



 , Yu Zhang 2, Xinge Huang 2, Wei Wang 2, Tao Chen 3,*
, Yu Zhang 2, Xinge Huang 2, Wei Wang 2, Tao Chen 3,* , Rui Chen 2,*
, Rui Chen 2,*1 Institute of Regenerative Medicine and Laboratory Technology Innovation, Qingdao University, 266003 Qingdao, Shandong, China
2 Department of Reproductive Medicine, The Affiliated Hospital of Qingdao University, 266000 Qingdao, Shandong, China
3 Department of Radiology, The University of Qingdao, 266971 Qingdao, Shandong, China
Abstract
Degeneration of the arcus tendineus fascia pelvis (ATFP) is often accompanied by compromise of the arcus tendineus levator ani (ATLA). ATFP is increasingly being recognized as a contributor to pelvic floor instability, particularly following childbirth and with advancing age. However, prevailing clinical phenotypes, such as pelvic organ prolapse and stress urinary incontinence, do not differentiate tendon-like connective tissue failure from muscular or fascial dysfunction. This review aims to define ATFP degeneration as a distinct biological substrate of pelvic floor dysfunction. Additionally, we present current evidence on the molecular and mechanobiological factors that control its initiation and progression.
We examine how mechanical overload can induce microdamage in the ATFP complex by integrating human imaging and tissue-based data with principles from tendon and fascia mechanobiology. This is particularly important during the peripartum period, when connective tissue undergoes rapid remodeling under high mechanical load. We also examine how extracellular matrix (ECM) homeostasis, mechanotransduction-dependent cell-state transitions, and stress response programs-modulated by factors such as inflammatory signaling, oxidative or mitochondrial stress, and systemic metabolic disturbance-can bias repair toward maladaptive remodeling and progressive loss of mechanical integrity.
Available evidence supports a convergent pathological model in which dysregulated ECM remodeling, characterized by altered collagen subtype balance, elastin loss, and impaired matrix turnover, represents the central endpoint of ATFP degeneration. Although the molecular mechanisms involved, such as matrix metalloproteinase activation, macrophage-mediated immune modulation, oxidative stress, and metabolic signaling, are well described in tendon biology, pelvic-specific human data remain limited. Current diagnostic and therapeutic approaches primarily target macroscopic support failure rather than the underlying degenerative biology, and most regenerative strategies are supported predominantly by non-pelvic or preclinical models.
ATFP degeneration should be conceptualized as a load-induced, biologically modulated failure of adaptive connective-tissue remodeling, rather than as an isolated inflammatory or structural defect. Progress in this area can be achieved by focusing on evidence specific to the human pelvis, combining multiple types of scientific approaches, and distinguishing well-supported findings from speculative ideas. Such a framework is key for developing mechanism-based risk stratification, optimizing the timing and targets of interventions, and advancing biologically aligned pelvic floor repair strategies.
Keywords
- pelvic tendinopathy
- arcus tendineus fascia pelvis (ATFP)
- arcus tendineus levator ani (ATLA)
- extracellular matrix remodeling
- mechanobiology
Tendon pathology includes both acute mechanical disruption and more slowly evolving degenerative change, the latter often described under the broad term of tendinopathy. Across different anatomical sites, these processes are accompanied by alterations in extracellular matrix (ECM) organization, changes in resident cell behavior, and a gradual decline in mechanical performance. Together, these may give rise to pain, swelling, and functional impairment [1]. Early work in this field was complicated by inconsistent terminology. Maffulli et al. [2] adopted the term “tendinopathy” as an inclusive descriptor for inflammatory and degenerative tendon conditions, including tendinitis, tenosynovitis, and tendinosis. Although this helped to standardize discussion, it did not resolve the underlying biological distinctions [2]. Nevertheless, this shift supported clearer clinicopathological communication and enabled more consistent comparisons across studies.
Subsequent research has made it clear that tendinopathy does not arise from a single causative factor. Instead, it can be influenced by internal factors, such as aging, the presence of systemic disease, genetic conditions, problems with nerves and muscles, and other pre-existing health issues. These interact with external factors that are mostly related to the body, such as repetitive movements, not recovering properly between sets, or training too hard [1, 3]. Importantly, these influences shape the background against which mechanical stress is experienced. Sometimes, a load that would usually be tolerable can be too much for the heart. This can lead to a lack of organization in how the body replaces old tissue, which can cause tissue to stop working properly.
The arcus tendineus fascia pelvis (ATFP) and the arcus tendineus levator ani (ATLA) are specialized connective tissue structures in the pelvic floor that serve as anchor points for the endopelvic fascia and the levator ani muscle complex [4, 5, 6].
Although these structures do not fit the usual definition of a tendon, they do share several features. For example, they are composed of a collagen-rich matrix and are very important for transmitting mechanical force. Damage or attenuation affecting the ATFP or ATLA has been associated with pelvic floor instability and symptom complexes such as pelvic organ prolapse (POP) [7], stress urinary incontinence (SUI) [8], and chronic pelvic pain [9]. However, these clinical labels offer little insight into the specific tissue responsible for mechanical failure.
This lack of anatomical specificity makes both mechanistic interpretation and clinical decision-making more difficult. Muscle dysfunction, fascial attenuation, and connective tissue degeneration are often discussed together, even though they are likely to have different mechanisms of injury and repair. To help discuss this more easily, the term “pelvic tendinopathy” will be used here to describe the condition in which the tendons in the pelvis become damaged over time due to improper use of the body. Within this usage, emphasis is placed on progressive ECM remodeling—such as collagen disorganization, shifts in collagen subtype expression, elastin loss, and impaired matrix turnover—rather than on acute rupture or inflammatory disease [10, 11]. Pelvic tendinopathy is used descriptively rather than diagnostically, and is intended to complement rather than replace the established clinical classifications.
This framework requires clear limits. The context does not consider sudden, serious injury needing immediate medical help, long-term inflammation in the connective tissue, or other types of pelvic pain, including endometriosis and interstitial cystitis [8, 9]. In earlier studies, the presence of ATFP or ATLA was often based on imaging findings, such as thickening, detachment, or reduced elastin-associated signal, as seen on ultrasound or magnetic resonance imaging (MRI) [12]. This was seen alongside signs of long-term tissue changes and measures of function, including reduced pelvic floor strength or difficulty controlling urination when under pressure [10]. Taken together, these observations do not define a discrete disease entity, but support a coherent biological narrative centered on the integrity of connective tissue.
