







 , Juan Yang 1,†, Jing Guo 2, Xuqin Wang 1,*
, Juan Yang 1,†, Jing Guo 2, Xuqin Wang 1,*
1 Department of Children’s Health, The Third Affiliated Hospital of Zunyi Medical University/The First People’s Hospital of Zunyi, 563002 Zunyi, Guizhou, China
2 Department of Gynecology and Obstetrics, The Third Affiliated Hospital of Zunyi Medical University/The First People’s Hospital of Zunyi, 563002 Zunyi, Guizhou, China
†These authors contributed equally.
Abstract
The incidence of gestational diabetes mellitus (GDM) has increased in China in recent years. Evidence suggests that gut microbiota composition plays a role in the development of GDM. This study aimed to characterize the gut microbiota in pregnant women with GDM using high-throughput sequencing methods.
A total of 60 pregnant women, comprising 30 diagnosed with GDM and 30 healthy controls, were enrolled at our hospital in this study, conducted from September 2021 to August 2022. Fecal samples were collected for DNA extraction, followed by amplification through polymerase chain reaction (PCR). High-throughput sequencing was then performed to analyze the 16S rDNA V3–V4 fragments in the fecal samples.
The diversity of the gut microbiota showed that the Ace, Chao, and Sobs indices were higher in the GDM group compared with those in the healthy control group (p < 0.05). The Shannon index was also significantly higher in the GDM group (p < 0.05). At the phylum level, the gut microbial communities in both groups predominantly comprised the following four phyla: Firmicutes, Actinobacteria, Proteobacteria, and Bacteroidota. Compared with the healthy control group, the relative abundance of Firmicutes and Bacteroidota was significantly higher in the GDM group (p < 0.05), whereas Actinobacteria and Proteobacteria were significantly lower (p < 0.05). At the genus level, the profusion of Shigella, Romboutsia, Paraprevotella, unclassified Clostridium UCG-014, and Trichospira increased significantly in the GDM group (p < 0.05).
During the third trimester, the intestinal microbial communities of diabetic pregnant women differ significantly from those of healthy pregnant women. In the intestinal flora of patients with GDM, the abundance of Firmicutes and Bacteroidota increased significantly, while that of Actinobacteria and Bifidobacterium decreased significantly. These findings suggest that the occurrence of GDM is accompanied by a reduction in the beneficial bacterium Bifidobacterium.
Keywords
- gestational diabetes mellitus
- gut microbiota
- case-control (α-diversity)
- high-throughput sequencing
Gestational diabetes mellitus (GDM), a prevalent complication in pregnancy, represents a disorder of glucose metabolism during pregnancy [1]. With economic development and the increasing number of people with overweight and obesity worldwide, approximately 7% of all pregnant women suffer from GDM. The incidence of GDM has escalated significantly in China, reaching 17.5%, partly due to the implementation of the two-child and three-child policies and partly because of the increasing incidence of geriatric pregnancy in recent years [2]. GDM has adverse outcomes for mothers, such as preeclampsia, abortion, cardiovascular disease, and type 2 diabetes mellitus (T2DM). In addition, it casts short- and long-term effects on the health of the offspring too [3], including macrosomia, preterm birth, deformity, overweight and obesity, insulin resistance, and neurocognitive decline. Therefore, pregnant women must undergo an oral glucose tolerance test (OGTT) during mid-pregnancy. Once diagnosed, doctors can immediately provide professional dietary and exercise guidance to such patients. Notably, patients with GDM tend to have good control of blood glucose. At the same time, there was no reduction in the incidence of adverse pregnancy outcomes, suggesting that other factors may also be responsible for adverse pregnancy outcomes. However, this is not the situation when patients have high blood glucose levels during their pregnancies.
Some recent studies based on metagenomics and metabolomics have shown that intestinal flora imbalance is related to many metabolic disorders, such as T2DM and GDM [4, 5]. Koren et al. [6] studied gut microbiota across different pregnancy stages in 91 healthy pregnant women and found that the overall content of Proteobacteria and Actinobacteria increased in late pregnancy while the overall abundance of the microbiota decreased. Such alterations may contribute to metabolic dysfunction and GDM. Crusell et al. [7] reported higher levels of Actinobacteria, Collinsella, Rothia, and Desulfovibrio during the third trimester of GDM compared to those in healthy controls.
However, a study has reported conflicting results regarding the gut microbiota in patients with GDM. One case-control study showed that, compared to pregnant women with normal blood sugar, those with GDM had decreased abundance of Bacteroidota and Actinobacteria [8]. In contrast, another study found no statistically significant difference in the gut microbiota of the two sets of pregnant women [9]. The previous research results are inconsistent or contradictory because of the significant differences in the design and sample size of the studies. Gut microbiota composition is influenced by various factors, including region, age, dietary habits, economic status, and gestational age. Therefore, in studies investigating the gut microbiota of patients with GDM, minimizing the impact of potential confounding factors is crucial for the accurate interpretation of results. Given the marked rise in the incidence of GDM in China, it is essential to understand the intestinal flora characteristics of patients with GDM.
In this study, we employed high-throughput sequencing technology to analyze the gut microbiota, aiming to compare the microbial composition between age-, body weight-, and gestational age-matched Chinese women with GDM and healthy pregnant controls in late pregnancy, providing a reference for subsequent interventional research on gut microbiota in pregnant women with GDM.
This prospective cohort study enrolled 30 GDM patients (GDM group) who delivered
at the Zunyi First People’s Hospital between September 2021 and August 2022. The
diagnostic criteria adhered to the Chinese Guidelines for the Diagnosis and
Treatment of Pregnancy Complicated with Diabetes (2014) [10]. To minimize the
influence of confounding factors, a control group was established comprising 30
healthy pregnant women who matched the GDM group in terms of age, pre-pregnancy
body mass index (BMI), and timing of pregnancy and who also underwent prenatal
examination and hospital delivery at the Third Affiliated Hospital of Zunyi
Medical University (the First People’s Hospital of Zunyi) during the same period.
