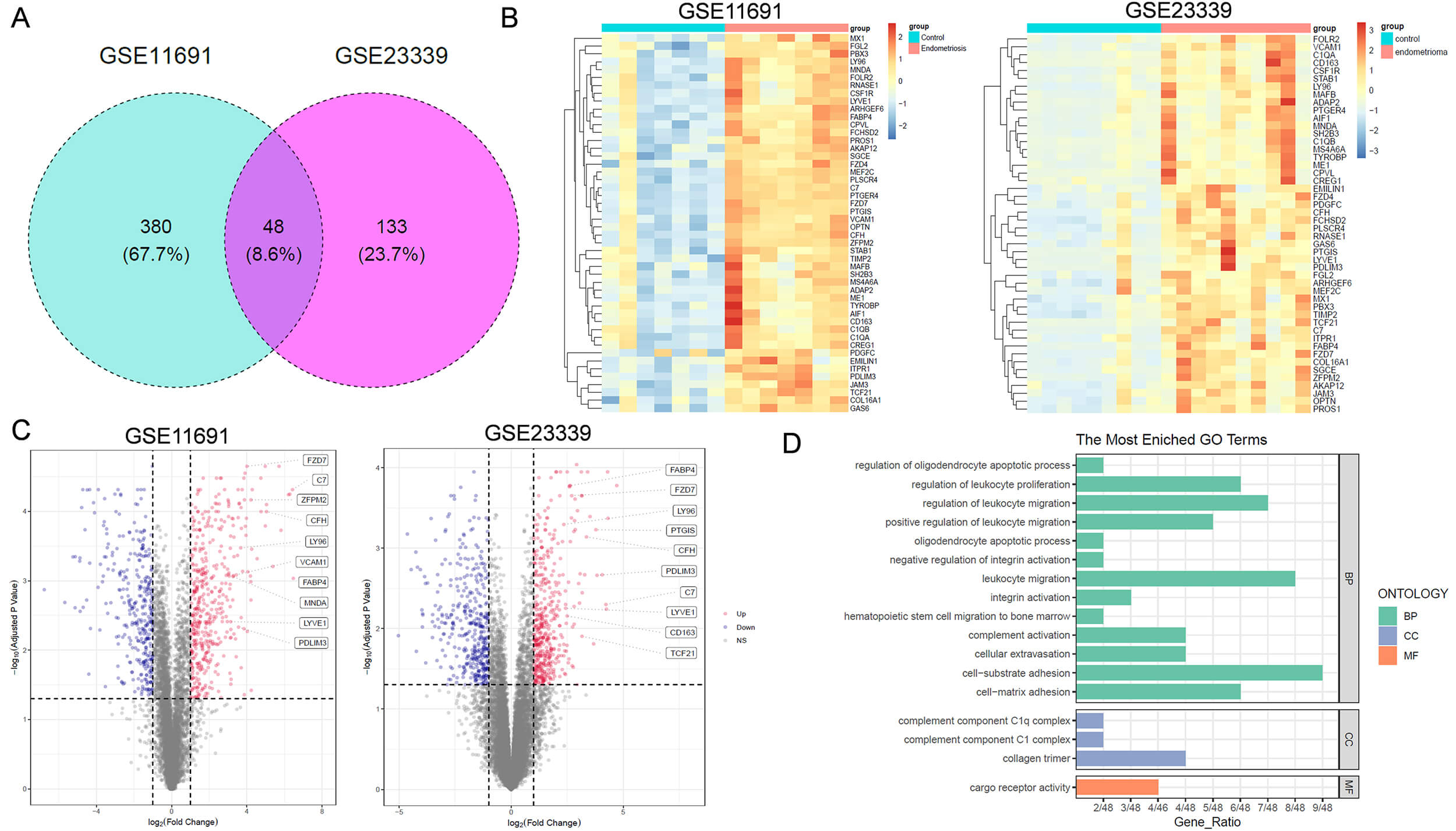 Fig. 2.
Fig. 2.