From a mechanical standpoint, involvement of the ATFP and ATLA may lead to distinct failure modes. Weakening of the ATFP is thought to compromise fascial support, which may contribute to compartmental prolapse. In contrast, degeneration of the ATLA is more likely to disrupt force transmission from the levator ani, potentially impairing continence during increases in intra-abdominal pressure [13]. Recognition of these distinctions may help to refine therapeutic strategies, for example, by tailoring pelvic floor muscle training (PFMT) toward dynamic support, or by directing surgical repair toward specific sites of connective tissue failure [10, 14].
Epidemiological data suggest that degeneration of pelvic connective tissue reflects the interaction between exposure and susceptibility factors [15]. Pregnancy and vaginal delivery, particularly when operative techniques such as forceps are used, subject the pelvic floor to substantial mechanical strain and have been associated with levator ani avulsion and fascial detachment [3, 16]. With aging, there are more risks due to factors that build up over time, as well as changes to the body [17, 18]. Such changes can also be linked to an increased prevalence of problems with the pelvic floor [19], supporting the notion of a period when the baby is at risk, both before and after birth. This is defined as the period during pregnancy and the early postpartum interval when connective tissue remodeling occurs under unusually high mechanical demand, potentially lowering the threshold for microdamage.
Despite the biomechanical importance of ATFP/ATLA, clinically meaningful injury or degeneration is frequently under-recognized, particularly when symptoms are subtle or develop gradually. Limitations in routine examination and variability in the interpretation of imaging results contribute to underdiagnosis. Advanced modalities—including dynamic MRI and three-dimensional ultrasound—have improved visualization of levator ani avulsion from the ATLA, and detachment or altered morphology of the ATFP, enabling more direct structure-function correlations in selected settings [12]. Nevertheless, clinically deployable tools for individualized risk prediction and mechanism-based stratification remain limited.
Current therapeutic options, such as PFMT, surgical repair, and biologic grafts, aim to restore support and function [10]. However, durable restoration of tendon-like connective-tissue competence remains challenging, reflecting the broader limitations of tendon repair biology and the incomplete understanding of pelvic floor-specific degeneration programs [20]. For this reason, inflammation, oxidative/mitochondrial stress, and metabolic disturbance are treated as phase-dependent modulators rather than uniform drivers. Their effects may differ across initiation (microdamage and early remodeling), propagation (persistent matrix turnover and altered cell state), and late-stage failure (scar-prone remodeling and mechanical decompensation). This phase-aware framing helps to reconcile apparently inconsistent findings across human, animal, and in vitro studies, as well as reducing overgeneralization from any single evidence class.
Accordingly, this review focuses on the molecular mechanisms of ATFP degeneration as the central scope. We prioritize ATFP/ATLA-specific evidence from human imaging and tissue-based studies where available, and we integrate non-human and in vitro mechanobiology data as a supportive, hypothesis-generating context. By organizing the literature around mechanotransduction, ECM homeostasis, and resident cell fate decisions—and by explicitly distinguishing established findings from inferential links—we aim to clarify the biological basis of ATFP failure and identify testable mechanistic nodes. This should enable better delineation of the constraints that currently limit translation to biology-aligned pelvic floor repair strategies.
The PubMed and Web of Science databases were used to find relevant literature published between 1 October 2015 and 1 October 2025. The search terms included “pelvis, pelvic, tendinopathy, pelvic tendinopathy, tendon, arcus tendineus levator ani, arcus tendineus fascia pelvis”, and related keywords. Comprehensive searches were performed using scientifically rigorous search strategies.
1. Study Types: Include original studies, systematic reviews, and meta-analyses related to “Pelvis, Pelvic, Tendinopathy, Fascia tendinopathy, Tendon, Arcus tendineus levator ani, Arcus tendineus fascia pelvis”, and related keywords.
2. Publications were released between 1 October 2015 and 1 October 2025.
3. Relevance: Studies must clearly include anatomical structures, pathological conditions, clinical symptoms, and ways to diagnose or treat the above keywords.
4. How complete is the data? Studies should provide sufficient data and information to allow verification of their results.
1. Publications that are the same: Only the most complete and credible version of a study was kept. Duplicate studies were ignored.
2. Studies not related to the above keywords were excluded.
3. Publications that were short reports, meeting summaries, or commentaries that did not include a full study report were excluded.
4. Specific Pathological Conditions: Studies on acute traumatic tendon ruptures (e.g., obstetric avulsions requiring emergency repair), inflammatory tendon diseases (e.g., rheumatoid arthritis involving pelvic tendons), and non-tendinous pelvic pain (e.g., endometriosis, interstitial cystitis) were excluded.
EndNote X9 (Clarivate Analytics; Stamford, CT, USA) was used to manage references and exclude duplicates. Two senior obstetrician-gynecologists independently participated in the study selection and data extraction process. Any differences were resolved through discussion with a third researcher. Ultimately, 47 articles were included in the study.
The ATFP and ATLA represent tendon-like connective-tissue specializations within the pelvic floor that distribute mechanical loads across the endopelvic fascia-levator ani continuum. Their ability to support the pelvic organs and maintain continence relies on a hierarchically organized ECM. This is integrated with a resident fibroblastic cell population that senses, transmits, and adapts to repetitive mechanical strain. Therefore, it is essential to define the anatomical configuration and molecular composition of ATFP and ATLA to understand why they are susceptible to degeneration during childbirth and during age-related changes in matrix turnover.
The ATFP is a band of tissue that runs from the ischial spine (part of the pelvis) to the pubic bone in the center and front of the pelvis. It is attached to the inner pelvic muscle (the obturator internus muscle) and holds up the pelvic organs, such as the bladder and rectum [21]. The ATLA, often referred to as the second white line, originates in association with the iliococcygeus muscle [22]. Although these two tendinous arches are mechanically coupled, they fulfil distinct roles. The ATFP primarily resists the sustained tensile loads associated with organ suspension. In contrast, the ATLA accommodates the cyclic deformation and force transmission generated by active muscle contraction.
At the molecular level, the ECM of both ATFP and ATLA is dominated by structural proteins that jointly determine stiffness, extensibility, and resistance to fatigue damage. Collagen is the main load-bearing component, with type I collagen (COLI) accounting for 85–90% of the total collagen content and providing high tensile strength under mechanical loading [1]. Type III collagen (COLIII) contributes to compliant behavior and is commonly associated with matrix remodeling following microtrauma [23]. Minor collagens, including types IV, V, and VI, are involved in the organization of basement membranes and the assembly of fibrils, thereby supporting cell–matrix interactions and matrix architecture [24]. Elastin, which typically comprises 1–2% of the ECM, provides recoil and elastic recovery, enabling adaptation to cyclic deformation during fluctuations in intra-abdominal pressure [25]. Meanwhile, glycoproteins such as fibronectin help to form fibrils and attach cells to each other. Proteoglycans, including decorin and versican, control the spacing of fibrils, the amount of water they contain, and their stretchiness [26]. These different components work together to provide a balance between stability and flexibility, which is important for supporting the body’s weight.