Matching tolerances were defined as follows:
The following were employed as the inclusion criteria for selecting patients for the GDM group: (1) the patients must meet the diagnostic criteria outlined by the Chinese Guidelines for Diagnosis and Treatment of Pregnancy Complicated with Diabetes (2014); (2) Han ethnicity, aged 20–45 years; (3) natural conception, single full-term pregnancy (37–40 weeks); (4) only dietary and exercise guidance needed to control blood glucose; and (5) did not use antibiotics or probiotic preparations in the past 2 weeks. The exclusion criteria were as follows: (1) diabetes, hypertension, psychosis, hyperthyroidism, cardiovascular, and kidney diseases before pregnancy; (2) presence of other complications besides GDM; (3) requirement of insulin therapy in addition to dietary and exercise guidance; and (4) a history of smoking, alcohol, and substance abuse.
Data related to variables such as age, length of pregnancy, pre-pregnancy BMI, education level, and economic income were collected from the outpatient and inpatient systems of the hospital. The BMI was determined by the following formula: weight/height2. All the patients were required to continuously record their dietary habits for the past three days, including the type, frequency, and intake of foods. We converted the food intake into nutrient intake based on the Chinese Food Composition Table 2004 and calculated the daily intake of energy, protein, fat, carbohydrates, and dietary fiber.
Fecal samples were procured from all participants on the morning of the second day after admission, collected in sterile tubes, and transported to the laboratory within 1 h for processing. Approximately 500 mg of each sample was placed into a sterile EP tube. Subsequently, the samples were stored in a –80 °C freezer for further analysis. A fecal DNA extraction kit (QIAGEN, Hilden, Germany) was used to extract the fecal bacterial DNA, following the manufacturer’s protocol. Agarose gel electrophoresis (1%) was utilized to identify impurities and degradation products in the genome, assessing its size and integrity. Concentration and purity of DNA samples were measured using a NanoDrop spectrophotometer (UV-1800, Shimadzu, Kyoto, Japan). The MiSeq sequencer (PE300, Illumina, San Diego, CA, USA) was employed to amplify the hypervariable region of 16S rDNA V3–V4 using the 338F/806R primers, followed by second-generation sequencing.
The raw sequencing data generated in this study have been deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under the BioProject accession number PRJNA1338062.
Analyses were executed using SPSS 29.0 (IBM Corp., Armonk, NY, USA), with
statistical significance set at p
Biological information of the gut microbiota was analyzed by processing
microbiota data using the Illumina MiSeq system. The sequencing raw data were
input into the QIIME2 software (version 2024.5, QIIME2 Development Team, https://qiime2.org) for processing and analysis.
Following quality filtration, the sequences were clustered into operational
taxonomic units with a 99% identity threshold, derived from the Greengenes
database (version 13.8, http://greengenes.microbio.me/greengenes_release/gg_13_8_otus/). Alpha, beta, and linear discriminant function analyses
(LEfSe) were conducted to identify differential foreign species at the genus
level. The analyses employed Mothur software (version 1.30.2, developed by the Mothur Project Team, https://mothur.org), QIIME2 software,
and LEfSe software (version 1.0, developed by the Segata Lab, http://huttenhower.sph.harvard.edu/lefse), where alpha and beta analyses with p
This prospective cohort study recruited 60 pregnant women in the late stage of
pregnancy. Of these 60 participants, 30 were placed in the GDM group and the
remaining 30 in the control group. Their age, duration of pregnancy,
pre-pregnancy BMI, third-trimester BMI, gestational weight gain, daily intake of
total energy, protein, fat, carbohydrates, and dietary fiber and education levels
were matched (p
| Variable | GDM (n = 30) | Control (n = 30) | p | |
| Age (years) | 29.52 |
29.40 |
0.935 | |
| Gestational age (weeks) | 39.02 |
39.17 |
0.531 | |
| BMI in the last trimester (kg/m2) | 29.70 |
29.36 |
0.729 | |
| Gestational weight gain (kg) | 14.32 |
14.01 |
0.687 | |
| Fetal birth weight (kg) | 3.53 |
3.23 |
0.015 | |
| Education level | 0.258 | |||
| Elementary and below | 5 (16.7%) | 5 (16.7%) | ||
| Secondary | 12 (40%) | 13 (43%) | ||
| High school | 9 (30%) | 6 (20%) | ||
| University and above | 4 (13%) | 6 (20%) | ||
| Fasting glucose (mmol/L) | 4.70 |
4.11 |
||
| Insulin (pmol/L) | 63.43 (58.05, 27.70) | 53.34 (53.95, 16.50) | 0.025 | |
| OGTT (mmol/L) | ||||
| OGTT 0 h | 5.05 |
4.56 |
||
| OGTT 1 h | 9.83 |
7.90 |
||
| OGTT 2 h | 8.28 |
6.88 |
||
| TC (mmol/L) | 6.65 (6.00, 9.00) | 6.00 (5.60, 8.55) | 0.373 | |
| TG (mmol/L) | 4.35 (2.93, 5.00) | 3.00 (2.43, 3.98) | 0.014 | |
| HDL-C (mmol/L) | 2.00 (1.83, 2.50) | 2.35 (2.00, 2.85) | 0.107 | |
| LDL-C (mmol/L) | 4.55 (3.73, 5.08) | 4.20 (3.68, 4.50) | 0.254 | |
| Total energy intake (kcal/day) | 1987.20 |
2083.12 |
0.144 | |
| Protein intake (g/day) | 89.55 |
90.50 |
0.843 | |
| Fat intake (g/day) | 71.90 |
74.45 |
0.301 | |
| Carbohydrate intake (g/day) | 181.50 |
188.50 |
0.134 | |
| Dietary fiber intake (g/day) | 32.40 |
30.60 |
0.203 | |
TC, cholesterol; TG, triglyceride; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; BMI, body mass index; GDM, gestational diabetes mellitus; OGTT, oral glucose tolerance test.
Two sets of fecal 16S rDNA samples were subjected to Illumina sequencing to obtain the paired-end (PE) reads. After processing, 11,158 amplicon sequence variants (ASVs) were generated, with an average of 16,456 effective sequences per sample and a sequencing depth of 99%. An analysis of alpha diversity pertaining to the gut microbiota was performed on the two groups of samples at the ASV level.
