



 , Vanesa Rayo-López 1, Isabel Campo-Gesto 3, Marta Calvo-Urrutia 1
, Vanesa Rayo-López 1, Isabel Campo-Gesto 3, Marta Calvo-Urrutia 11 Human Reproduction Unit, Department of Obstetrics and Gynecology, Hospital Clinico San Carlos, 28040 Madrid, Spain
2 Department of Maternal and Child Health and Public Health, Complutense University, 28040 Madrid, Spain
3 Ultrasound and Prenatal Diagnosis Unit, Department of Obstetrics and Gynecology, Hospital Clinico San Carlos, 28040 Madrid, Spain
Abstract
To provide an updated synthesis of the current knowledge on the epidemiology, pathophysiology, genetic basis, diagnostic strategies, and management of recurrent hydatidiform mole (RHM), incorporating recent molecular and clinical findings.
We conducted a narrative review of peer-reviewed literature, focusing on genetic, epigenetic, molecular, and clinical studies addressing the pathogenesis, diagnostic strategies, and clinical management of RHM.
Mutations in maternal-effect genes, primarily nucleotide-binding oligomerization domain, leucine-rich repeat and pyrin domain-containing 7 (NLRP7) and KH domain-containing 3-like (KHDC3L), account most familial cases, while other subcortical maternal complex (SCMC) genes, including peptidyl arginine deiminase 6 (PADI6), nucleotide-binding oligomerization domain, leucine-rich repeat and pyrin domain-containing 5 (NLRP5), transducin-like enhancer of split 6 (TLE6), zygote arrest 1 (ZAR1), and oocyte-expressed protein (OOEP), have also been implicated. Histological features, such as villous edema, circumferential trophoblastic hyperplasia, and the presence or absence of embryonic or fetal tissue, remain crucial in diagnosis, complemented by p57 immunohistochemistry (IHC) and short tandem repeat (STR) genotyping. Although many cases can be explained by genetic mutations, others may result from epimutations, mosaicism, or polygenic inheritance. Reproductive counseling now incorporates molecular diagnostics. While in vitro fertilization with intracytoplasmic sperm injection (IVF-ICSI) or preimplantation genetic testing (PGT) may reduce recurrence risk, donor oocytes remain the only definitive option for women with confirmed mutations.
RHM represents a unique model of imprinting disorders in which defective oocyte biology leads to abnormal conceptions. Integration of molecular diagnostics with clinical management offers a precision medicine approach, while future research may identify new avenues for prevention and targeted interventions.
Keywords
- hydatidiform mole
- recurrent hydatidiform mole
- FRCM
- diploid biparental mole
- genomic imprinting
- maternal effect genes
- subcortical maternal complex
- DNA methylation
- epigenetics
- mosaicism
Hydatidiform mole (HM) is a gestational trophoblastic disease (GTD) characterized by abnormal proliferation of trophoblastic tissue and hydropic degeneration of chorionic villi, occurring in the context of absent embryonic development in complete hydatidiform mole (CHM) or defective embryonic development in partial hydatidiform mole (PHM) [1]. While most cases resolve after appropriate management, a small subset of women experience recurrence, which may occur in up to 1–2% after a single episode and up to 15–20% in cases with prior recurrent hydatidiform moles (RHMs) [2]. RHM is a rare and challenging condition with significant clinical, reproductive, and psychological implications [3]. The etiology of RHM is complex and remains incompletely understood. While sporadic cases are often associated with chromosomal abnormalities, familial cases have been linked to maternal-effect mutations. Early diagnosis and careful monitoring are critical to preventing malignant forms of gestational trophoblastic neoplasia (GTN) [2].
In this narrative review, we aim to provide an updated synthesis of current knowledge on RHM, including its pathophysiology, management strategies, and future reproductive considerations.
