

 , Hong Liao 2,3,†, Xin Liu 1,2, Mei Yang 1,2, Xinlian Chen 1,2, He Wang 1,2, Hanbing Xie 1,2,*
, Hong Liao 2,3,†, Xin Liu 1,2, Mei Yang 1,2, Xinlian Chen 1,2, He Wang 1,2, Hanbing Xie 1,2,* , Shanling Liu 1,2,*
, Shanling Liu 1,2,*1 Department of Medical Genetics/Prenatal Diagnostic Center, West China Second University Hospital, Sichuan University, 610041 Chengdu, Sichuan, China
2 Key Laboratory of Birth Defects and Related Diseases of Women and Children (Sichuan University), Ministry of Education, 610041 Chengdu, Sichuan, China
3 Department of Obstetrics and Gynecology, West China Second University Hospital, Sichuan University, 610041 Chengdu, Sichuan, China
†These authors contributed equally.
Abstract
Roberts–SC phocomelia syndrome (RBS; OMIM #268300) is a rare autosomal recessive disorder caused by biallelic variants in the sister chromatid cohesion N-acetyltransferase 2 (ESCO2) gene. It is characterized by tetraphocomelia and craniofacial anomalies. Prenatal cases are often lethal, and molecular confirmation is crucial for diagnosis and recurrence prevention.
We investigated a Chinese family with recurrent fetuses that presented severe tetraphocomelia. Whole-exome sequencing (WES) of both fetuses and their parents was performed, followed by Sanger validation, protein modeling using AlphaFold2, and Western blot analysis in transfected human embryonic kidney 293 (HEK293) cells. Two fetuses carried novel compound heterozygous nonsense variants in ESCO2, c.754A>T (p.Lys252*) and c.1276G>T (p.Glu426*). Structural modeling predicted truncated proteins lacking the C-terminal acetyltransferase domain. Western blot analysis showed that p.Lys252* caused complete loss of ESCO2, whereas p.Glu426* produced a truncated protein. Based on these findings, PGT-M was performed, identifying a mutation-free embryo for transfer. Prenatal testing confirmed the absence of ESCO2 variants, and a healthy child was delivered.
We report novel ESCO2 variants associated with severe prenatal RBS and provide functional and clinical evidence supporting their pathogenicity. This study expands the ESCO2 genotype–phenotype spectrum and underscores the value of molecular diagnosis and PGT-M in preventing recurrence of lethal autosomal recessive disorders.
Keywords
- Roberts-SC phocomelia syndrome
- whole-exome sequencing
- ESCO2
- preimplantation genetic testing
- prenatal diagnosis
Roberts syndrome is a rare autosomal recessive disorder first described by Roberts in 1919 [1]. Similar but milder phenotypes reported later are referred to as SC phocomelia. These conditions are now considered part of a continuous spectrum, collectively termed Roberts–SC phocomelia syndrome (RBS; OMIM #268300) [2]. Clinical features vary and may include tetraphocomelia (symmetrical limb reduction), pre- and postnatal growth retardation, craniofacial anomalies (microcephaly, cleft lip and palate), intellectual disability, congenital heart defects, and renal malformations [3]. Severe RBS often results in fetal or early neonatal death, whereas milder cases may survive to adulthood with varying degrees of intellectual disability [4]. The prenatal ultrasound features of severe RBS, such as tetraphocomelia and craniofacial abnormalities, can overlap with those of other conditions, including Cornelia de Lange syndrome and thrombocytopenia-absent radius syndrome. This overlap underscores the need for molecular diagnosis to enable accurate prenatal differentiation.
Biallelic variants in establishment of sister chromatid cohesion N-acetyltransferase 2 (ESCO2; OMIM #609353) cause RBS [5]. ESCO2 comprises 11 exons and encodes an N-acetyltransferase of the Eco1/Ctf7 family. It contains a Cys2His2 (C2H2) zinc-finger–like motif and a conserved C-terminal acetyltransferase domain. This acetyltransferase activity is essential for the establishment of sister chromatid cohesion [6].
Herein, we describe three fetuses with severe tetraphocomelia from a Chinese
family. Two fetuses carried novel compound heterozygous ESCO2 variants
(c.754A
Genomic DNA extracted from muscle tissue of the two fetuses and from peripheral blood from their parents was analyzed by WES. Fetal DNA samples underwent short tandem repeat (STR) analysis using quantitative fluorescent polymerase chain reaction (QF-PCR) to exclude maternal cell contamination. Exome enrichment was performed using the Nano WES Human Exome V2 kit (Berry Genomics, Guangzhou, Guangdong, China). Paired-end sequencing with 150-bp reads was performed on an Illumina NovaSeq 6000 platform (Illumina Inc., San Diego, CA, USA). Reads were aligned to the GRCh38/hg38 reference genome using the Burrows-Wheeler Aligner (BWA, Broad Institute, Cambridge, MA, USA). Variants were annotated using ANNOtate VARiation (ANNOVAR, University of Pennsylvania, Philadelphia, PA, USA) and the Enliven Variant Annotation and Interpretation System (Berry Genomics, Guangzhou, Guangdong, China). Population databases (gnomAD, 1000 Genomes) were used to filter rare variants. PubMed, ClinVar, OMIM, HGMD, and HGVS resources were consulted to evaluate variant pathogenicity. Variants were classified according to the American College of Medical Genetics and Genomics (ACMG) guidelines [7].