Ace, Chao, and Sobs indices are typically used for assessing microbial richness,
whereas Shannon and Simpson indices primarily reflect community diversity. The
findings demonstrated that the abundance indices of gut microbiota Ace, Chao, and
Sobs in the GDM group were markedly elevated compared to those in the control
group (p
| Variable | GDM (n = 30) | Control (n = 30) | p |
| Ace | 422 |
309 |
0.010 |
| Chao | 409 |
299 |
0.011 |
| Sobs | 356 |
270 |
0.016 |
| Shannon | 3.84 |
3.52 |
0.009 |
| Simpson | 0.06 |
0.07 |
0.087 |
Ace, Chao, Sobs: Assess the abundance of the microbial community; Shannon and Simpson: Assess the diversity of the microbial community.
Principal coordinate analysis (PCoA) was performed on two groups of samples to explore similarities and dissimilarities in their gut microbiota. The Adonis test was carried out to analyze the differences between the two groups. The analysis revealed that the interpretation rates for principal component 1 (PC1) and principal component 2 (PC2) along the coordinate axis were 17.82% and 12.53%, respectively, demonstrating that the two groups of samples were effectively clustered and discriminated. Furthermore, the separation distance between the two sample groups was substantial, indicating a significant differentiation in the composition of the gut microbiota of the two groups (R = 0.3742, p = 0.001, Fig. 1).
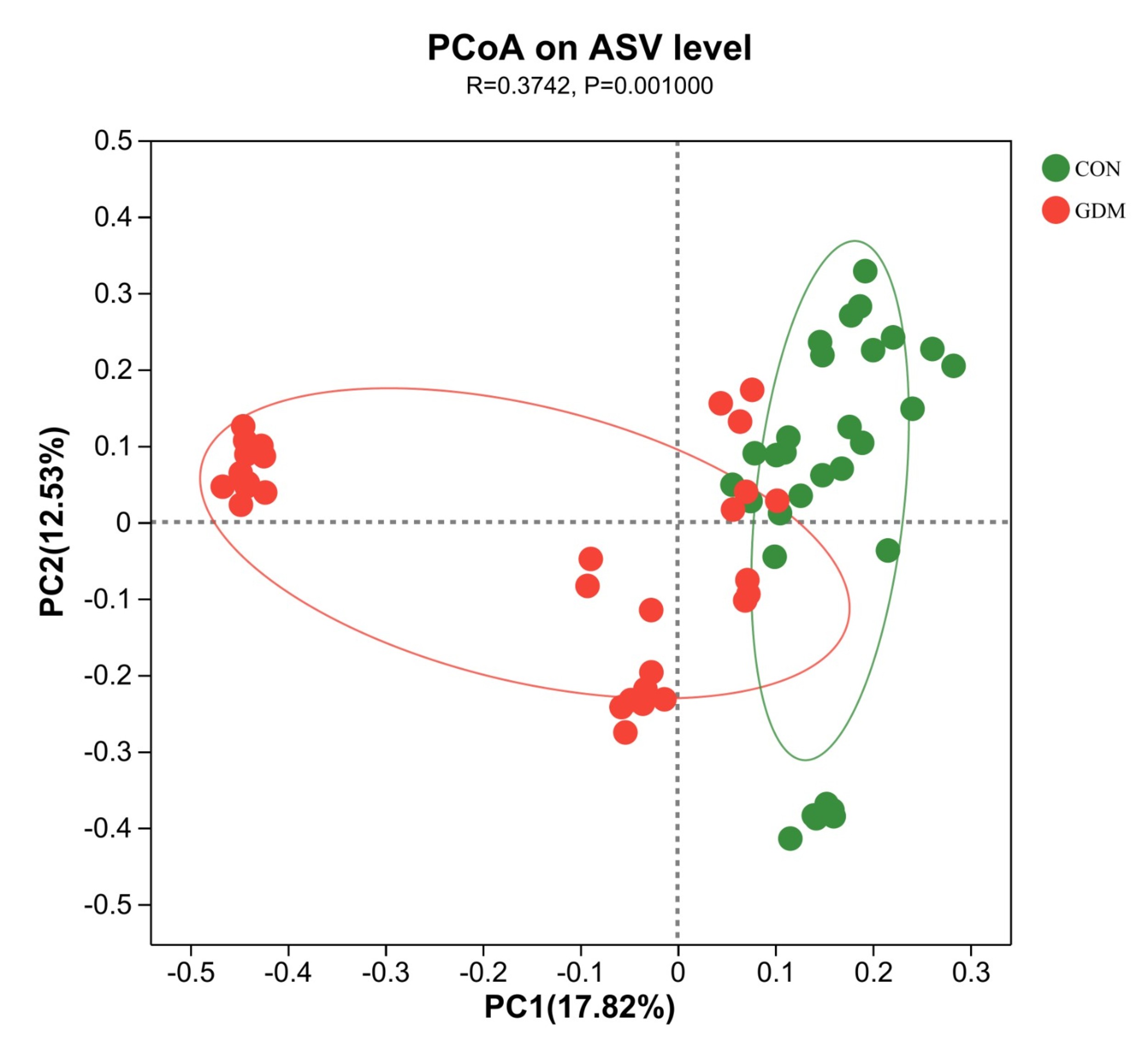 Fig. 1.
Fig. 1.
Principal component analysis of the gut microbiota of healthy pregnant women and pregnant women with gestational diabetes mellitus. PCoA, principal coordinate analysis; ASV, amplicon sequence variant; PC1, principal component 1; PC2, principal component 2; CON, control; GDM, gestational diabetes mellitus.
The Venn diagram illustrates the similarities and intersection of species composition within the fecal samples. The analysis revealed that the microbial communities in the control and GDM groups contained 46 and 41 unique microbial communities, respectively, and 209 overlapping microbial communities. Variations in species distribution between the two sample groups are shown in Fig. 2.