Functional analysis of key DEGs associated with endometriosis. (A) Venn diagram showing key DEGs related to endometriosis in both GSE11691 and GSE23339 datasets. (B) Heatmap of 48 DEGs across samples. (C) Volcano plot of differentially expressed genes in patients with endometriosis. The boxes on the right in each panel label the top 10 upregulated genes positively correlated with endometriosis. (D) Gene Ontology (GO) analysis of the 48 DEGs. BP, Biological Process; CC, Cellular Component; MF, Molecular Function.
Among the 48 critical DEGs, FABP4 has been reported to play a role in embryonic
implantation [9]. Khanaki et al. [20] reported that a high
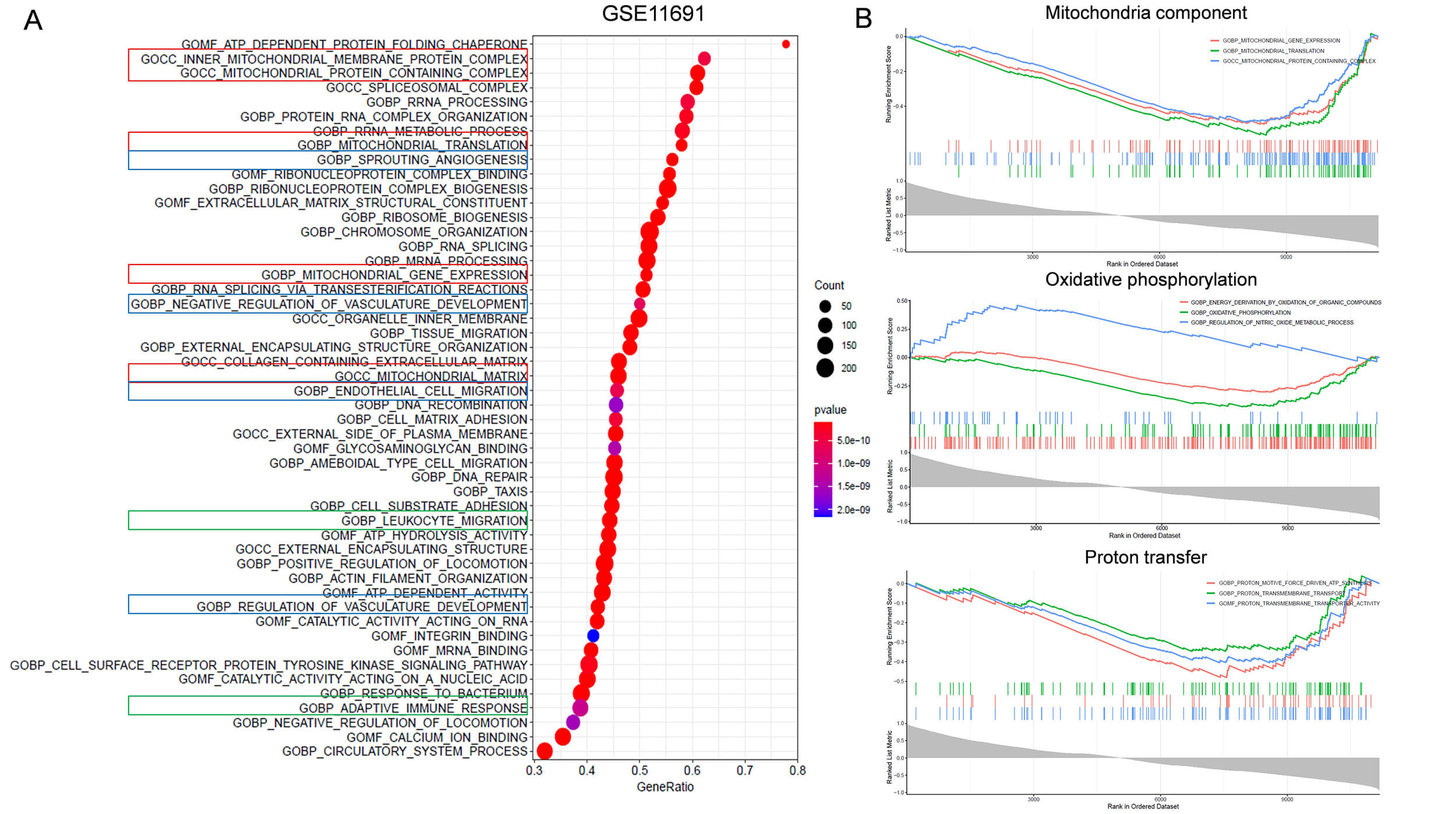
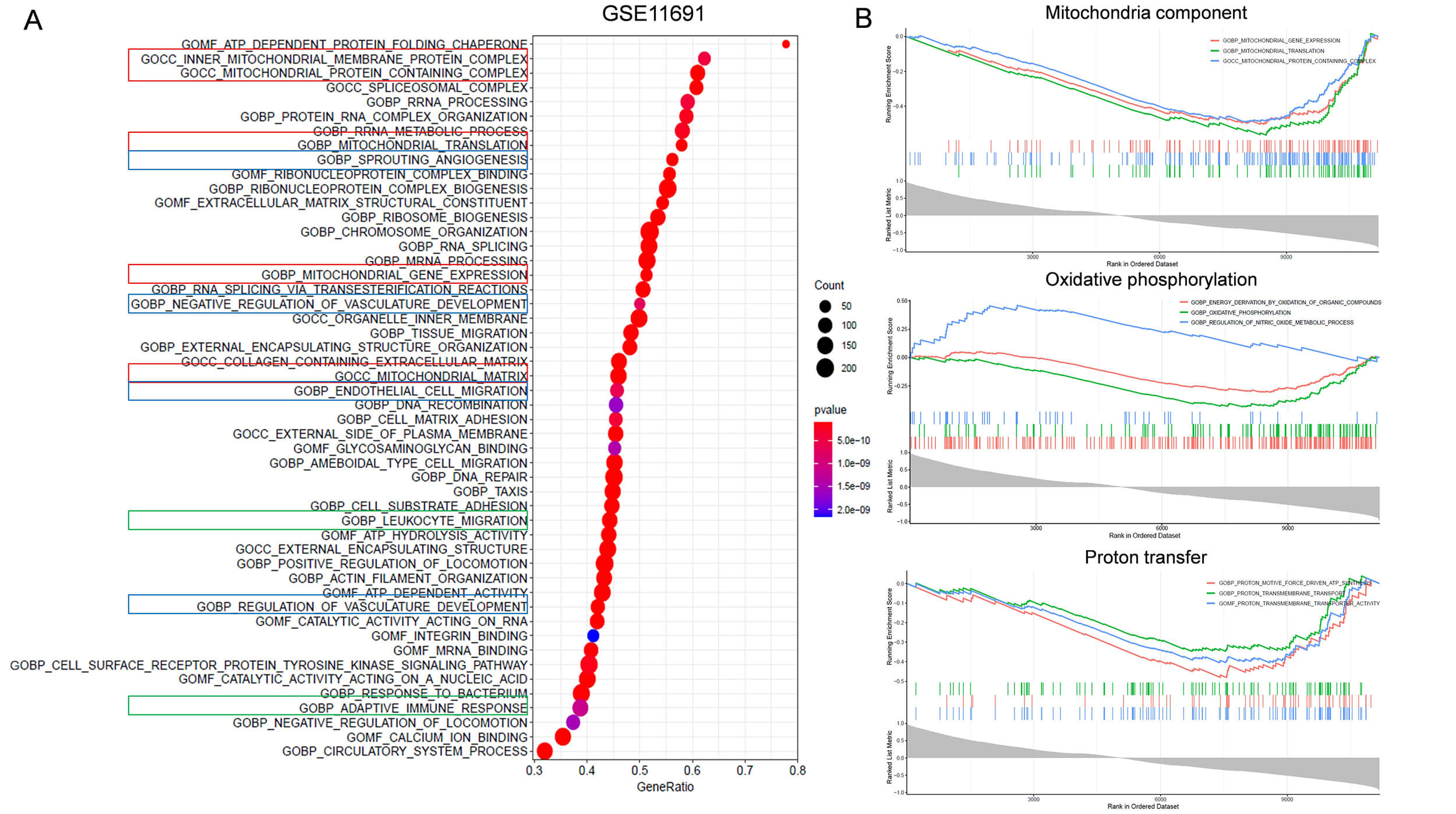 Fig. 3.
Fig. 3.