We predicted three-dimensional (3D) structures of wild-type (WT) and variant
ESCO2 proteins using AlphaFold2
(https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb).
A TM-score
Human ESCO2 cDNA (NM_001017420.3, 1806 bp) with an N-terminal His-tag
was cloned into pcDNA3.1. Variants were generated using a high-fidelity PCR mix
(#TSE101; Tsingke Biotechnology, Beijing, China). Primers were designed
following standard site-directed mutagenesis principles. The target nucleotide
substitution was positioned in the middle of the primer. Each side was flanked by
15–20 bp of perfectly matching sequence to ensure efficient and specific
amplification. Primer design was performed using PrimerX to ensure optimal
melting temperature, GC content, and minimal secondary structures. The c.754A
Human embryonic kidney 293 (HEK293) cells (GNHu-43; Chinese Academy of Sciences Cell Bank, Shanghai, China) were cultured in DMEM (#11,965,092, Gibco, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS) (#SFBS-NZ, Bovogen Biologicals Pty Ltd, Melbourne, Victoria, Australia), 100 U/mL penicillin, and 100 µg/mL streptomycin (#C0222, Beyotime Biotechnology, Shanghai, China), at 37 °C in 5% CO2. Cells were transiently transfected in 6-well plates with 3 µg plasmid (WT or variant) or co-transfected with 1.5 µg of each variant plasmid using Lipofectamine 2000 (#11668019, Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions.