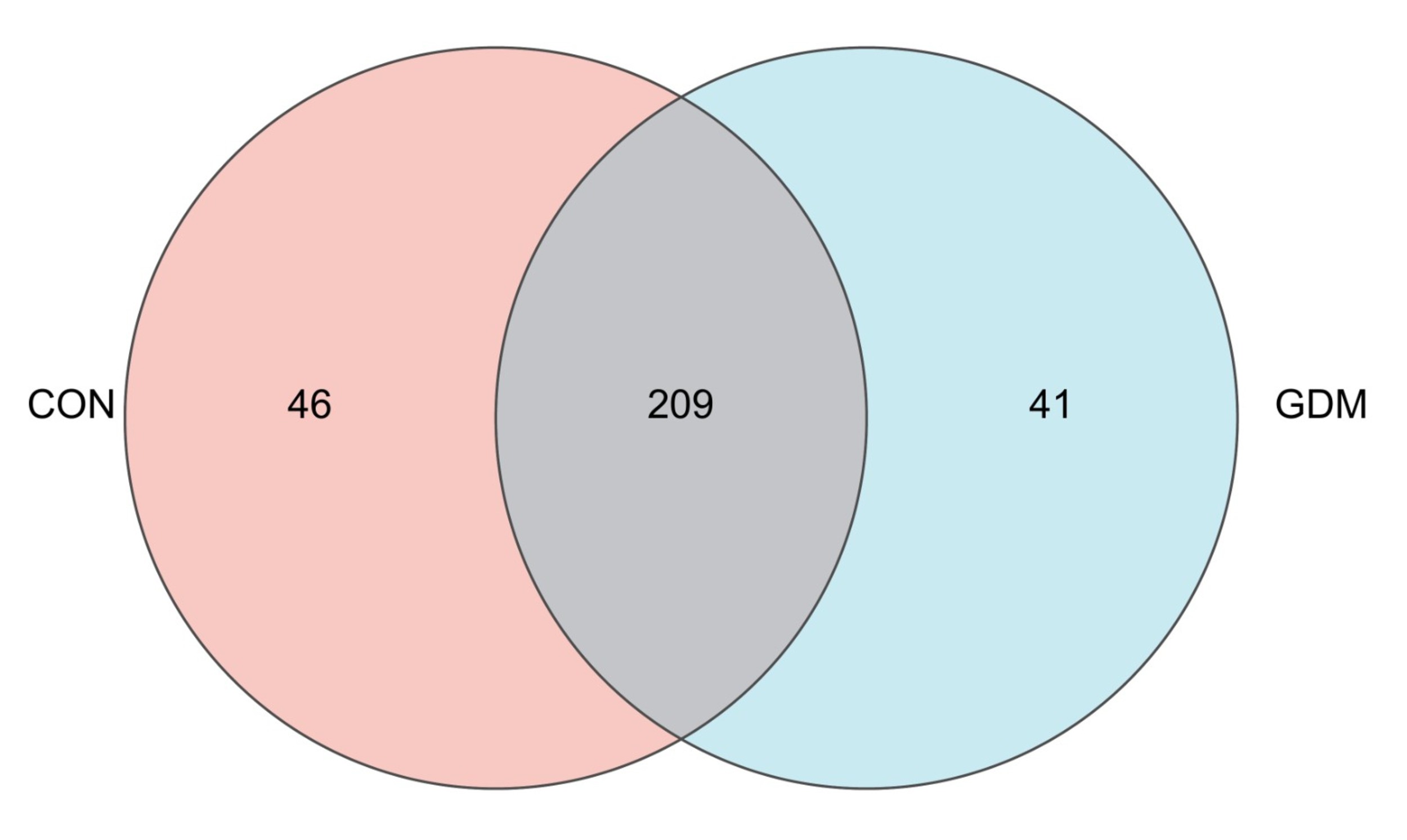 Fig. 2.
Fig. 2.
Venn diagrams at the genus level showing taxonomic differences between healthy pregnant women and pregnant women with gestational diabetes mellitus.
A bar plot analysis was undertaken to examine the species distribution within the two groups. The findings indicated that, at the phylum level, the microbial communities in both groups primarily consisted of Firmicutes, Actinobacteria, Proteobacteria, and Bacteroidota. Notably, a significant difference in abundance was observed between the two groups of samples. In the control group, the proportions of Firmicutes, Actinobacteria, Proteobacteria, and Bacteroidota were 64.75%, 22.54%, 11.81%, and 0.56%, respectively, whereas, in the GDM group, the corresponding proportions of the bacterial community were 75.39%, 10.48%, 8.61%, and 4.76% (Fig. 3). At the genus level, the distribution of bacterial genera between the two groups of samples showed both similarities and uniqueness. Blautia, Bifidobacterium, Escherichia–Shigella, Faecalibacterium, Romboutsia, Collinsella, Eubacterium_hallii_group, and Subdolibranulum were abundant in both groups. This result suggests that the microbial community structure mainly comprises bacterial genera belonging to the phyla Firmicutes, Actinobacteria, and Proteobacteria, consistent with the results at the phylum level (Fig. 4).