The reported incidence varies depending on geographic location, population characteristics, and the diagnostic criteria used. The baseline risk of molar pregnancy in the general population is approximately 1 in 1000 pregnancies. HM represents the most benign end of the GTD spectrum but carries the potential to progress to invasive mole or GTN, particularly in complete forms, which occurs in 15–20% of cases compared to 1–5% in PHM. After a single CHM, the recurrence risk rises to approximately 1–2%, and following two previous molar pregnancies, the risk increases significantly, up to 15–20%.
Geographic variability exists, with higher baseline rates of molar pregnancy,
and consequently of RHM, reported in Southeast Asia, the Middle East, and parts
of Latin America compared with Europe and North America [1]. These differences
may reflect variations in genetic background, maternal age, nutritional status,
and access to healthcare, as well as differences in reporting practices [2].
Extremes of maternal age (
Overall, the epidemiology of RHM reflects a complex interplay between genetic susceptibility and environmental factors. The rarity of the condition makes precise quantification challenging, underscoring the need for large, multicenter registries and genetic studies to refine risk estimates and improve patient counseling. The major epidemiological and clinical risk factors associated with RHM, along with the current strength of these associations, are summarized in Table 1.
| Risk factor | Description | Association |
| Genetic mutations (NLRP7, KHDC3L, PADI6) | Maternal effect gene mutations leading to abnormal embryonic development | Strong |
| History of prior molar pregnancy | 1–2% recurrence risk after one event; 15–20% after two or more events | Strong |
| Maternal age ( |
Associated with higher incidence of initial molar pregnancy; nuclear impact on recurrence | Moderate |
| Ethnic/geographic background | Higher incidence in Southeast Asia, Middle East, parts of Latin America | Moderate |
| Nutritional deficiencies (low carotene, animal fat) | Observed association with initial molar pregnancy; limited recurrence-specific data | Weak |
| Environmental/socioeconomic factors | Delayed access to prenatal care and diagnostis; role in recurrence unclear | Weak |
These data indicate that maternal-effect gene mutations are the strongest predictors of recurrence, while environmental and demographic factors also modulate risk, underscoring the multifactorial nature of RHM. RHM, recurrent hydatidiform mole; NLRP7, nucleotide-binding oligomerization domain, leucine-rich repeat and pyrin domain-containing 7; KHDC3L, KH domain-containing 3-like; PADI6, peptidyl arginine deiminase 6.
From a biological perspective, HM results from an imbalance in parental genomic contributions to the conceptus. HMs are subclassified as CHM or PHM based on clinical, histological, cytogenetic, and molecular criteria.
CHM arises from fertilization of an anucleated oocyte by either a single haploid sperm that duplicates, resulting in a 46,XX of entirely paternal origin, or, more rarely, by two sperm, producing a 46,XY diploid androgenetic conceptus. Histologically, they are characterized by diffuse villous edema, absence of embryonic or fetal tissue, and circumferential trophoblastic hyperplasia. In contrast, PHM results from fertilization of a normal oocyte by two sperm (dispermy) in approximately 90% of cases, producing a triploid genome (69,XXY; 69,XXX; or 69,XYY) with biparental origin. These PHMs may contain fetal or embryonic remnants and display focal villous edema with irregular trophoblastic hyperplasia. While these mechanisms explain sporadic moles, they do not account for familial or recurrent forms, in which a maternal genetic factor plays a primary code.