Maintenance and remodeling of the ECM is predominantly mediated by tenocytes (specialized fibroblasts aligned along collagen fibrils) that coordinate matrix synthesis, organization, and turnover in response to mechanical cues [3, 27]. Interfascicular tenocytes help to connect tissues at the points where they meet, while intrafascicular tenocytes control fiber alignment and strength of the tissues. Tendon stem/progenitor cells (TSPCs) are found in special areas around blood vessels. They can create new cells and also become different types of cells. They are distinguished from mature tenocytes (a type of connective tissue cell) by their expression of markers such as scleraxis (Scx) and tenomodulin (Tnmd). When TSPCs are under pressure or have been injured, they can move towards the damaged area and transform into cells that help to repair the tissue [28]. However, when conditions worsen, the cell function changes, the matrix composition is altered, or stressors in the body induce incorrect repair of the cell populations, this can lead to problems with the ECM.
Although ATFP and ATLA share core ECM constituents, the available evidence suggests their molecular composition is adapted to their distinct mechanical demands. The ATFP anchors the endopelvic fascia to the pelvic sidewall and is enriched in COLI and fibronectin, consistent with it being a suspensory structure optimized for resisting sustained tensile loads [29]. The ATLA has more elastin and proteoglycan than other similar tissues, helping it to support the pelvic floor during contraction [5]. At the levator ani attachment zone, fibrochondrocytes embedded within a collagen-rich matrix may facilitate graded load transfer between muscle contraction and the tendon-like arch. In contrast, the ATFP-endopelvic fascia interface is dominated by fibroblasts that maintain fascial integrity and flexibility by continuously producing ECM [30]. A comparative summary of the anatomical, molecular, and biomechanical features of ATFP and ATLA is provided in Table 1 (Ref, [4, 5, 6, 14, 15, 16, 19, 21, 22, 23, 27, 31, 32, 33, 34]).
| Category | ATFP | ATLA |
| Location | Extends from the ischial spine to the pubic bone along the obturator internus muscle [4, 6]. | Originates from the iliococcygeus muscle, serving as the attachment site for the levator ani muscles [5, 21]. |
| Function | Anchors the endopelvic fascia, supports pelvic organs (e.g., bladder, uterus), and maintains pelvic suspension [6, 16]. | Provides dynamic support during pelvic floor contractions, stabilizes the pelvic floor under intra-abdominal pressure, and aids in continence [21, 22]. |
| Structural Features | Fibrous tendineus arch rich in dense collagen fibers [6]. | More elastic tendineus arch to accommodate dynamic movements [5]. |
| Tightly connected to the obturator internus for static support [4]. | Directly linked to the levator ani muscles for force transmission [21]. | |
| Extracellular Matrix (ECM) | Higher collagen type I (COLI) (85–90% of collagen content). | Collagen type I (COLI) (85–90% of collagen content). |
| Collagen type III (COLIII) [5, 15]. | Higher collagen type III (COLIII) [5, 15]. | |
| Minor collagen types IV, V, VI [15, 16]. | Minor collagen types IV, V, VI. | |
| Higher Fibronectin [15, 31]. | Higher Elastin [15, 16]. | |
| Elastin. | Proteoglycans (e.g. decorin, versican) [14, 32]. | |
| Proteoglycans (e.g. decorin, versican) [14, 32]. | ||
| Cellular Components | Dominated by tenocytes, which are specialized fibroblasts [15, 16, 19]. | Dominated by tenocytes, which are specialized fibroblasts [15, 16, 19]. |
| Fibro-chondrocytes (at muscle-tendon junctions, adapting to transitional stress) [15, 21]. | Fibro-chondrocytes (at muscle-tendon junctions, adapting to transitional stress) [15, 21]. | |
| Tendon stem/progenitor cells (TSPCs) involved in repair [19]. | Tendon stem/progenitor cells (TSPCs) involved in repair [19]. | |
| Biomechanical Properties | High tensile strength (15–20 MPa). | Moderate tensile strength (8–12 MPa). |
| Low elasticity. | High elasticity (elastic modulus: 5–8 MPa). | |
| Stress-strain curve characterized by steep linear phase (resistance to static loading) [14]. | Stress-strain curve with prominent elastic phase (adaptation to cyclic contraction) [5, 32]. | |
| Vulnerability | Prone to overstretching or avulsion during childbirth (e.g., instrumental delivery, macrosomia) and collagen degradation due to aging [33, 34]. | Susceptible to levator ani muscle detachment during vaginal delivery, chronic inflammation, or oxidative stress-induced loss of elasticity [23, 27, 33]. |
The ATFP primarily functions as a static suspensory structure for pelvic organs, with mechanical competence largely determined by its collagen I–rich extracellular matrix that confers high tensile strength. In contrast, the ATLA supports dynamic pelvic floor function by transmitting levator ani contractile forces and exhibits greater elastic adaptability, consistent with its relatively higher elastin and proteoglycan content. Despite these functional differences, both structures share key regulatory features, including sensitivity to hormonal remodeling (relaxin and estrogen), responsiveness to mechanical loading, and modulation by systemic metabolic state. These shared properties provide a mechanistic basis for their vulnerability to load-induced injury during the peripartum susceptibility window and to progressive degeneration with aging.
ATFP and ATLA are both tendon-like pelvic structures with collagen I–dominant ECMs whose organization and cellular composition support load transmission and adaptive remodeling under physiological strain [1, 22, 26]. The extent to which the reported compositional differences between ATFP and ATLA reflect intrinsic specialization rather than inter-individual, methodological, or age-related variability remains unclear [5]. Research that examines the structure of these tissues, their component cells, and how they behave, and the forces acting on them, may reveal a direct link between the tissue architecture and how easily ATFP may be damaged compared to ATLA.
Progressive ECM remodeling is the central pathological process in pelvic tendinopathy, affecting the ATFP and ATLA. There is now substantial evidence that ATFP degeneration is not caused by a single factor, but instead by a combination of mechanical overload and biological modifiers that influence how tissues respond, and how quickly they repair themselves. These modifiers (hormonal remodeling, inflammatory signaling, oxidative stress, and systemic metabolic disturbance) affect matrix homeostasis and cell fate, explaining why similar mechanical exposures can lead to very different structural and clinical outcomes.