At 24–36 hours post-transfection, cells were lysed in radio-immunoprecipitation
assay (RIPA) buffer (#P0013B, Beyotime Biotechnology, Shanghai, China)
supplemented with protease inhibitors (#P1005, Beyotime Biotechnology, Shanghai,
China). 30 µg of protein lysate were separated by 8% sodium dodecyl
sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), transferred to
nitrocellulose membranes, and blocked in 5% nonfat dry milk. Membranes were
incubated with the following primary antibodies: anti-6X His-tag antibody (mouse
monoclonal, 1:1000, #ab18184, Abcam, Cambridge, UK) and anti-
Western blotting was used for qualitative assessment of protein truncation and relative expression levels. Experiments were repeated in three independent replicates to ensure reproducibility.
Approximately 5–8 trophectoderm (TE) cells were biopsied from each blastocyst
for whole-genome amplification (WGA) using multiple displacement amplification
(REPLI-g Single Cell MDA kit, QIAGEN, Venlo, Limburg, Netherlands). WGA products
from parental oral mucosa and TE cells were analyzed by Sanger sequencing for
validation of variant sites. Single nucleotide polymorphisms (SNP)-based
haplotyping within a 1 Mb flanking region was performed using next-generation
sequencing (NGS). NGS-based copy-number variants (CNVs) analysis screened for
deletions and duplications
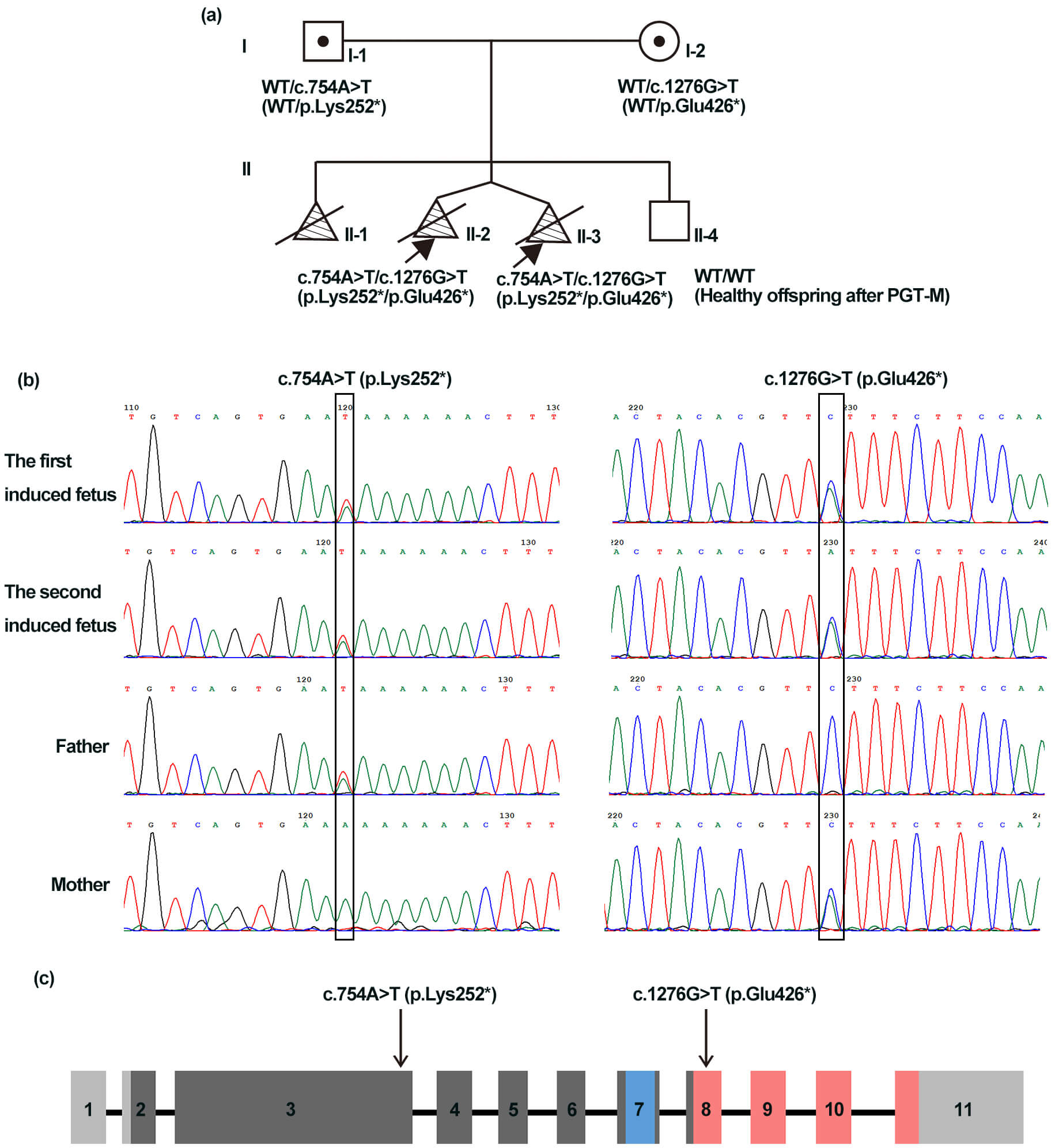
A 31-year-old woman (Fig. 1a,I-2) was referred for genetic counseling. In her first pregnancy, second-trimester ultrasound revealed tetraphocomelia, cleft lip and palate, and micrognathia, after which the pregnancy was terminated without prenatal diagnosis. A subsequent pregnancy in this couple was carefully monitored. At 12 weeks of gestation, ultrasound examination revealed dichorionic twins. One fetus presented nuchal translucency (NT) thickness of 3.3 mm, and the lengths of the upper and lower limbs were 5.7 mm and 3.8 mm, respectively. The other fetus also exhibited an increased NT (4.2 mm), with the upper and lower limbs lengths of 6.2 mm and 3.9 mm, respectively. Both fetuses lacked or had severely shortened humeri, ulnae, radii, femurs, tibiae, and fibulae. The pregnancy was terminated, and postmortem examination confirmed the ultrasound findings. No related clinical manifestations were observed in the parents or grandparents (pedigree shown in Fig. 1a).