Accurate classification of HM subtypes remains a diagnostic challenge, particularly in early gestational stages, when morphological features are not fully developed. Approximately 50% of true PHMs cannot be reliably identified through conventional histopathological methods due to the absence of definitive histological criteria and inter-observer variability, even among experienced pathologists [5, 6]. Similarly, conventional cytogenetics, once considered the gold standard of genetic research, cannot distinguish a 46,XX or 46,XY CHM from a 46,XX or 46,XY non-molar conceptus [7]. Precise subclassification of HMs is crucial for guiding appropriate clinical management and follow-up. Consequently, evaluation of p57 expression by immunohistochemistry (IHC), combined with polymerase chain reaction (PCR)-based genotyping via short tandem repeat (STR) markers, has become an essential tool for the accurate classification of HMs [8, 9].
p57, a cyclin-dependent kinase inhibitor encoded by the cyclin-dependent kinase inhibitor 1C (CDKN1C) gene on chromosome 11p15.5, is a maternally expressed and paternally imprinted gene, detectable in the nuclei of cytotrophoblasts and villous stromal cells during early gestation. Immunohistochemical assessment of p57 has become a widely adopted tool in diagnostic pathology, providing a reliable method to differentiate triploid PHM from androgenetic CHM [10]. In practice, triploid PHMs, which retain a maternal genomic contribution, show positive p57 staining, whereas androgenetic CHMs, lacking maternal genetic material, consistently demonstrate absent expression.
However, p57 cannot distinguish dispermic from monospermic androgenetic CHMs or triploid PHMs from diploid arrested products of conception (POC), some of which some may exhibit molar features. In such cases, genotyping of the POC is required. Additionally, rare cases show aberrant p57 expression occurs, such as in rare CHMs retaining maternal chromosome 11, reinforcing the need for molecular confirmation. The most commonly used genotyping method in clinical histopathology is multiplex analysis of STR in the POC. STRs are repetitive DNA sequences that are highly polymorphic in the population and are analyzed using PCR amplification. In this technique, alleles in villous tissue are identified as either paternal or maternal. STR genotyping assesses highly polymorphic microsatellite regions distributed across multiple chromosomes, allowing comparison of allelic patterns between molar tissue and maternal, and in some cases paternal, DNA. This methodology reliably differentiates the main genotypic variants of HMs, including diploid androgenetic monospermic, diploid androgenetic dispermic, triploid dispermic, triploid diandric, and diploid biparental complete hydatidiform mole (BiCHM). Accurate classification holds significant clinical relevance, as CHMs carry a markedly higher risk of progression to GTN compared with PHMs. Furthermore, among CHMs, the dispermic androgenetic subtype demonstrates a greater propensity for malignant transformation compared with the monospermic form.
A recent study [11] has reinforced the clinical utility of STR genotyping in distinguishing HM subtypes. In a cohort of 31 PHMs and 23 hydropic abortions, Meng et al. (2023) [11] demonstrated that STR analysis consistently identified one maternal and two paternal alleles across informative loci, confirming the diandric triploid origin of PHMs. Comparison with decidual tissue from the same patients further validated the biparental control pattern. Importantly, STR-based classification showed excellent concordance with histology and p57 immunostaining, underscoring its reliability. These findings highlight that STR genotyping not only refines diagnostic accuracy in morphologically ambiguous cases but also provides direct evidence of parental contribution, thereby reducing the risk of misclassification between PHMs and hydropic abortions.