Genetic and transcriptomic observations support ECM dysregulation and fibroblast survival as core elements of pelvic floor connective-tissue failure. In the first exome chip study of POP, Kluivers et al. [35] identified four interrelated biological processes operating across epithelial cells, fibroblasts, and the surrounding ECM of the female urogenital tract: epithelial–mesenchymal transition, immune response activation, ECM modulation, and fibroblast survival/apoptosis. Since these processes are very similar to the degenerative cascades described in other load-bearing connective tissues, ATFP degeneration can be viewed as a failure of adaptive remodeling rather than as an isolated structural rupture.
Pregnancy and childbirth can lead to hormonal and molecular changes that overlap with features seen in pelvic floor problems, such as immune system activation and changes in how the body regulates connective tissue [36, 37]. During the peripartum susceptibility window—defined here as the interval from late pregnancy through early postpartum, and characterized by accelerated connective-tissue remodeling under high mechanical demand—elevated relaxin and estrogen levels promote matrix softening and extensibility to accommodate parturition [38]. The matrix metalloproteinases (MMPs) showing the strongest association with POP are MMP-1, MMP-2, and MMP-9. MMP-1 is an interstitial collagenase that can cleave COLI, while MMP-2 and MMP-9 are gelatin-degrading enzymes that can also break down the cleavage products of MMP-1 [39]. Relaxin has been shown to activate MMPs, particularly MMP-1 and MMP-13, causing collagen to break down and reduce skin strength [40].
It is very important to consider the context in studies of pelvic tendinopathy. Ferlin et al. [40] showed that relaxin activates a specific protein in tendon fibroblasts. Estrogen also makes tissue more elastic and increases the flexibility of connective tissue. These changes help the body cope with childbirth, but can also make it more difficult to resist excessive strain under high mechanical loads [41]. Substantial inter-individual variability exists, and not all women with elevated levels of peripartum relaxin develop pelvic tendinopathy. This suggests the presence of modifying factors, such as genetic variation (e.g., COL1A1 polymorphisms [42]) and the quality of the baseline matrix. Notably, much of the mechanistic evidence is derived from animal or non-pelvic tendon models [40], indicating a need for longitudinal human studies that directly examine ATFP responsiveness across the pregnancy and postpartum recovery periods.
Of all the contributing factors, mechanical overload is the most direct and consistently implicated trigger of ATFP and ATLA injury. Vaginal delivery can expose these tendinous arches to forces that exceed physiological tolerance, resulting in microtears, partial- or full-thickness disruption, or avulsion injuries, particularly at the levator ani attachment to the ATLA [15, 16]. Prolonged labor, fetal macrosomia, and operative deliveries using forceps or vacuum extraction further increase the strain magnitude and duration, and hence the risk of structural compromise.
Experimental studies in animals demonstrate that excessive mechanical loading suppresses gene programs associated with orderly wound healing, while activating stress-responsive and inflammatory mediators such as prostaglandin endoperoxide synthase 2 (Ptgs2), leading to impaired repair [43]. Complementary in vitro and in vivo data show that mechanically stressed tendon cells release extracellular vesicles that promote macrophage recruitment and amplify local immune signaling [44]. While these observations derive primarily from non-pelvic tendon systems, they offer insights into how acute overload can trigger a cascade of maladaptive responses in the ATFP, particularly when it coincides with the hormone-sensitive peripartum state.
The absence of a classic acute inflammatory infiltrate does not exclude a role for inflammation in pelvic tendinopathy [45]. Instead, inflammation is increasingly being conceptualized as a phase-dependent modulator whose influence varies throughout the processes of injury initiation, propagation, and chronic degeneration. This concept reconciles conflicting views in the literature. While some researchers emphasize inflammation as a key driver of tendinopathy, others characterize chronic disease as primarily degenerative [46].
Molecular evidence suggests that inflammatory signaling is most prominent during
the early, often asymptomatic stages of repetitive microtrauma [21].
Pro-inflammatory cytokines (e.g., interleukin-1 beta [IL-1
Macrophages play a key regulatory role in this phase-dependent framework.
Classically activated M1 macrophages are important for early inflammatory
responses, whereas alternatively activated M2 macrophages are important for
resolving inflammation and repairing tissue damage. Research into the damage and
repair of animal tendons has revealed that the ‘macrophage’ cell type affects the
speed with which the tendon can be restored and how well it regenerates after
damage [48, 49]. M1 macrophages release cytokines (e.g., IL-1
Oxidative stress, characterized by excessive production of reactive oxygen species (ROS), acts as a secondary amplifier of tendon degeneration by impairing cellular viability and matrix integrity [55]. ROS activate pathways that respond to stress, including c-Jun N-terminal kinase (JNK) phosphorylation, leading to increased MMP activity and accelerated collagen breakdown [56]. At the same time, ROS-induced cell death (apoptosis) reduces the viability of tenocytes and progenitor cells, while senescent cells begin to produce cytokines that perpetuate inflammatory and matrix-degrading signals [32]. The molecular pathogenesis described above is depicted in Fig. 1.