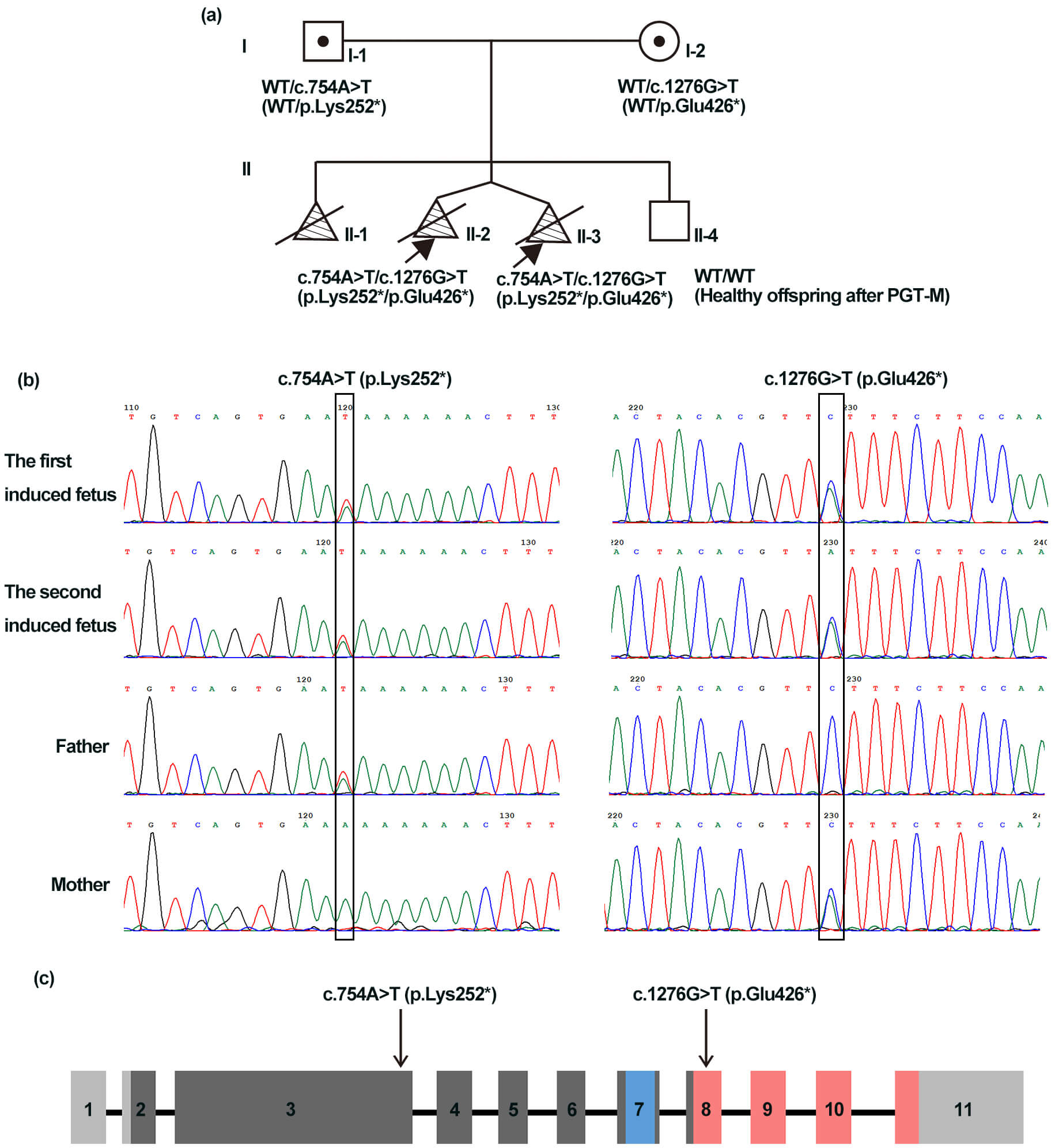 Fig. 1.
Fig. 1.
Clinical characteristics of the family and schematic of the
establishment of sister chromatid cohesion N-acetyltransferase 2
(ESCO2). (a) Pedigree that shows compound heterozygous ESCO2
variants (c.754A
WES was performed on muscle tissue from the twins and peripheral blood from
their parents. WES identified two novel compound heterozygous ESCO2
variants in the two probands: c.754A
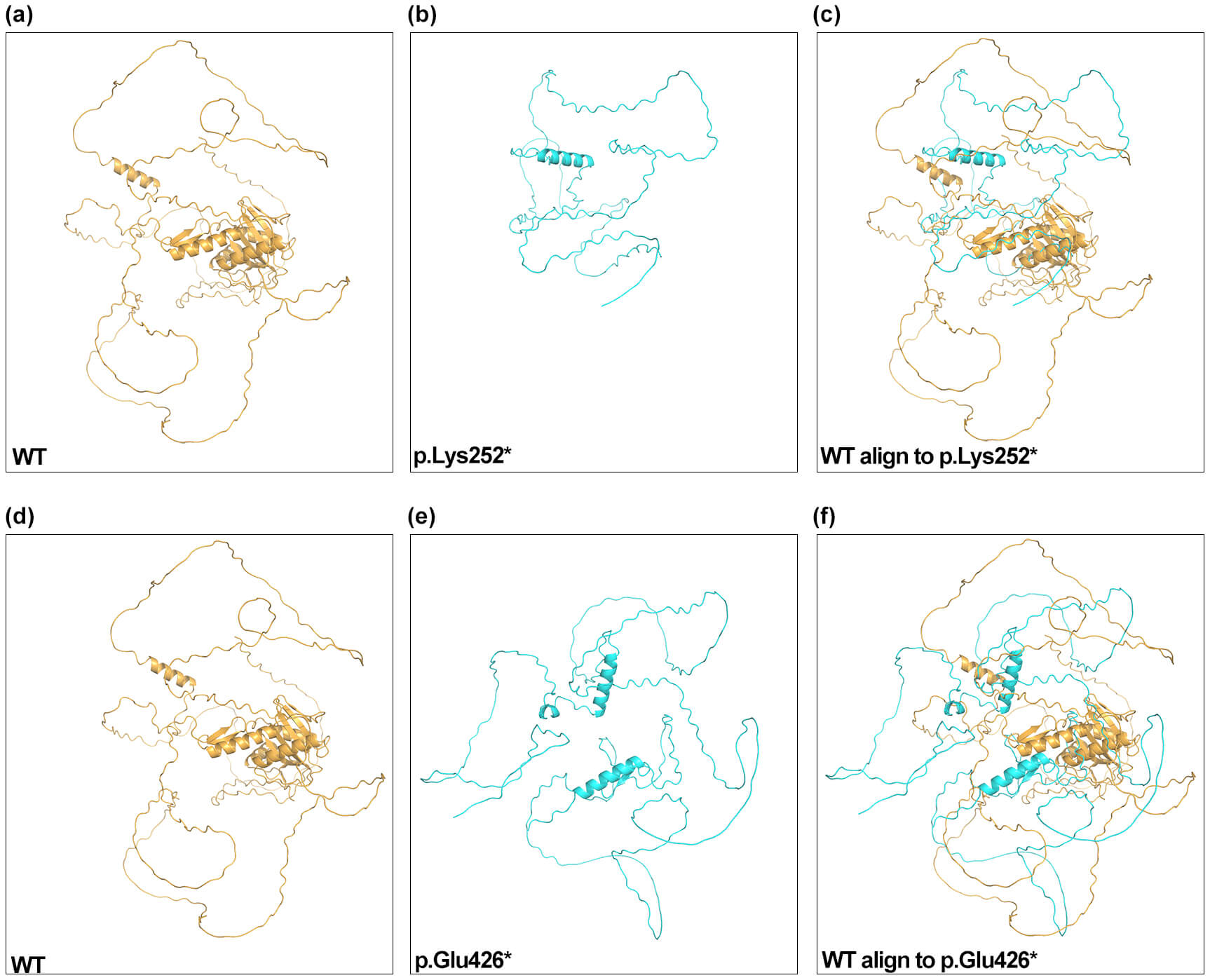
AlphaFold2 predicted truncated ESCO2 structures for the p.Lys252* and p.Glu426* variants relative to the WT protein (Fig. 2). The highest-ranked p.Lys252* model aligned poorly with WT structure (TM-score 0.03; RMSD 0.66), and the p.Glu426* model also showed low similarity (TM-score 0.08; RMSD 16.63). The markedly reduced TM-scores indicate that both truncating variants lack the native global fold of ESCO2. RMSD values represent the extent of structural deviation within aligned regions and are influenced by alignment length. The elevated RMSD observed for the p.Glu426* variant reflects significant conformational alteration.
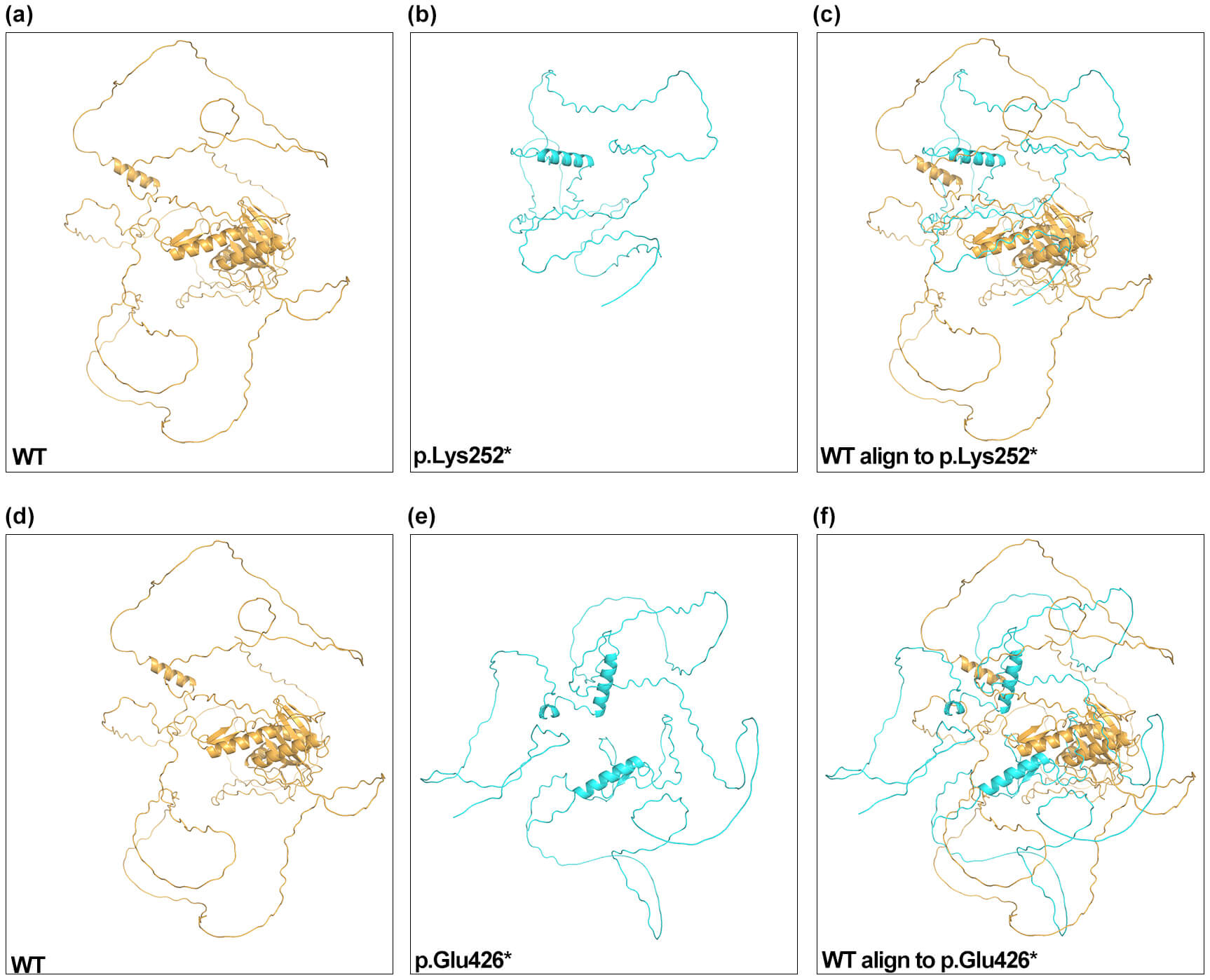 Fig. 2.