Identification of FABP4 related biological functions using the
GSE11691 dataset. (A) Gene set enrichment analysis (GSEA) was used to identify
enriched GO terms of genes correlated with FABP4. Bubble plot shows the top 50
enriched GO terms of genes associated with FABP4. Red box indicates GO terms
related to mitochondria function. Blue box indicates terms related to
angiogenesis. Green box labels terms related to immune response. (B) GSEA shows
enrichment of genes related to FABP4 in mitochondrial related functions (p
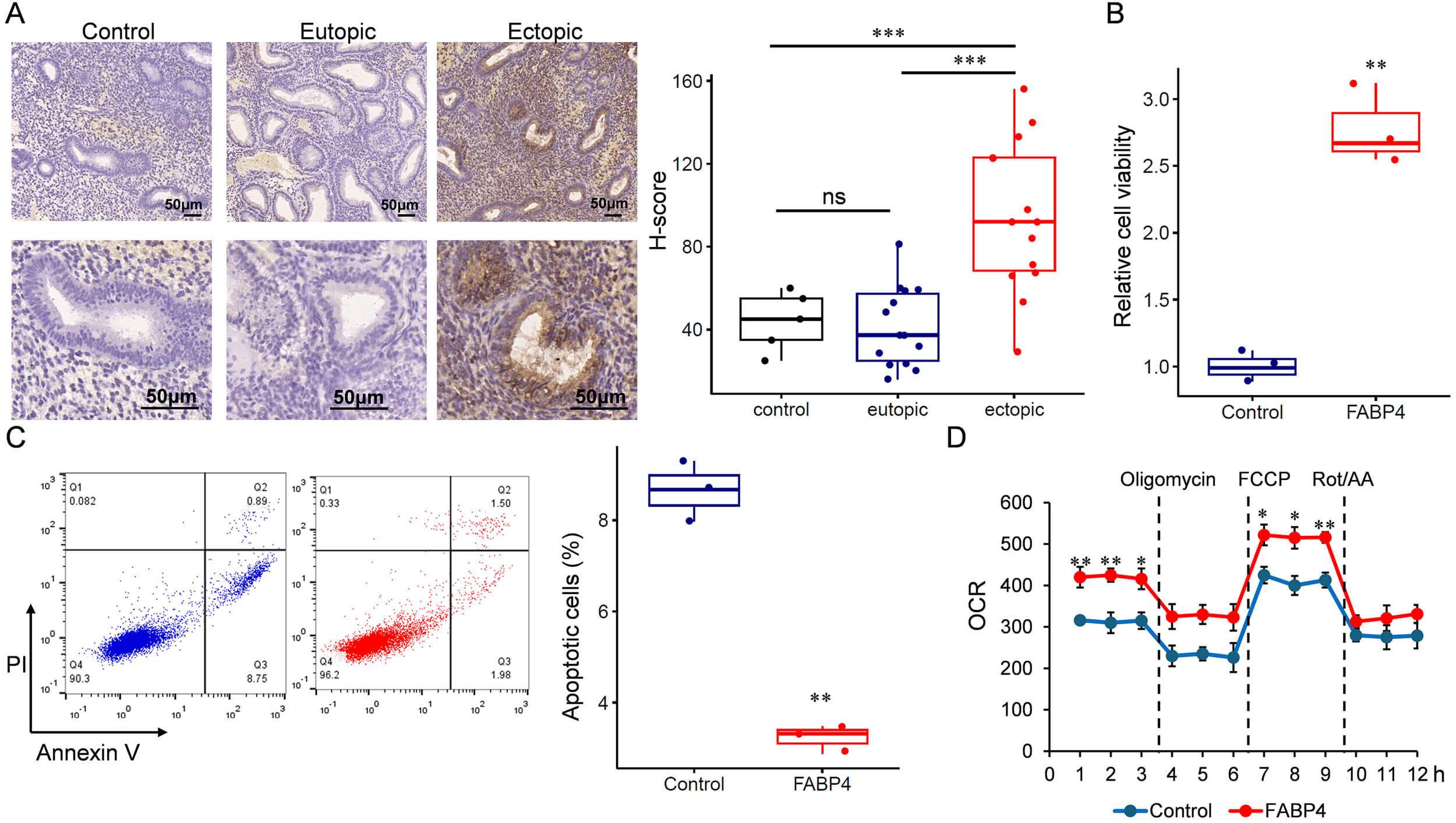
To investigate the role of FABP4 in endometriosis, we recruited a cohort comprising 14 patients with endometriosis. FABP4 expression was assessed by IHC, confirming its upregulation in ectopic endometrium compared with eutopic tissue (Fig. 4A). Subsequently, primary ESCs were isolated and transfected to achieve transient overexpression of FABP4 (Supplementary Figs. 3,4). FABP4 overexpression promoted endometrial stromal growth (Fig. 4B) and inhibited apoptosis (Fig. 4C). We then examined the impact of FABP4 on mitochondrial function by measuring the OCR. As shown in Fig. 4D, cells overexpressing FABP4 exhibited a significantly increased OCR, indicating altered mitochondrial activity.