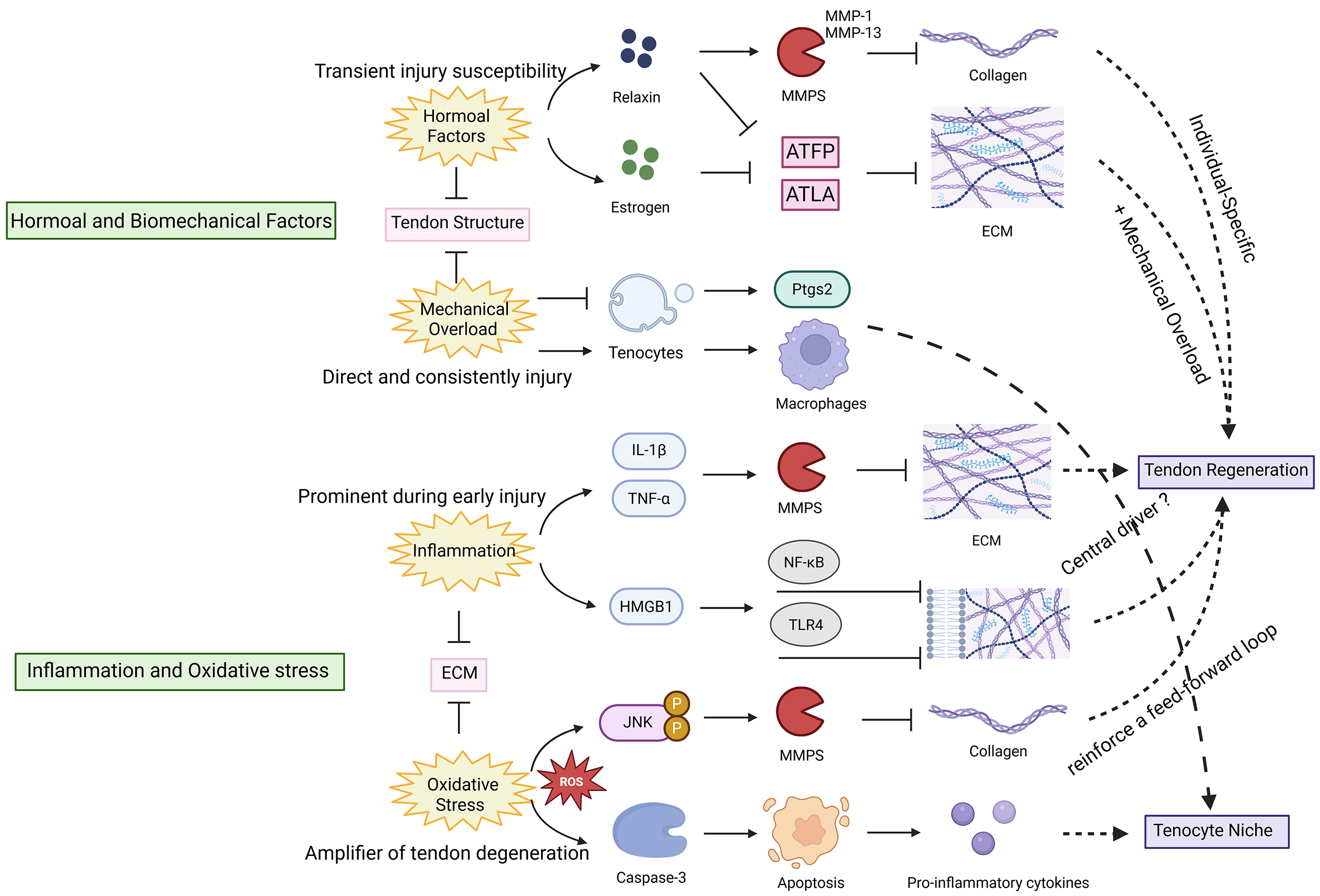 Fig. 1.
Fig. 1.
Hormonal, biomechanical, inflammatory, and oxidative stress
pathways impair ATFP/ATLA regeneration. Tenocyte niche: the microenvironment
supporting tenocyte survival and function. Relaxin activates matrix
metalloproteinases (MMPs), degrading collagen and reducing tendon strength, while
estrogen increases ATFP/ATLA elasticity, predisposing to overextension.
Mechanical overload induces prostaglandin endoperoxide synthase 2 (Ptgs2) and
tendon cell-derived extracellular vesicles, promoting macrophage recruitment and
injury. interleukin-1 beta (IL-1
Systemic metabolic disorders, including diabetes mellitus and
hypercholesterolemia, can also influence tendon homeostasis and repair capacity
[56]. An in vitro study found that high glucose levels induce
mitochondrial dysfunction and ferroptosis—an iron-dependent form of regulated
cell death—in TSPCs, impairing their capacity to regenerate the ECM [57].
Activation of the receptor for advanced glycation end products
(RAGE)/
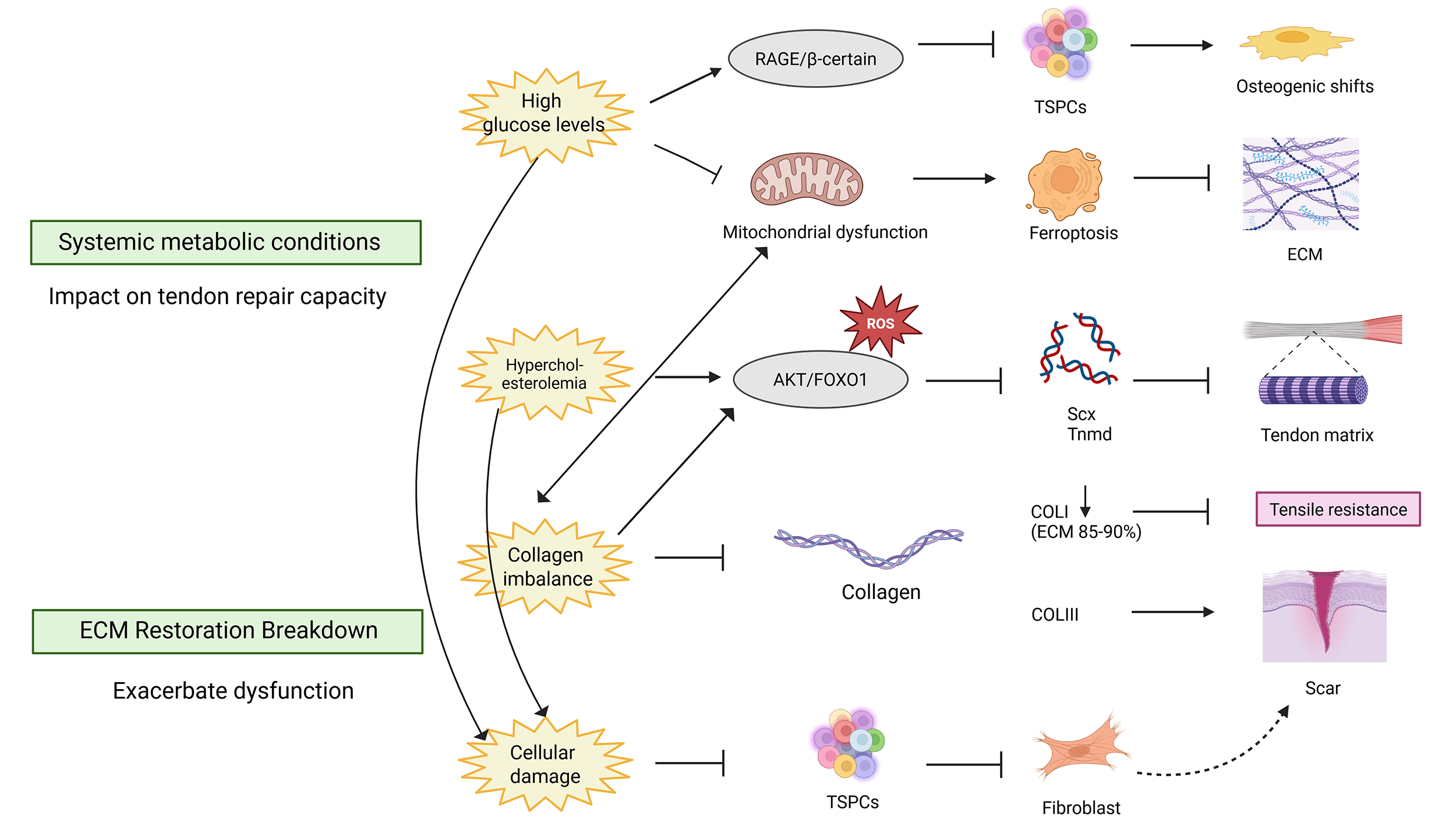 Fig. 2.