Fig. 2.
AlphaFold2 structural models of ESCO2. Predicted structures for WT (a,d), p.Lys252* (b), p.Glu426* (e), and structural alignments (c,f) are shown. Two views of the WT structure are displayed for clarity. WT, wild-type.
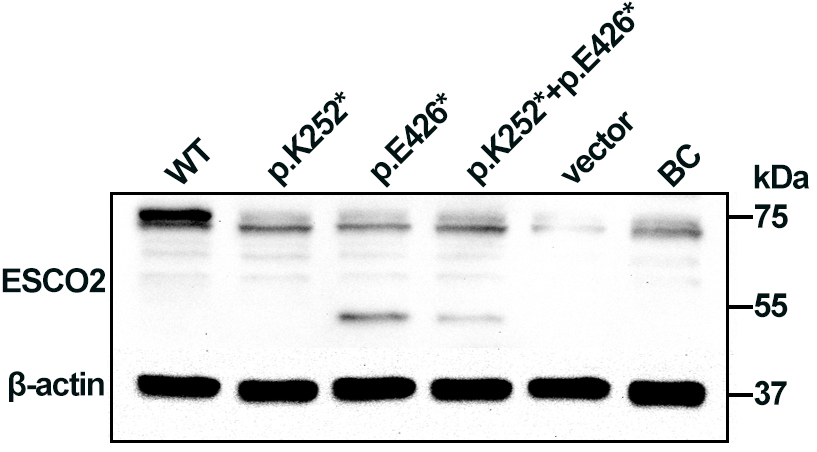
Western blotting analysis of HEK293 cell lysates showed a 72 kDa band corresponding to full-length ESCO2 in cells transfected with WT plasmid. A 50 kDa truncated band was detected in cells transfected with the p.Glu426* plasmid and in cells co-expressing both variants (p.Lys252* + p.Glu426*), albeit at low levels, suggesting partial escape from nonsense-mediated decay (NMD). No full-length or truncated ESCO2 was detected in cells expressing p.Lys252*, consistent with NMD-mediated loss of that transcript (Fig. 3). The p.Lys252*, located in exon 3, lies upstream of the acetyltransferase domain, whereas p.Glu426* (located in exon 8) truncates the protein within the acetyltransferase domain (Fig. 1c). Loss of ESCO2 acetyltransferase activity is likely central to RBS pathogenesis.
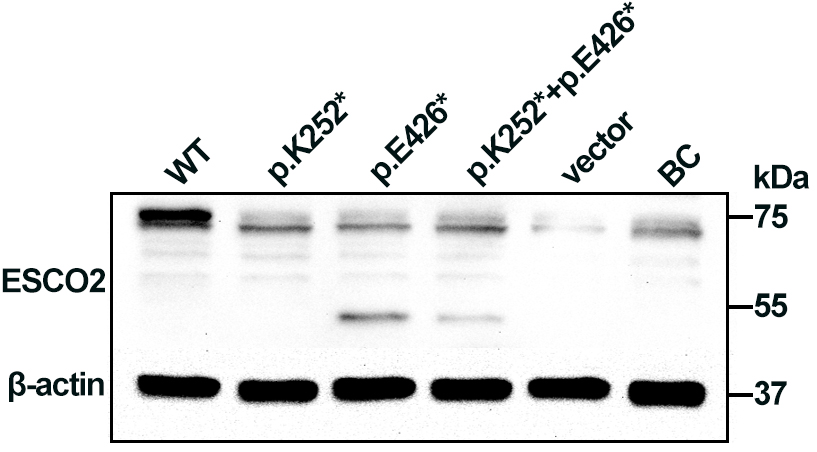 Fig. 3.
Fig. 3.
Western blot analysis of ESCO2 expression in
human embryonic kidney 293 (HEK293) cells. Cell extracts were probed with an
anti-6X His tag antibody. A 72 kDa band corresponding to full-length
ESCO2 was observed in cells transfected with the WT plasmid. A 50 kDa
truncated band was detected in cells transfected with p.Glu426* and with the
combined (p.Lys252* + p.Glu426*) constructs.