This combined approach facilitates precise differentiation among various types of conceptuses (Table 2), including [12, 13]:
| CHM | PHM | Recurrent mole | |
| Genetic origin | Androgenetic diploidy (90%: 46,XX/10%: 46,XY); | Diandric triploidy (69,XXX/XXY/XYY); | Biparental diploidy but with maternal-effect gene defects |
| Loss of maternal DNA. | Two paternal sets and one maternal set. | (NLRP7, KHDC3L, PADI6, NRLP5). | |
| Mechanism | Absence of maternal imprinting. | Extra paternal genoma; | Imprinting failure; |
| Imbalance of parental contribution. | Epimutations; | ||
| Mosaicism; | |||
| Polygenic inheritance. | |||
| Histology | Villous edema; | Hydropic villa; | Variable, often mimic CHM. |
| Circumferential trophoblastichyperplasia; | Focal trophoblastic proliferation; | ||
| No fetal tissue. | Presence of fetal tissue. | ||
| Diagnostic markers | p57: negative; | p57: positive; | STR: biparental contribution. |
| STR: androgenetic. | STR: diandric triplody. | ||
| Incidence | ~1:1000 pregnancies; Higher in Southeast Asia, Middle East. | Less common than CHM ~1:2000–3000 pregnancies. | After 1 mole: 1–2%; |
| After 2 moles: 15–20%; Familial forms: rare (autosomal recessive). | |||
| Progression to GTN | 15–20% of cases; Dispermic subtype: higher risk than monospermic. | 1–5% of cases; Significantly lower than CHM. | Variable, depends on molecular subtype BiCHM: similar risk to CHM. |
| Recurrence risk | After 1 CHM: 1–2%; After 2 CHM: 15–20%. | Lower recurrence than CHM. | With confirmed mutations (NLRP7, KHDC3L): virtually 100%; |
| Mutation-negative: variable. |
BiCHM, biparental complete hydatidiform mole; CHM, complete hydatidiform mole; PHM, partial hydatidiform mole; GTN, gestational trophoblastic neoplasia; STR, short tandem repeat; NLRP5, nucleotide-binding oligomerization domain, leucine-rich repeat and pyrin domain-containing 5.
It is essential to integrate results from multiple diagnostic methods while accounting for the limitations of each. When discrepancies or unexpected findings arise, they should be addressed by repeating the discordant test or using alternative approaches. No single method should be used to completely override another, as each had inherent limitations.
The relationship between p57 expression and the morphologic features of HM remains incompletely understood and is not solely determined by the presence or absence of the maternal genome. Indeed, p57 also acts as a negative regulator of cell proliferation and as a marker of terminal differentiation.
In conclusion, findings from morphology, p57 IHC, and genotyping should be thoroughly described, with conclusions supported by clear reasoning and accompanied by a discussion of the limitations of each method. It is also important to recognize that molar pregnancies are inherently abnormal conceptions, in which unusual genotypic configurations such as chimerism and mosaicism, can occur and have been reported.
Although most cases of HM are sporadic and result from errors during fertilization, a minority of women experience RHM, often caused by genetic and epigenetic defects in the oocyte. The identification of maternal-effect mutations in these cases has transformed our understanding of HM, revealing it not merely as a cytogenetic aberration but as a complex epigenetic disorder originating in oocyte biology.
RHM is defined as two or more consecutive HMs, predominantly CHMs. The risk of a third HM is strongly associated with CHM. While most HMs are sporadic, a minority are attributed to familial recurrent complete mole (FRCM), a rare autosomal recessive condition associated with a markedly reduced likelihood of achieving a viable pregnancy (~1.8%) [14]. A history of previous molar pregnancies suggests that a third mole is most likely androgenetic in origin; however, the possibility of FRHM cannot be excluded [15].
Over the past decade, understanding of RHM has expanded significantly, particularly regarding the role of maternal-effect genes and genomic imprinting. While most HMs are sporadic, a small but clinically relevant subset represents familial forms, often associated with biallelic mutations in key maternal genes involved in oocyte epigenetic regulation [16]. The underlying molecular mechanism involves epigenetic imprinting errors, specifically hypomethylation of maternal imprints during oocyte maturation [3, 17]. These errors result in biallelic expression or silencing of critical imprinted genes, leading to failed embryogenesis and abnormal trophoblastic development [18], often resembling an androgenetic CHM even when the conceptus is biparental.
Several key genes implicated in RHM have been described [19]. The subcortical maternal complex (SCMC) is a group of proteins localized in the oocyte cortex that regulates essential processes during the oocyte-to-embryo transition. By regulating spindle positioning, chromosome segregation, and early cleavage divisions, the complex ensures genomic stability and developmental competence. Disruption of these processes leads to embryonic arrest, abnormal imprinting, and reproductive loss.