Fig. 2.
Systemic metabolic dysregulation and impaired ECM restoration
compromise ATFP/ATLA function. Hyperglycemia activates the receptor for advanced
glycation end products (RAGE)/
Conventional management of tendinopathy includes non-steroidal anti-inflammatory or antioxidant agents, mechanically-based interventions (e.g., shock-wave therapy [60, 61]), and selected biological adjuncts [62]. However, these primarily mitigate symptoms rather than reversing the underlying degenerative biology. Degeneration of the ATFP within the pelvic floor is better understood as a disorder characterized by impaired load adaptation and maladaptive ECM remodeling rather than as an isolated inflammatory condition. This change in perspective shifts the therapeutic focus from short-term symptom suppression to strategies that address the underlying mechanisms. Such strategies aim to preserve or restore the organization of the ECM while respecting the constraints imposed by tissue mechanics and intrinsic repair capacity.
Within this framework, inflammation, oxidative stress, and metabolic disturbance are best considered as phase-dependent modulators that influence the course of degeneration and repair following a mechanically-induced injury, particularly during the defined peripartum susceptibility period. Fig. 3 illustrates how these modulatory processes intersect with existing and emerging therapeutic concepts. It is important to note that most of the evidence for these treatments comes from early studies or studies on non-pelvic tendons. Therefore, caution is needed when applying this evidence to ATFP pathology.
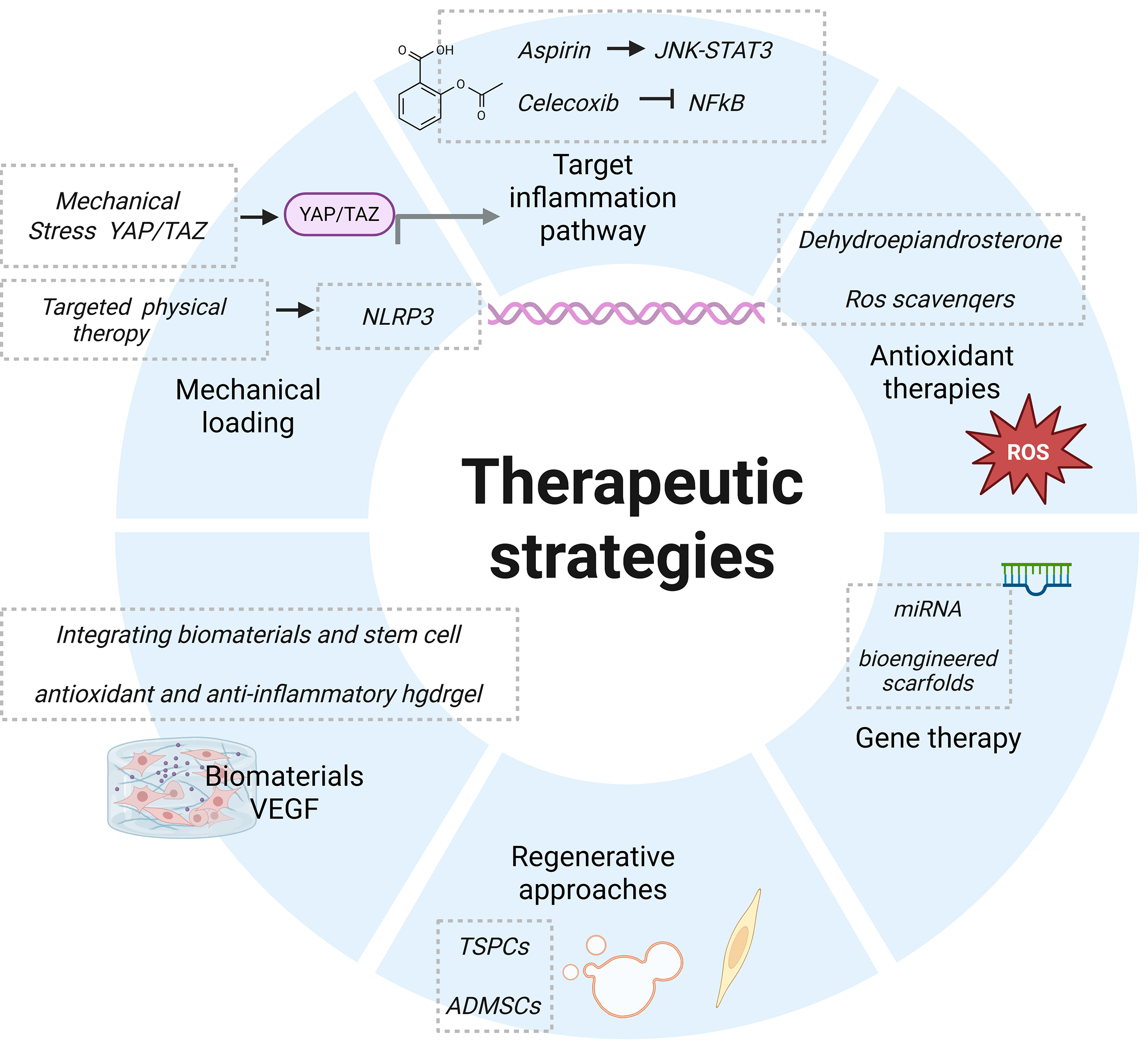 Fig. 3.
Fig. 3.
Future research directions and innovative therapeutic strategies. Promising therapeutic strategies include: targeted anti-inflammatory approaches, antioxidant therapies, regenerative medicine techniques, novel biomaterials, gene therapy, and optimized mechanical loading regimens. VEGF, vascular endothelial growth factor; YAP,yes-associated protein; TAZ, transcriptional coactivator with PDZ-binding motif; NLRP3, NOD-like receptor family, pyrin domain containing protein 3; ADMSCs, adipose-derived mesenchymal stem cells (Created in BioRender. (2025) https://BioRender.com/4tb6cma).