Based on the family’s known ESCO2 variants, PGT-M with concurrent CNV screening was performed. One blastocyst, graded 5BC, was free of both familial ESCO2 variants and showed no clinically significant CNVs; this embryo was selected and transferred. Second-trimester prenatal diagnosis, including Sanger sequencing of targeted sites, CMA, and karyotype, confirmed the absence of the familial ESCO2 variants and chromosomal abnormalities. Prenatal ultrasound findings remained unremarkable. At the last follow-up, the child was three years old and showed no features observed in the affected probands (Fig. 1a,II-4).
We identified compound heterozygous ESCO2 variants (c.754A
RBS exhibits wide clinical variability. ESCO2 encodes an N-acetyltransferase that contains a zinc-finger–like motif and a C-terminal acetyltransferase domain. In this study, structural predictions yielded extremely low TM-scores for both truncating variants. This finding indicates a complete loss of the native ESCO2 structural framework and suggests severe disruption of the functional acetyltransferase domain [9]. Western blot analysis supported these predictions and revealed distinct molecular consequences for the two nonsense variants. The p.Lys252* variant resulted in undetectable ESCO2 protein, which is consistent with NMD, whereas the p.Glu426* variant produced a truncated protein, suggesting partial escape from NMD. Despite these differences in protein expression, both variants are expected to severely compromise acetyltransferase function. This provides a plausible molecular mechanism for the RBS phenotype observed in this family.
To explore genotype–phenotype correlations, we reviewed ESCO2 variants reported in HGMD and the literature (accessed October 2025; Table 1, Ref. [4, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33]). Approximately 43 likely pathogenic or pathogenic ESCO2 variants have been reported to date. The variant spectrum comprises small deletions/insertions that cause frameshifts (25/43; 58.14%), splicing variants (7/43; 16.28%), nonsense variants (5/43; 11.63%), missense variants (5/43; 11.63%), and in-frame deletions (1/43; 2.33%). Most pathogenic variants are null alleles that truncate the protein or cause mRNA instability, and many occur upstream of or within the acetyltransferase domain [10]. However, genotype or variant location alone does not reliably predict clinical severity, as both severe prenatal phenotypes and milder postnatal presentations have been observed across similar classes of variants. Consistent with previous reports, no clear genotype–phenotype correlation has been established for ESCO2 variants [11, 12].
| ID | Variants | Exon | Tetraphocomelia | Growth retardation | Craniofacial abnormalities | Cleft palate/lip | Mental retardation | Others |
| 1 | c.505C |
ex 3 | NA | NA | NA | NA | NA | [13] |
| 2 | c.604C |
ex 3 | + | + | + | – | hemangioma [14] | |
| 3 | c.1166G |
ex 7 | NA | NA | NA | NA | NA | [15] |
| 4 | c.1220A |
ex 7 | + | + | + | – | + | cerebrovascular diseases [16] |
| 5 | c.1269G |
ex 8 | NA | NA | NA | NA | NA | [17] |
| 6 | c.1489A |
ex 9 | NA | NA | NA | NA | NA | [18] |
| 7 | c.1567C |
ex 10 | NA | NA | NA | NA | NA | [19] |
| 8 | c.1615T |
ex 10 | NA | NA | NA | NA | NA | [10, 20, 21] |
| 9 | c.1654C |
ex 10 | + | + | – | + | + | seizure [22] |
| 10 | c.1741G |
ex 11 | + | NA | NA | NA | NA | [11] |
| 11 | c.1131+1G |
int 6 | + | + | + | NA | prominent eyes [12] | |
| 12 | c.1132-7A |
int 6 | + | + | + | – | NA | [12] |
| 13 | c.1263+1G |
int 7 | NA | NA | NA | NA | NA | [17] |
| 14 | c.1354-18G |
int 8 | NA | NA | NA | NA | NA | [10] |
| 15 | c.1673+1G |
int 10 | + | + | – | – | – | [23] |
| 16 | c.1674-2A |
int 10 | NA | NA | NA | NA | NA | [10] |
| 17 | c.166_170del(p.Val56fs) | ex 3 | – | + | – | – | cardiac abnormalities [4] | |
| 18 | c.252_253del(p.Ser85fs) | ex 3 | + | NA | NA | NA | NA | [20] |
| 19 | c.294_297del(p.Arg99fs) | ex 3 | NA | NA | NA | NA | NA | [10] |
| 20 | c.307_311del(p.Lys103fs) | ex 3 | NA | NA | NA | NA | NA | [13] |
| 21 | c.308_309del(p.Lys103fs) | ex 3 | NA | NA | NA | NA | NA | [10] |
| 22 | c.417del(p.Lys139fs) | ex 3 | + | + | + | – | + | [24] |
| 23 | c.745_746del(p.Val249fs) | ex 3 | + | + | – | – | NA | [25] |
| 24 | c.752del(p.Lys253fs) | ex 3 | + | + | + | – | hemangioma [14] | |
| 25 | c.760del(p.Thr254fs) | ex 3 | NA | NA | NA | NA | NA | [13] |
| 26 | c.764_765del(p.Phe255fs) | ex 3 | + | + | + | + | + | [26] |