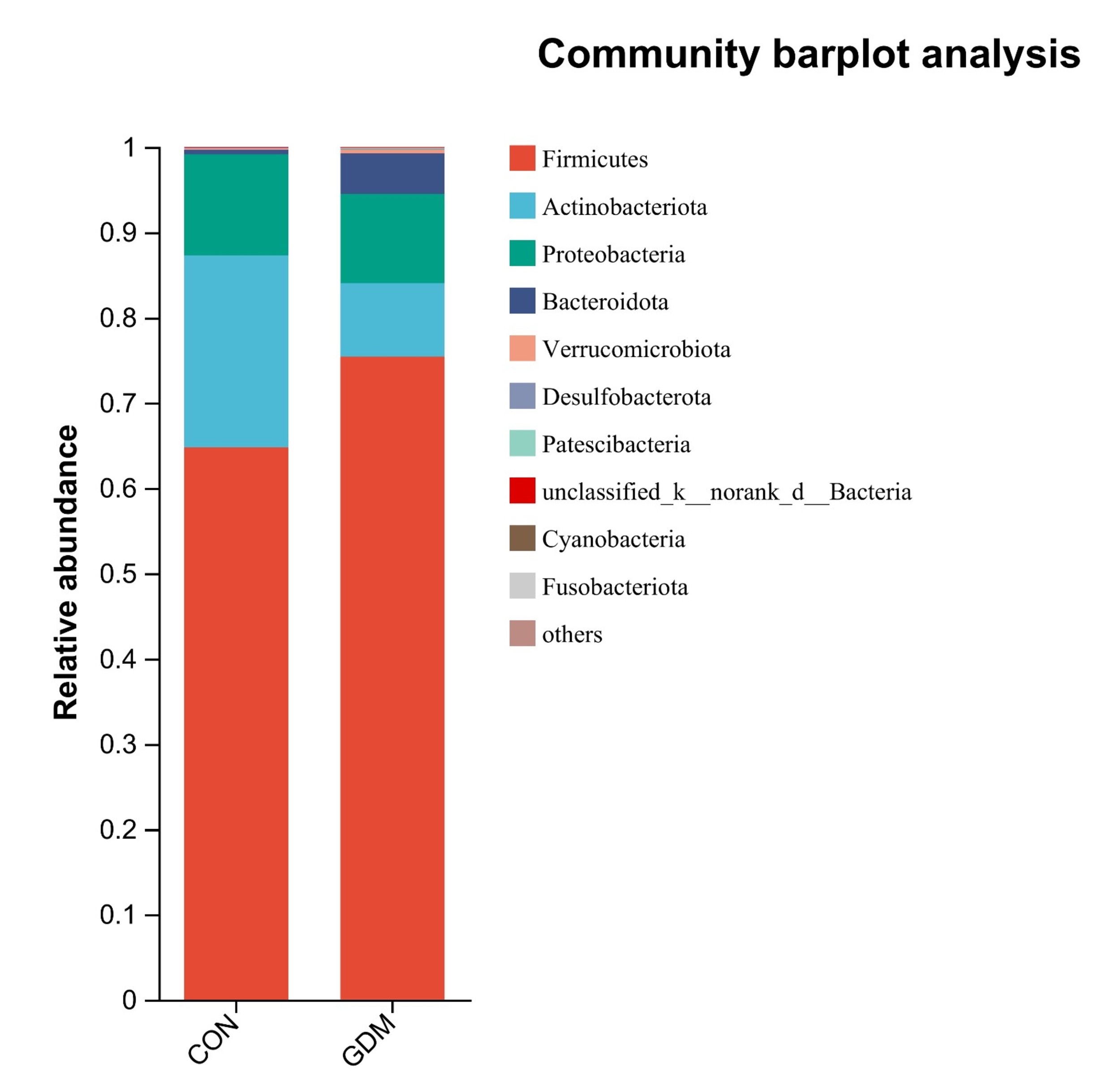 Fig. 3.
Fig. 3.
Compositional profiles of the gut microbiota at the phylum level between healthy pregnant women and pregnant women with gestational diabetes mellitus.
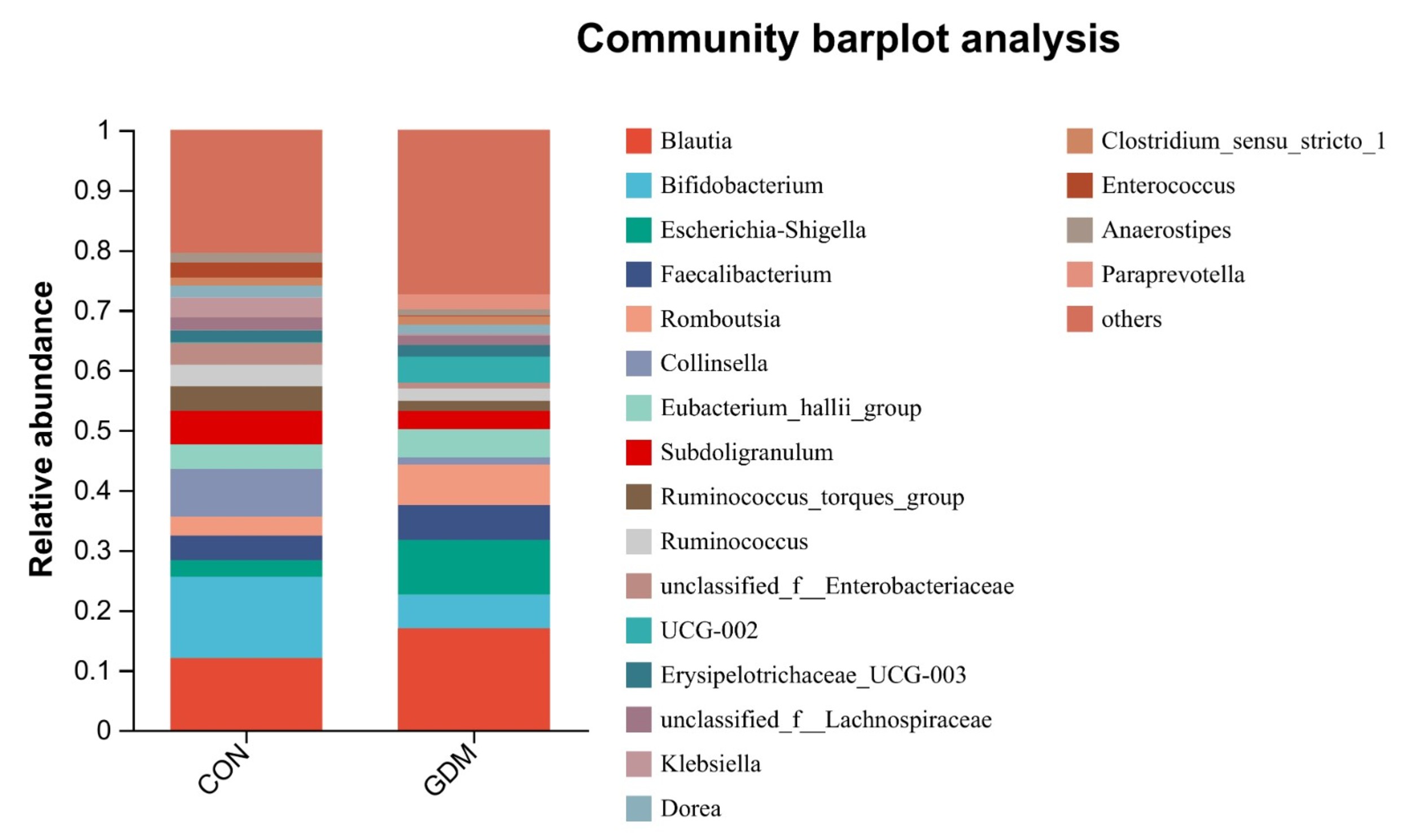 Fig. 4.