The efficacy of pharmacological modulation of inflammatory signaling has been demonstrated in experimental tendinopathy models. This is consistent with the idea that inflammation regulates matrix turnover rather than being the sole driver of disease. Non-steroidal anti-inflammatory drugs (NSAIDs), such as aspirin, attenuate JNK/signal transducer and activator of transcription 3 (STAT3) signaling and have been reported to reduce inflammation, scar formation, and the risk of re-rupture in injured tendons [63]. Beyond conventional NSAIDs, pristimerin—a quinone methide triterpenoid—has shown superior efficacy to indomethacin in preclinical tendinopathy models by enhancing biomechanical recovery and suppressing inflammation through absent in melanoma 2 (AIM2) inflammasome–PYCARD/apoptosis-associated speck-like protein (ASC) modulation via selective autophagy [64, 65]. Moreover, most studies are based on animal models or small-scale human trials. Large randomized controlled trials (RCTs) to confirm the efficacy of NSAIDs in pelvic tendinopathy are still lacking.
Despite these encouraging mechanistic findings, their relevance to pelvic tendinopathy remains uncertain. Long-term use of NSAIDs can prolong the time needed for tendons to heal, and may also cause problems in other parts of the body. Most studies supporting the use of NSAIDs are based on animal experiments or on small numbers of people.
Oxidative stress worsens degeneration after mechanical injury, which is why antioxidant treatments are useful. Agents such as dehydroepiandrosterone and ROS scavengers protect tendon cells from damage and reduce calcium accumulation in experimental tendinopathy models [61]. However, most supporting studies rely on animal models or limited human cohorts rather than pelvic-specific randomized trials.
Oxidative stress acts as an amplifier of degeneration once mechanical injury has occurred, providing a rationale for antioxidant-based interventions. Agents such as dehydroepiandrosterone and ROS scavengers protect tenocytes from oxidative damage and reduce ectopic calcification in experimental tendinopathy models [55]. Epigallocatechin gallate has been shown to mitigate oxidative injury, prevent non-tumorigenic differentiation, and promote ECM repair. However, its effects are dose-dependent, and a high concentration may actually inhibit tenocyte proliferation [66]. These observations underscore the challenge of attenuating pathological ROS accumulation without disrupting the physiological redox signaling required for normal repair processes [67].
A multi-center study found that physical labor, vaginal delivery, diabetes, and a family history of POP were all important factors in weakening of the pelvic floor muscles. On the other hand, PFMT and sexual activity were found to have a protective effect [68]. Mechanical loading represents another non-pharmacological lever with dual effects on tendon biology. Excessive or poorly distributed loading promotes degeneration, whereas controlled mechanical stimulation supports collagen alignment and matrix reorganization [69]. Mechanical loading is another way to treat tendon problems without using drugs. It has two effects on tendon biology. Too much or too little loading can cause problems, but controlled mechanical stimulation can help to align collagen and reorganize the matrix [69]. The interaction between yes-associated protein (YAP)/transcriptional coactivator with PDZ-binding motif (TAZ) and other proteins is important for maintaining chromosome structure and preventing chromosome breakage under stress [42]. Non-surgical rehabilitation remains the most important treatment, and new methods such as percutaneous needle electrolysis have been reported to activate the NOD-like receptor family, pyrin domain containing protein 3 (NLRP3) inflammasome and stimulate collagen-mediated regeneration in experimental studies [70].
However, the optimal loading parameters for pelvic tendinopathy—including intensity, frequency, and duration—have yet to be defined. The application of too much pressure on the pelvic floor can worsen degeneration, but the application of insufficient pressure could mean that it takes longer to recover [71]. Personalized mechanotherapy, possibly guided by imaging or sensor feedback, is a promising yet underexplored strategy.
The two strategies that aim to repair torn tendons are ‘regenerative’ and ‘molecularly targeted’. Both aim to repair the tissue around the torn tendon and to restore cell function within the tissue. However, with regard to degeneration of the Achilles tendon fascicle, these methods are still largely in the experimental stage. Consequently, the discussion here is confined to their mechanistic relevance and biological feasibility, rather than clinical endorsement. It is important to note that most regenerative concepts have been developed in non-pelvic tendon systems, where the loading environment, tissue geometry, and repair constraints differ substantially from those of the pelvic floor.
Cell-based strategies have primarily focused on increasing matrix synthesis and preserving tenogenic cell populations. TSPCs contribute to the restoration of the ECM by depositing collagen and fibronectin at injury sites [72]. Encapsulating TSPCs within ECM-mimetic hydrogels enhances cell retention and activates transcriptional programs linked to repair and matrix organization [73]. Similarly, adipose-derived mesenchymal stem cells (ADMSCs) and their extracellular vesicles have demonstrated the capacity to improve biomechanical properties, reduce apoptosis, and restore mitochondrial function in oxidatively stressed tendon models [74]. However, donor age, cell heterogeneity, and variability in delivery systems can all markedly influence outcomes, highlighting the need for rigorous standardization [74]. Furthermore, subcellular mechanisms, such as the transfer of mitochondria from mesenchymal stem cells to tenocytes, demonstrate how cell-based interventions can enhance survival without directly inducing regeneration [75]. Although a systematic review of 11 clinical studies reported architectural improvement in injured tendons following cell therapy [76], pelvic-specific evidence is still lacking, thus limiting direct extrapolation to ATFP degeneration.
Gene-based modulation offers a theoretically precise means of targeting
degenerative pathways, but faces substantial translational barriers. MicroRNAs
such as miR-146a have been shown to protect tenocytes from senescence through
suppression of interleukin-1 receptor-associated kinase 4 (IRAK-4)/TNF
receptor-associated factor 6 (TRAF6)/NF-
Bioactive materials and advanced delivery platforms aim to combine mechanical support with biological modulation. Scaffolds made from a type of plastic called Poly (lactic-co-glycolic acid) (PLGA) can alter macrophage behavior and help to reduce inflammation in models of tendon injury [79]. Decellularized ECM scaffolds can be adjusted according to their mechanical properties and the speed at which they break down, providing early support for tissue as it changes shape [80, 81].
Adding antioxidant or anti-inflammatory hydrogel microspheres allows control of specific stressors that depend on phase, such as oxidative and inflammatory signaling [67, 73]. Growth factor-functionalized scaffolds, including those incorporating vascular endothelial growth factor (VEGF) or platelet-derived growth factor (PDGF), enhance blood vessel formation and repair. However, too many new blood vessels may lead to scarring rather than helping the body to heal, highlighting the need for controlled blood vessel growth [82, 83].