| 27 | c.876_879del(p.Asp292fs) | ex 4 | + | + | + | + | – | [27] |
| 28 | c.879_880del(p.Arg293fs) | ex 4 | + | + | – | + | – | [20] |
| 29 | c.955+2_955+5del | int 4 | NA | NA | NA | NA | NA | [13] |
| 30 | c.1156del(p.Ala386fs) | ex 7 | NA | NA | NA | NA | NA | [28] |
| 31 | c.1359_1361del(p.Glu453del)) | ex 7 | NA | NA | NA | NA | NA | [11] |
| 32 | c.1461_1462del(p.Arg487fs) | ex 9 | + | NA | NA | NA | NA | [20] |
| 33 | c.1562del(p.Ala521fs) | ex 10 | + | + | + | – | + | cerebrovascular diseases [16] |
| 34 | c.244_245dup(p.Thr83fs) | ex 3 | + | + | + | + | + | blue sclera [26] |
| 35 | c.417dup(p.Pro140fs) | ex 3 | NA | NA | NA | NA | NA | [29] |
| 36 | c.416_417dup(p.Pro140fs) | ex 3 | NA | NA | NA | NA | NA | [29] |
| 37 | c.636dup(p.Val213fs) | ex 3 | NA | NA | NA | NA | NA | [20] |
| 38 | c.751dup(p.Glu251fs) | ex 3 | NA | NA | NA | NA | NA | [30] |
| 39 | c.760dup(p.Thr254fs) | ex 3 | + | + | + | + | – | cardiac anomalies [26, 31] |
| 40 | c.877dup(p.Arg293fs) | ex 4 | NA | NA | NA | NA | NA | [32] |
| 41 | c.1111_1112insG(p.Thr371fs) | ex 6 | NA | NA | NA | NA | NA | [10] |
| 42 | c.1111dup(p.Thr371fs) | ex 6 | + | + | + | + | NA | [33] |
| 43 | c.1597dup(p.Cys533fs) | ex 10 | NA | NA | NA | NA | NA | [10] |
Abbreviation: Ex, exon; Int, intron; +, feature is present; –, feature is
absent;
Data sources: Variants were collated from the HGMD professional database and the published literature (up to October 2025).
Nevertheless, when variant distribution and functional domains are considered together, a consistent pathogenic mechanism emerges. Regardless of variant type (e.g., frameshift, nonsense, splicing, or missense), most pathogenic ESCO2 variants are expected to impair or abolish acetyltransferase activity, either through NMD, protein truncation, or direct disruption of the catalytic domain. This convergence on loss of acetyltransferase function suggests that impaired ESCO2-mediated acetylation represents the core pathogenic mechanism underlying RBS, even in the absence of a strong genotype–phenotype correlation. The phenotypic variability may therefore reflect differences in residual activity and modifying genetic or environmental factors, rather than variant class or position alone.
Prenatal diagnosis of RBS is challenging because tetraphocomelia and growth restriction overlap with several other syndromes (e.g., Cornelia de Lange syndrome, CHARGE syndrome, thrombocytopenia-absent radius syndrome). Therefore, genetic testing is essential for accurate prenatal diagnosis [34]. In this family, recurrent severe tetraphocomelia prompted WES, which identified causative ESCO2 variants and enabled PGT-M and prenatal diagnosis. These measures successfully prevented recurrence and resulted in the birth of a healthy infant.
A limitation of this study is the lack of additional functional assays that assess ESCO2 acetyltransferase activity. Such experiments would provide more direct mechanistic evidence linking the observed nonsense variants to loss of enzymatic function and RBS pathogenesis.
In summary, we describe novel compound heterozygous ESCO2 variants associated with severe prenatal RBS and demonstrate the value of molecular diagnosis to guide reproductive decision-making and prevent recurrence.
The data and materials that support the findings of this study are available from the corresponding authors upon reasonable request.
Conceptualization: SL, HX, HW, XC, MY, PW, HL, XL; Methodology: PW, XL; Writing-original draft preparation: PW, HL; Writing-review and editing: SL, HX, HW; Funding acquisition: SL; Supervision: SL, HX. All authors contributed to critical revision of the manuscript for important intellectual content. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The study was carried out in accordance with the guidelines of the Declaration of Helsinki. The study was approved by the ethical committee of West China Second University Hospital, Sichuan University (Medical Research Ethics Review 2024 No. 228). In addition, the family signed written informed consent to participate.
Not applicable.
This study was supported by the National Key Research and Development Program of China (2022YFC2703302).
The authors declare no conflicts of interest.
During the preparation of this work, the authors used ChatGPT-3.5 in order to check spelling and grammar. After using this tool, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/CEOG47712.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.