Fig. 4.
Compositional profiles of the gut microbiota at the genus level between healthy pregnant women and pregnant women with gestational diabetes mellitus.
At the phylum level, a statistically significant increase (p
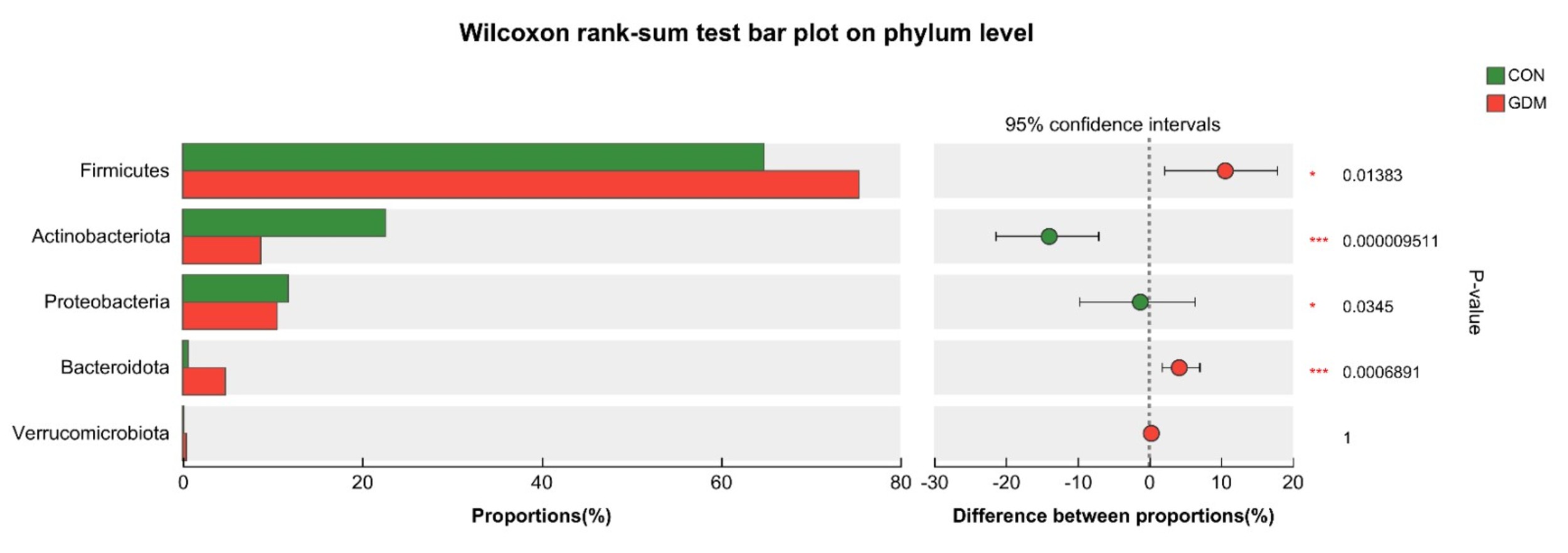 Fig. 5.
Fig. 5.
Species differences at the phylum level between the two groups. * 0.01
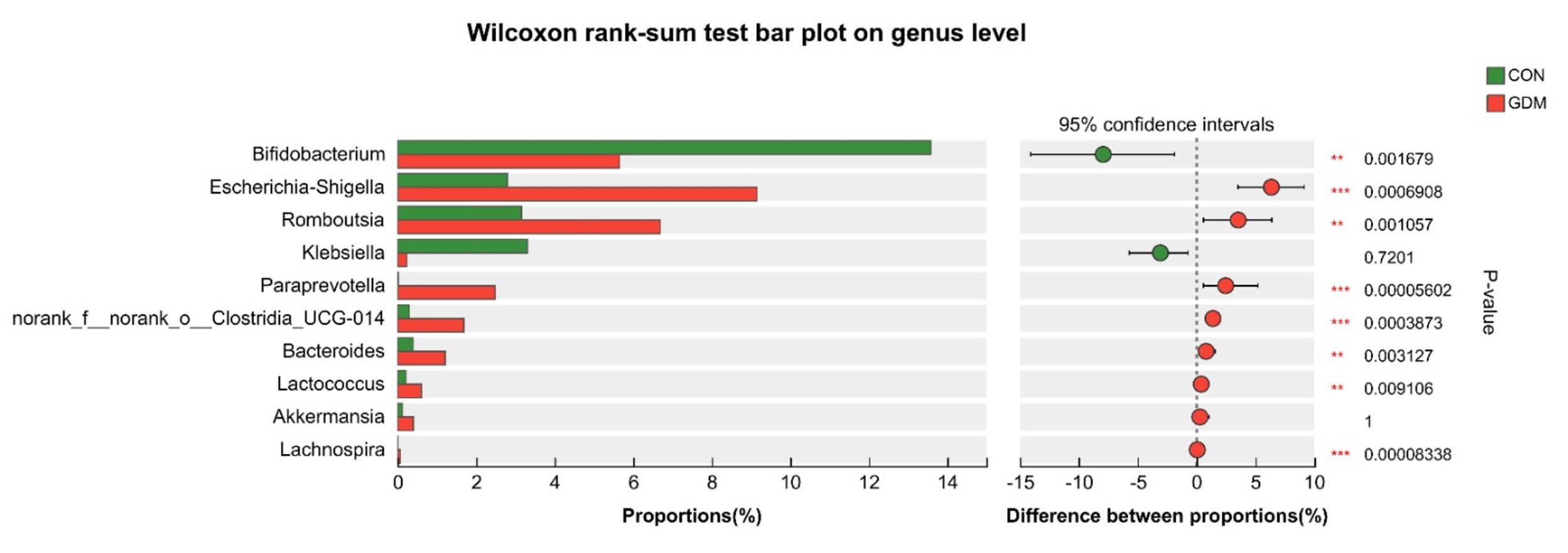 Fig. 6.
Fig. 6.