Nanoparticle-based systems can help by releasing the drug over a longer period, reaching deeper into the tendon, and causing less tissue damage when injected [84, 85, 86]. Notably, lipid nanoparticles carrying SMAD family member 3 (SMAD3) siRNA and collagen I mRNA have shown combined antifibrotic and regenerative effects in human tenocyte models, demonstrating the potential of dual-function molecular payloads [87].
Taken together, these findings reinforce the principle that mechanical overload is the main cause of ATFP degeneration. Hormonal remodeling during the peripartum susceptibility window, along with other phase-dependent factors, then shapes the subsequent repair trajectories. Progress in this field is therefore unlikely to result from the widespread application of regenerative technologies in isolation. Instead, meaningful advances will depend on the precise alignment of intervention timing, molecular target selection, and the tissue-specific mechanical context with the underlying biology of ATFP degeneration.
Cell-, gene-, and biomaterial-based strategies can modulate ECM organization, cell survival, and inflammatory tone in experimental tendon models. However, the safety, durability, and biomechanical compatibility of these approaches in the pelvic floor, particularly in the ATFP, remain unproven. To evaluate the progression of this condition using regenerative or molecularly targeted strategies, preclinical studies with pelvic models must include physiological loading and long-term functional outcomes.
A major limitation of the current review is the heavy reliance on mechanistic evidence derived from non-pelvic tendon models, such as the Achilles tendon, or preclinical animal systems. These models provide a basic understanding of general tendinopathy. However, the way the body moves, the shape of the tissue, and the way it is repaired are very different in the pelvic floor compared to the extremities. This means that we cannot take what we learn about matrix turnover and cellular signalling in the human ATFP and just apply it elsewhere without careful thought first. The unique loading patterns and specific biological modifiers in the pelvis may significantly alter the observed degenerative pathways.
There is also a considerable scarcity of pelvic-specific human data. In particular, there is little information on longitudinal molecular and architectural transitions during the peripartum susceptibility window or throughout ageing. Prevailing clinical phenotypes, including pelvic organ prolapse and stress urinary incontinence, do not currently differentiate tendon-like connective tissue failure from broader muscular or fascial dysfunction. This lack of anatomical specificity in the clinical literature complicates the identification of the ATFP as a distinct biological substrate. It also hinders the development of clinically deployable tools for mechanism-based risk stratification, early detection, and objective monitoring of tissue condition.
A substantial translational gap persists between emerging regenerative strategies and validated clinical outcomes. Molecularly targeted approaches, including cell-based therapies, gene modulation and bioactive scaffolds, have shown promise in experimental settings. However, the long-term safety, durability and biomechanical compatibility of these approaches within the human pelvic floor remain to be proven. There is a lack of large-scale randomised controlled trials and multicentre registries in this field, which would be necessary to verify the efficacy of these interventions and establish standardised structural endpoints for repair. Without this clinical validation, many of the advanced repair strategies discussed here remain hypothetical rather than forming part of established treatment modalities.
ATFP degeneration constitutes a clinically important yet frequently under-recognized structural substrate underlying pelvic floor instability and related symptom phenotypes. Mechanistic evidence supports a model in which mechanical overload, most prominently during childbirth, initiates microdamage. A definable peripartum susceptibility window lowers the threshold for maladaptive repair. This spans the period from late pregnancy through to early postpartum and is distinguished by fast connective-tissue remodeling under high mechanical demand. Disease progression is subsequently determined by phase-dependent modulators that bias matrix turnover and cell-state transitions, including inflammatory signaling, oxidative and mitochondrial stress, and systemic metabolic disturbance. Across these stages, a convergent pathological endpoint emerges: dysregulated ECM remodeling accompanied by progressive loss of mechanical competence.
Despite advances in imaging that provide better visualization of pelvic tendinous arches, clinically deployable tools for mechanism-based risk stratification, early detection, and longitudinal monitoring remain limited. Current interventions, including rehabilitation approaches and surgical repair, primarily address symptoms or macroscopic support failure rather than directly correcting the underlying degenerative biology or restoring durable tissue-level mechanics. Accordingly, the most immediate translational priority is not the broad application of regenerative technologies, but rather the generation of pelvic-specific evidence linking ATFP structural alterations to molecular programs and functional outcomes. Such linkage is key to determining when, where, and in whom the specific treatments might be effective.
Future progress will depend on integration across evidence tiers. Human imaging and tissue-based studies should be prioritized to establish ATFP-specific degeneration signatures and reduce the current reliance on extrapolation from non-pelvic tendon models. Mechanistic insights from animal and in vitro systems remain essential for elucidating causality and pathway interactions, but should be regarded as hypothesis-generating unless validated in pelvic tissues. The convergence of multi-omics profiling, advanced biomechanical modeling, and mechanobiological experimentation should enable the identification of testable nodes within matrix homeostasis, mechano-transduction, and stress-response pathways. These approaches may inform both prophylactic measures (such as intrapartum protection and postpartum load optimization) and biology-aligned repair concepts. Nonetheless, the translational gap remains substantial. Many candidate interventions show promise in preclinical settings, but lack strong clinical validation. In addition, heterogeneity in age, obstetric exposure, and comorbidities continues to complicate generalization.
To move the field forward in a systematic and clinically meaningful manner, we propose three priorities: (1) establishment of multicenter registries that integrate longitudinal clinical phenotyping with standardized imaging of ATFP/ATLA integrity; (2) development and validation of biomarkers capable of detecting early degeneration and predicting progression, ideally anchored to tissue-based molecular correlates; and (3) implementation of comparative effectiveness studies of existing management strategies that incorporate mechanism-informed stratification and objective structural endpoints.
RC and TC contributed to the design of this work. RC, TC, QT, YZ, XGH and WW reviewed the literature. RC, TC and WW organized the work. RC, TC and YZ created the figures. WW, RC, TC, QT, YZ and XGH drafted the work. RC, TC, QT and YZ revised critically for important intellectual content. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We thank BioRender (https://app.biorender.com/) for providing the platform used to create the graphical abstract and figures. The illustrations were created with BioRender.com under an academic license.
This work was supported by the National Natural Science Foundation of Qingdao Municipality (24-4-4-zrjj-119-jch), the National Natural Science Foundation of Shandong Province (ZR2024QH209).
The authors declare no conflict of interest.
During the preparation of this work the authors used ChatGpt-3.5 in order to check spell and grammar. After using this tool, the authors reviewed and edited the content as needed and takes full responsibility for the content of the publication.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.