Maternal recessive mutations in nucleotide-binding oligomerization domain, leucine-rich repeat and pyrin domain-containing 7 (NLRP7) and KH domain-containing 3-like (KHDC3L), both encoding SCMC components, are the primary cause of BiCHM [20].
Other SCMC-related genes, such as peptidyl arginine deiminase 6 (PADI6), nucleotide-binding oligomerization domain, leucine-rich repeat and pyrin domain-containing 5 (NLRP5), transducin-like enhancer of split 6 (TLE6), zygote arrest 1 (ZAR1), and oocyte-expressed protein (OOEP), have also been implicated in atypical RHM and other forms of reproductive failure, including recurrent implantation failure and early embryonic arrest. These observations support the concept that RHM is part of a continuum of disorders caused by disruption of the SCMC.
Although biallelic mutations in maternal-effect genes explain many familial RHMs, a substantial proportion of cases show no detectable pathogenic variants, suggesting that additional mechanisms contribute to the phenotype. This observation has prompted researchers to explore epigenetic and non-Mendelian mechanisms that could disrupt normal embryogenesis and trophoblastic development [19]:
Finally, a polygenic model has been proposed. Some RHMs may result from the additive or synergistic effects of multiple mild genetic variants that individually are insufficient to cause disease but collectively disrupt oocyte maturation or imprinting. This polygenic model may explain familial clusters of RHMs where no single pathogenic mutation is identified. PADI6, NLRP5, TLE6, ZAR1, and OOEP, which are involved in SCMC integrity, DNA methylation, or chromatin remodeling, have been reported in patients with atypical RHMs, recurrent implantation failure, and early embryonic arrest [27]. These findings support the hypothesis that RHM may fall within a broader spectrum of SCMC-related reproductive disorders [28, 29].
Overall, current evidence indicates that RHM is not solely a consequence of cytogenetic abnormalities but rather a complex reproductive disorder arising from the interplay between single-gene defects, epigenetic deregulation, mosaicism, and polygenic contributions.
The occurrence of a third HM is rare and may indicate possible FRCM. In patients with two molar pregnancies, genotyping of the conceptus is recommended following histological and immunohistochemical review to determine whether it is androgenetic or biparental in origin, thereby guiding reproductive counseling. Because BiCHM are rare, most women with recurrent complete moles are likely to have androgenetic CHMs [30].
In cases of BiCHM, mutational screening of NLRP7 is the first-line approach. If the results are negative, KHDC3L sequencing should follow. If no pathogenic variants are identified, analysis can be extended to other SCMC genes or methylation profiling [31].
In recurrent androgenetic CHMs, in vitro fertilization with intracytoplasmic sperm injection (IVF-ICSI) can ensure monospermic fertilization and prevent dispermic fertilization. Preimplantation genetic testing (PGT) may be considered [32] to prevent the transfer of 46,XX embryos, thereby avoiding CHMs resulting from the fertilization of an inactive oocyte by a haploid X-bearing sperm, which subsequently undergoes duplication [33]. PGT may reduce the risk but does not eliminate it. Similarly, although ICSI can reduce the risk of fertilization anomalies, it does not fully prevent risk, as it does not address underlying epigenetic anomalies [32].
An overview of the management approach for patients with two or more HMs is shown in Fig. 1.