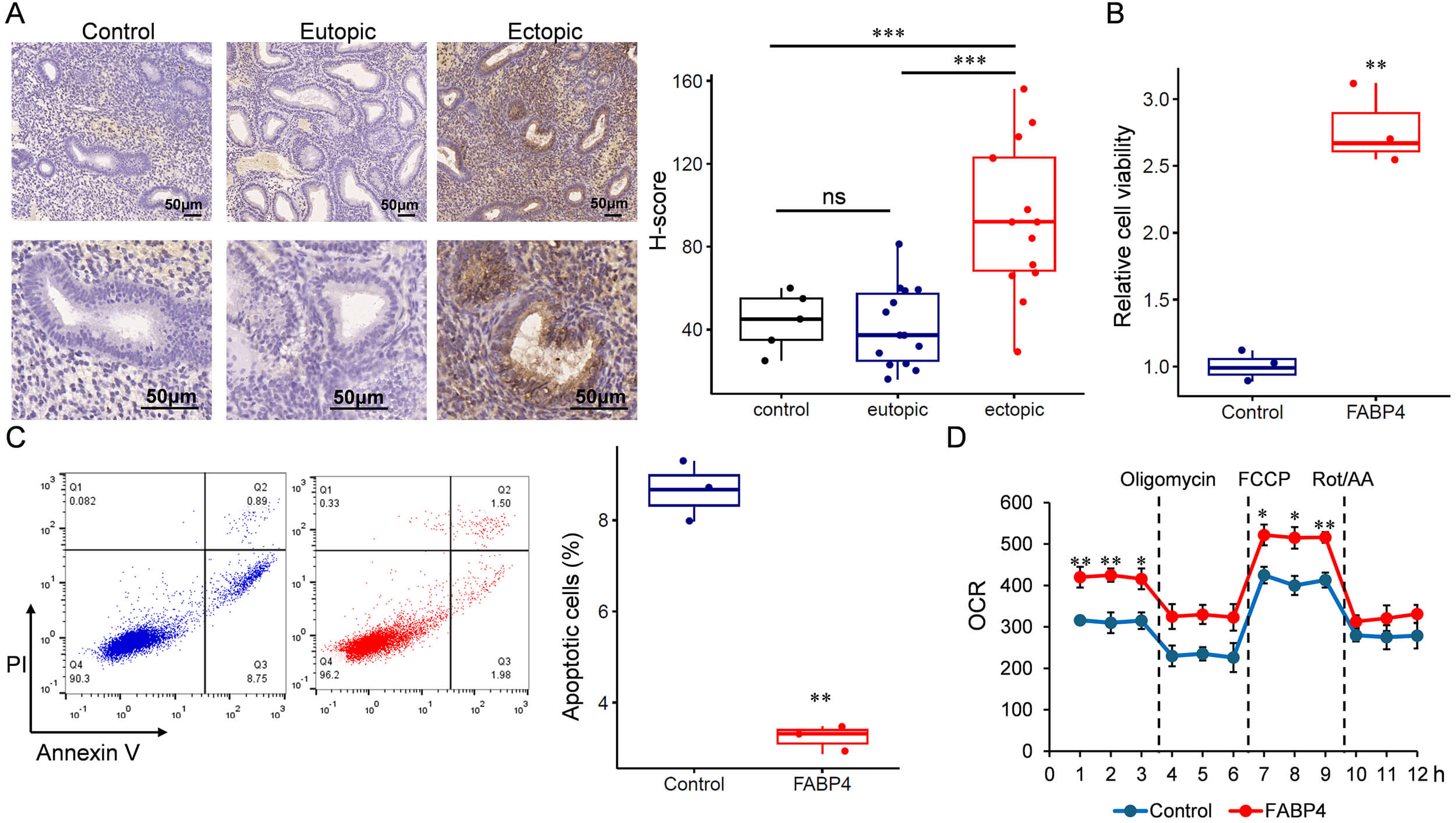 Fig. 4.