Species differences between the two groups at the genus
level. ** 0.001
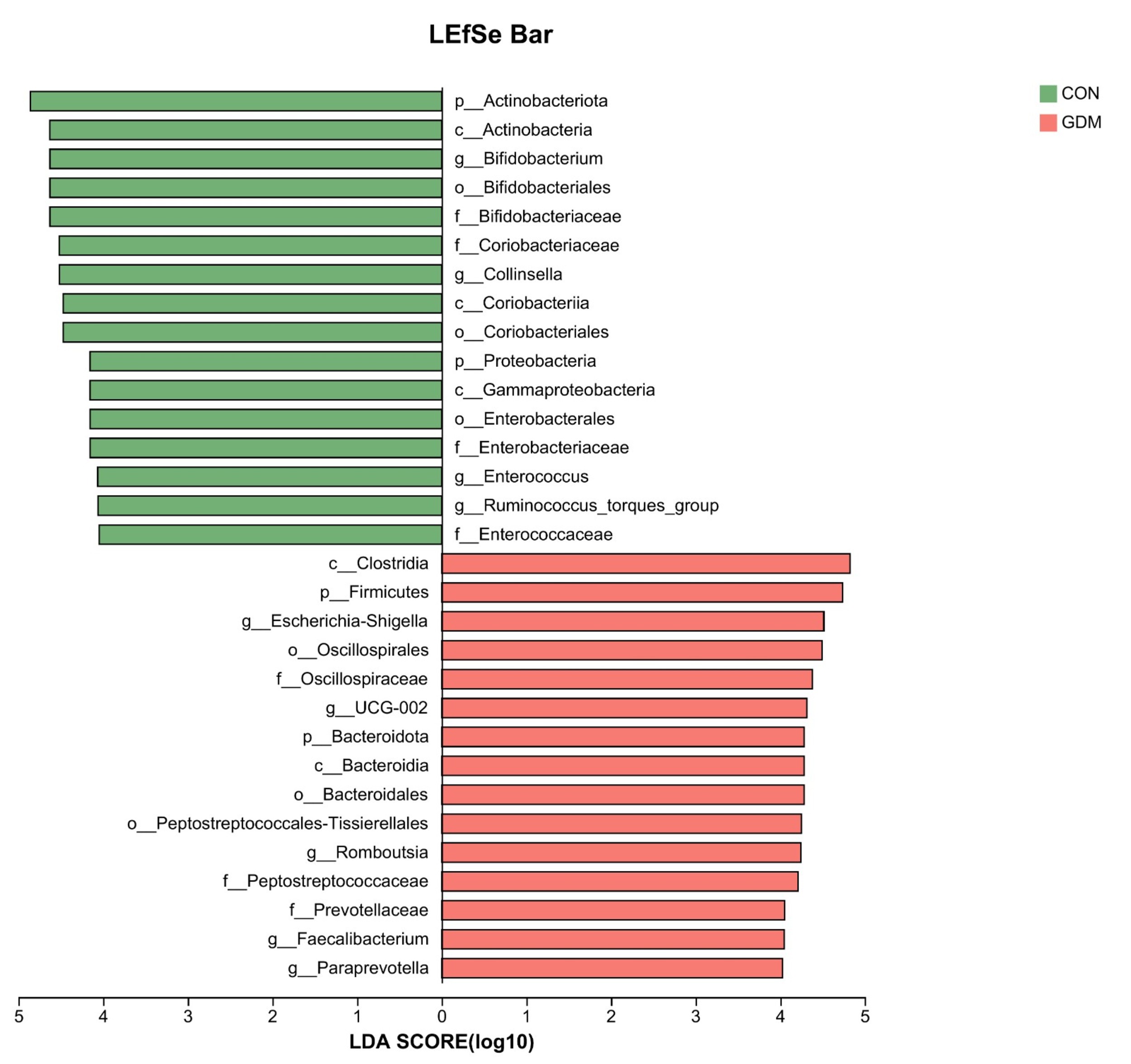 Fig. 7.
Fig. 7.
LEfSe analysis of the two groups. LDA, linear discriminant analysis; LEfSe, linear discriminant function analysis.
In this study, we analyzed the composition of the gut microbiota in women with GDM and healthy pregnant women during late pregnancy. Distinct differences were observed between the two groups. Specifically, GDM patients exhibited increased abundances of Firmicutes, Bacteroidota, Escherichia–Shigella, Romboutsia, and Paraprevotella, alongside a decreased abundance of Bifidobacterium. These alterations suggest that the aforementioned taxa may play pivotal roles in the onset and progression of GDM.
Currently, community diversity is most commonly assessed using diversity
indices, with
This study undertook a comparative analysis of the composition of the microbial
community between the group affected by GDM and the healthy control group,
revealing that at the phylum level, there existed 209 microbial communities
common to both groups. A subsequent cluster analysis elucidated that the gut
microbiota of both groups mainly comprised the following four phyla: Firmicutes,
Proteobacteria, Actinobacteria, and Bacteroidota. Blautia,
Bifidobacterium, Escherichia, Shigella,
Faecalibacterium, Romboutsia, Collinella,
Eubacterium_hallii_group, and Subdoligranulum are the
predominant genera within the two sample groups, with Brucella,
Clostridium difficile, and Romboutsia being notably dominant.
These results align with those of the majority of current studies on the
composition of dominant microbial communities [14, 15]. Ma et al. [16]
identified the following four as the predominant bacterial phyla: Firmicutes,
Bacteroidota, Proteobacteria, and Tenericutes. In the current study, the
relative abundance of Firmicutes was observed to be
Previous studies regarding metabolic disorders, such as obesity, GDM, and metabolic syndrome, have reported inconsistent findings concerning disparities in gut microbiota species. Firmicutes and Bacteroides have been linked with obesity and T2DM [17]. Empirical evidence has demonstrated that a high-fat diet can elevate the abundance of Firmicutes, whereas a fiber-rich diet can diminish it, thereby reducing obesity and weight gain [18]. The phylum Bacteroidota is identified as the dominant gut microbiota in humans and other mammals. In terms of polysaccharide metabolism, Bacteroidota have a potent fermentation capacity, potentially supplying an additional 10–15% energy to the human body. Bäckhed et al. [19] determined that inoculating rats with Bacteroides resulted in a 23% increase in body fat and the onset of insulin resistance, which suggests that this microbial community may promote fat accumulation. This may be related to its proficiency in decomposing plant polysaccharides to facilitate nutrient absorption and energy reserves of the host. Collado et al. [20] also ascertained that Bacteroidota are prevalent among pregnant women that are overweight or have obesity. It is well-recognized that being overweight or obese is a risk factor for GDM. Thus, based on the discussion so far, we can suggest that a boom in the abundance of Firmicutes and Bacteroidota may be associated with the emergence of GDM.