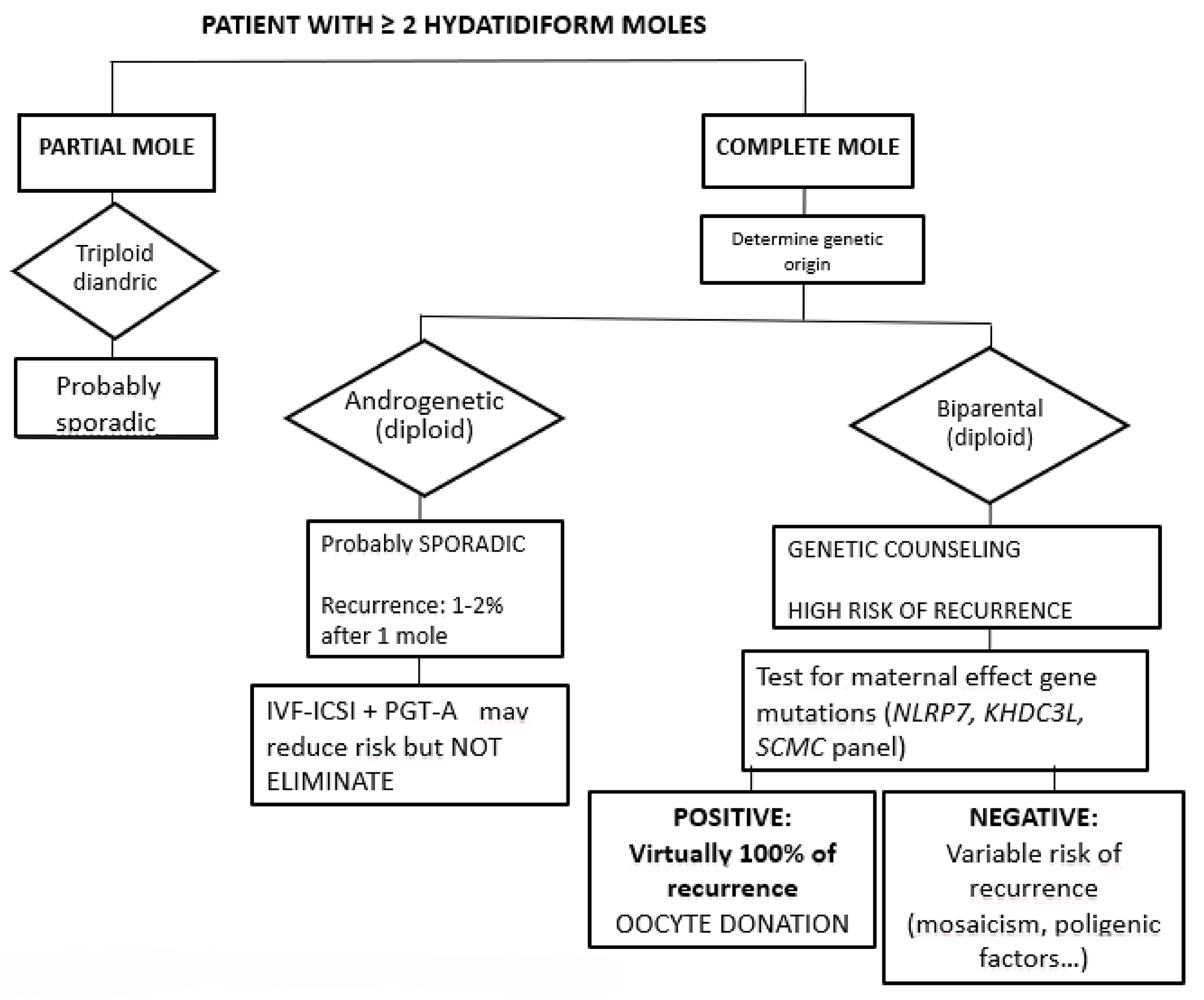 Fig. 1.
Fig. 1.
Initial approach in patient with
As previously described, even in the absence of identifiable mutations, FRCM cannot be excluded, warranting histopathological review and consideration of additional gene testing. In women with confirmed mutations in NLRP7, KHDC3L, or related genes, recurrence is virtually inevitable [34]. Consequently, genetic counseling must clearly communicate that natural conception is unlikely to result in a viable pregnancy and IVF with donor oocytes remains the only effective reproductive strategy, as the epigenetic abnormality resides in the maternal DNA of the oocyte, and ovum replacement entirely prevents the disease [35, 36]. However, in mutation-negative cases, reproductive outcomes can vary, supporting the need for individualized, risk-based counseling.