Fig. 4.
FABP4 is upregulated in ectopic endometrium and related to
increased oxygen consumption and upregulated cell proliferation. (A) IHC
detection of FABP4 in eutopic and ectopic endometrium. Scale bar: 50 µm.
(B) Cell viability analysis. (C) Apoptotic cells detected by flow cytometry
analysis. (D) Oxygen consumption assay. IHC, Immunohistochemistry. *p
Endometriosis is a multifactorial disease characterized by the presence of endometrial-like tissue outside the uterine cavity. Key pathogenic mechanisms involve alterations in cell proliferation, apoptosis, cell adhesion, and inflammation [21, 22]. Herein, we utilized GEO gene expression datasets for DEG analysis and WGCNA. This approach identified 48 key DEGs. Functional enrichment analysis revealed that these genes are primarily involved in processes such as complement activation and cell adhesion, both of which contribute to the pathogenesis of endometriosis. Notably, we identified FABP4, a gene associated with mitochondrial dysfunction in ESCs, as one of the most significantly upregulated genes in ectopic endometrium. To the best of our knowledge, this preliminary study is the first to characterize the role of FABP4 in human ESCs and to propose it as a candidate therapeutic target for endometriosis.
FABP4 is an intracellular lipid chaperone that is primarily expressed in adipocytes and macrophages [23, 24]. It is commonly dysregulated in metabolic disorders, including obesity and metabolic syndrome [25]. Abnormal FABP4 expression has also been reported in several cancer types [26]. Within the reproductive system, FABP4 acts as an important regulator of proliferation, migration, and invasion in endometrial epithelial cells, and it also contributes to the regulation of endometrial receptivity [8, 9]. In this study, we found that FABP4 upregulation is positively correlated with endometriosis. To our knowledge, this study is the first to confirm that FABP4 overexpression promotes cell viability and regulates mitochondrial function. Through analysis of gene expression profiling data, we also observed that FABP4 may regulate angiogenesis and adaptive immune responses. The multifunctional role of FABP4 may contribute to the rapid cellular growth and inflammatory phenotypes of endometriosis, although further validation is required.
This study had several limitations: (1) Our findings are based on bioinformatic analyses and in vitro cellular experiments; in vivo studies with animal models are necessary to validate the role of FABP4 in endometriosis. (2) Although this study confirms the role of FABP4 in the regulation of mitochondrial function, its potential effects on angiogenesis and immune responses require further investigation.
In conclusion, we identified a functional role for FABP4 dysregulation in endometriosis and propose it as a potential therapeutic target for endometriosis.
All data generated or analyzed during this study are included in this article. Further inquiries can be directed to the corresponding authors.
YH and XW are responsible for the conceptual idea. GS and DM conducted the experiments and contributed equally to the work. YJ, CG, JY, TZ, HZ and XZ helped with the experimental procedure. GS and YH did the statistical analysis. YH wrote the manuscript. YH revised and edited the final manuscript. All authors contributed to critical revision of the manuscript for important intellectual content. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. The study was approved by the Bioethics Committee of The Fourth Hospital of Shijiazhuang (Approval number: 20220039). Written informed consent was obtained from all participants.
We thank all the staff in the center for reproductive medicine of The Fourth Hospital of Shijiazhuang.
Hebei Province Medical Science Research Key Project (20231650); Youth Fund of Beijing Shijitan Hospital, Capital Medical University, China (2021-q05).
The authors declare no conflicts of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/CEOG48214.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.