This study demonstrated a marked increase in the prevalence of Firmicutes and Bacteroidota within the GDM cohort. Within the phylum Bacteroidota, both Bacteroides and prevotella showed a significant rise in the GDM cohort relative to that in healthy pregnant women. Concurrently, the levels of fasting glucose, insulin, OGTT and triglycerides were considerably elevated in the GDM cohort compared to those in healthy pregnant women. These findings further suggest that the pronounced proliferation of Firmicutes and Bacteroidota may be associated with dysregulated glucose and lipid metabolism. In fact, it was reported that those who eat plenty of protein and animal fats have Bacteroides bacteria predominantly [21]. However, our comparative analysis of three-day average dietary intake revealed no significant differences in total fat or protein consumption between the two groups. This may be attributed to dietary management interventions for patients with GDM post-diagnosis, potentially obscuring the authentic dietary patterns of both groups. Given that long-term dietary habits are strongly associated with the gut microbiome composition [22], this confounding factor deserves careful consideration. Our findings are in line with a parallel study examining the correlations between the GDM gut microbiota and blood glucose levels [23]. In contrast, another study revealed a diminished abundance of Firmicutes in the GDM group, as the study participants were in the early pregnancy stage [24]. Actinobacteria, encompassing filamentous Gram-positive bacteria, comprise various beneficial short-chain fatty acid-producing bacteria. In this study, the gut microbiota of patients with GDM showed a significant reduction in Actinobacteria and the genus Bifidobacterium compared to that in the healthy group, indicating that a decline in the beneficial Bifidobacterium coincides with the onset of GDM. An increase in the proportion of actinomycetes within the altered gut microecology of patients with GDM has also been reported [25]. This might be ascribed to the augmented presence of Collinsella bacteria within actinomycetes, which exerts a potentially adverse impact on the regulation of glucose metabolism during mid- and late-pregnancy stages.
Our study demonstrated that Shigella, Clostridium, and
Romboutsia exhibited increased prevalence in the GDM cohort. Shigella,
a member of the phylum Proteobacteria, primarily produces lipopolysaccharide
(LPS) as its toxic agent. The LPS shows its effect by activating the Toll-like
receptor-4 along its downstream pathways, thereby facilitating the translocation
of endotoxins into the circulatory system and initiating persistent inflammatory
reactions. Additionally, the LPS can upregulate the expression of
pro-inflammatory cytokines and chemokines within the adipose tissue, impairing
normal cellular functions in humans and contributing to metabolic endotoxemia,
obesity, and insulin resistance [26, 27]. Two independent studies have reported
enrichment of LPS biosynthesis pathways in women with GDM [28, 29]. Enhanced gut
permeability allows LPS to enter the systemic circulation, eliciting inflammatory
responses that impair insulin signaling and lead to glucose intolerance during
pregnancy. Romboutsia is a Gram-positive bacterium capable of
compromising intestinal mucosal integrity. A study has established a positive
correlation between the prevalence of this bacterium in the gut microbiota and
ulcerative colitis [30]. The highly virulent exotoxins and invasive enzymes
synthesized by Clostridium are also capable of inducing inflammatory
processes [31]. Prolonged and subclinical inflammatory states may compromise the
architecture and functionality of pancreatic islet
This study has several limitations. Firstly, the fecal samples were collected only at a single time point during the late gestation period. As a result, dynamic shifts in gut microbiota over the course of pregnancy could not be observed. Moreover, the relatively small cohort size and single-center methodology of the study diminish its generalizability. Secondly, our study primarily described compositional differences in gut microbiota between GDM patients and healthy controls, without elucidating the corresponding functional alterations. Consequently, the specific mechanisms through which microbial dysbiosis influences host metabolic pathways—such as short-chain fatty acids (SCFAs) production, bile acid metabolism, and inflammatory signaling—remain unclear. Future research integrating multi-omics approaches, including metagenomics and metabolomics, combined with validation in animal models, will be essential to uncover the mechanistic and causal relationships between gut microbiota alterations and GDM pathogenesis.
This preliminary investigation focused on bacterial colonies. A diversity analysis indicated that gut microbiota abundance was heightened in the GDM cohort. The study demonstrated a comparatively greater abundance of gut microbiota in women with GDM. In addition, it reported notable distinctions in the gut microbiota of women with GDM and healthy pregnant women. Future research should integrate multi-omics data—such as metagenomics and metabolomics—and leverage longitudinal cohorts as well as animal models to further elucidate the causal relationships and specific mechanisms linking these differential microbial communities to the pathogenesis of GDM.
ASV, amplicon sequence variant; BMI, body mass index; GDM, gestational diabetes mellitus; IDF, International Diabetes Federation; LDA, linear discriminant analysis; LEfSe, linear discriminant function analysis; LPS, lipopolysaccharide; OGTT, oral glucose tolerance test; PCoA, Principal component analysis; PE, paired-end; T2DM, type 2 diabetes mellitus.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA1338062.
BZ and JY participated in study design, data acquisition, statistical analysis, and literature search and also drafted the manuscript. JG participated in study design, performed the research, and analyzed the data. XQW conceived the study and participated in its design and fund collection. All authors contributed to editorial changes in the manuscript. All the authors have read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The study was conducted in accordance with the Declaration of Helsinki. The research protocol was approved by the Ethics Committee of The Third Affiliated Hospital of Zunyi Medical University (The First People’s Hospital of Zunyi) (Ethic Approval Number: 2021-1-77), and all of the participants provided signed informed consent.
We would like to express our gratitude to the Department of Gynecology and Obstetrics of the First People’s Hospital of Zunyi, Guizhou, China, for their assistance in data collection, as well as to all the participants in the study.
This study was supported by the Scientific Research Project of the Guizhou Provincial Health Commission (zwkj2021-390).
The authors declare no conflict of interest.
During the preparation of this work the authors used ChatGpt-3.5 in order to check spelling and grammar. After using this tool, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.