Future directions in this field likely include the development of non-invasive biomarkers for early detection of abnormal imprinting. Notably, advances in cell-free DNA (cfDNA) and cfRNA technologies [37] may enable the identification of epigenetic signatures of defective trophoblast development before clinical manifestations arise, a principle already being explored in placental disorders such as preeclampsia [38]. Applying these methodologies to RHM could significantly improve early detection and monitoring in high-risk pregnancies.
Standard genotyping and methylation arrays may fail to detect subtle, transient, or cell-type-specific methylation errors. To address undiagnosed familial cases, there is increasing interest in using expanded genetic panels combined with comprehensive epigenomic profiling. This approach includes whole-genome bisulfite sequencing (WGBS) and single-oocyte methylome analysis [19], which enables single-base resolution mapping of DNA methylation across the entire genome. Long-read single-oocyte epigenomic profiling provides information on both methylation and chromatin state in individual oocytes, capturing heterogeneous or mosaic epigenetic patterns. These technologies may identify cryptic imprinting defects or dynamic methylation changes in oocytes that predispose to RHM, expanding our understanding of non-classical pathogenic mechanisms.
The establishment of international RHM registries could significantly improve our understanding of long-term reproductive outcomes and potential oncologic risks [39]. While comprehensive registries for GTD, and molar pregnancy in particular, exist in regions such as the United Kingdom, globally coordinated databases could facilitate large-scale epidemiologic and genotype–phenotype studies.
Finally, clustered regularly interspaced short palindromic repeats (CRISPR)-based genome and epigenome editing tools offer a transformative platform for modeling maternal imprinting disorders in vitro [40]. In particular, the CRISPR/Cas9 system has opened new opportunities to dissect the genetic and epigenetic mechanisms underlying HM. CRISPR has been successfully applied to generate cellular and animal models that replicate defects in maternal-effect genes and imprinting regulation, providing insights into trophoblastic development and disease progression. However, it is important to emphasize that CRISPR has not yet been translated into therapeutic interventions for molar pregnancy. Its current role remains primarily in the field of mechanistic research and disease modeling, where it serves as a powerful tool to test hypotheses and explore potential molecular targets for future applications.
These strategies reflect a shift toward a precision medicine framework in RHM. This integrated approach may enable earlier diagnosis, improved risk stratification, and the development of targeted interventions tailored to the molecular basis of each case.
HM and its recurrent forms represent a unique model of human disease in which defective genomic imprinting and maternal-effect gene mutations converge to disrupt embryonic development and trophoblastic differentiation. While most cases are sporadic and clinically manageable, RHM highlights the critical role of maternal genetic and epigenetic factors, particularly involving NLRP7, KHDC3L, and other components of the SCMC. The integration of histopathology, p57 IHC, and DNA genotyping has markedly improved diagnostic accuracy; however, a significant proportion of cases remain unexplained, suggesting contributions from epimutations, mosaicism, and polygenic inheritance.
Future progress will likely stem from high-resolution epigenomic profiling, development of non-invasive biomarkers using cfDNA and cfRNA, and functional CRISPR-based models, which together may transform early detection and mechanistic understanding. At the clinical level, comprehensive genetic counseling and long-term reproductive follow-up are essential, and the establishment of international registries will be pivotal to defining recurrence risk, guiding patient care, and informing fertility planning.
TG-H was responsible of conceptualization, design of the review, extensive literature search, drafting of the initial manuscript, and integration of sections. VR-L and IC-G: Literature review, organization of references, preparation of tables/figures, and contribution to manuscript writing. MC-U: Contribution to the conception and design of the review, interpretation of the literature. All authors contributed to critical revision of the manuscript for important intellectual content. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We would like to express our gratitude to all those who helped us during the writing of this manuscript. Thanks to all the peer reviewers for their opinions and suggestions.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.