




 , Zitong Xu 1, Xianjue Zheng 1, Haojie Pan 1, Yimin Wang 2, Haiying Chen 3, Zhenzhen Zheng 3, Hongping Zhang 2,3, Jiayong Zheng 1,2,*
, Zitong Xu 1, Xianjue Zheng 1, Haojie Pan 1, Yimin Wang 2, Haiying Chen 3, Zhenzhen Zheng 3, Hongping Zhang 2,3, Jiayong Zheng 1,2,*
1 Department of Medical Genetics, Wenzhou People’s Hospital, 325000 Wenzhou, Zhejiang, China
2 Wenzhou City Key Laboratory of Gynecology and Obstetrics, 325000 Wenzhou, Zhejiang, China
3 Department of Gynecology and Obstetrics, Wenzhou People’s Hospital, 325000 Wenzhou, Zhejiang, China
Abstract
Caroli disease is a rare ductal plate malformation. While most polycystic kidney and hepatic disease 1 (PKHD1)-related biliary phenotypes arise from compound-heterozygous variants, the prenatal implications of homozygous variants remain unclear. Reporting the first prenatal diagnosis linked to a homozygous PKHD1 variant, along with the diagnostic workflow, has direct implications for genetic counseling and recurrence prevention strategies.
A 25-year-old woman, gravida 2, presented with isolated fetal intrahepatic bile duct dilatation at 22+5 weeks of gestation. Ultrasound showed arborizing, avascular tubular channels; and fetal magnetic resonance imaging (MRI) confirmed diffuse intrahepatic involvement with normal renal anatomy. Karyotype analysis and chromosomal microarray were normal. Trio-exome sequencing identified a homozygous PKHD1 c.2507T>C (p.Val836Ala) variant, and Sanger sequencing confirmed parental heterozygosity. Interval surveillance documented enlargement to 47 × 37 × 24 mm. Following multidisciplinary counseling, the pregnancy was electively terminated, and no autopsy was performed. The early, isolated hepatobiliary presentation contrasts with previously reported compound-heterozygous cases.
Combined ultrasound, fetal MRI, and trio-exome sequencing established an etiologic prenatal diagnosis and refined the differential diagnosis from choledochal cyst and cystic biliary atresia. The homozygous c.2507T>C variant likely confers a dosage-dependent, more severe fetal phenotype, thereby expanding the PKHD1-associated spectrum and strengthening genotype–phenotype correlations. These findings provide direct clinical utility and educational value by highlighting key imaging features, outlining a stepwise genomic diagnostic workflow, and emphasizing the utility of preimplantation genetic testing to prevent recurrence.
Keywords
- PKHD1 variants
- Homozygote variants
- Caroli
- prenatal diagnosis
Caroli disease is a rare congenital malformation of the ductal plate characterized by non-obstructive, cystic dilatation of the intrahepatic bile ducts; when congenital hepatic fibrosis or cirrhosis co-occurs, the phenotype is termed Caroli syndrome [1]. Biallelic variants in polycystic kidney and hepatic disease 1 (PKHD1), which encodes fibrocystin/polyductin expressed in hepatobiliary and renal tubular epithelia, anchor Caroli phenotypes within the hepatorenal fibrocystic disease spectrum. PKHD1 is the established cause of autosomal recessive polycystic kidney disease (ARPKD), in which biliary abnormalities and congenital hepatic fibrosis are frequent components [2, 3].
Across published cohorts, most PKHD1-related biliary presentations are compound heterozygous. By contrast, the fetal consequences of homozygous PKHD1 variants, especially regarding whether they can manifest as an isolated hepatobiliary phenotype without renal involvement, remain poorly defined. This uncertainty has practical consequences for prenatal care. When fetal imaging shows intrahepatic bile-duct dilatation with normal kidneys, clinicians must distinguish Caroli disease from mimics such as choledochal cyst and cystic biliary atresia, and interpret exome findings in support of accurate counseling, perinatal planning, and recurrence-risk management.
Here we describe a fetus with diffuse intrahepatic bile-duct dilatation and
normal kidneys in whom trio-based whole-exome sequencing (WES-trio) identified a
homozygous PKHD1 c.2507T
A 25-year-old gravida 2, para 0 (G2P0) woman was referred to our fetal medicine unit after a suspected hepatobiliary anomaly was noted on the second-trimester anatomic survey. She and her partner were non-consanguineous Han Chinese. Medical history and medications were unremarkable aside from prenatal vitamins; there was no tobacco, alcohol, or illicit drug exposure. First-trimester screening at 13+2 weeks showed a nuchal translucency of 1.5 mm (within normal limits), and cell-free DNA screening at 13+2 weeks was low risk for common aneuploidies. A previous pregnancy was electively terminated at 32 weeks after detection of fetal intrahepatic bile duct dilatation; no cytogenetic/genomic testing or post-delivery external examination or autopsy was performed. Fetal sex in the current pregnancy was not disclosed.
At 22+5 weeks, targeted fetal ultrasound demonstrated a mixed-echogenic lesion
in the right hepatic lobe measuring 27
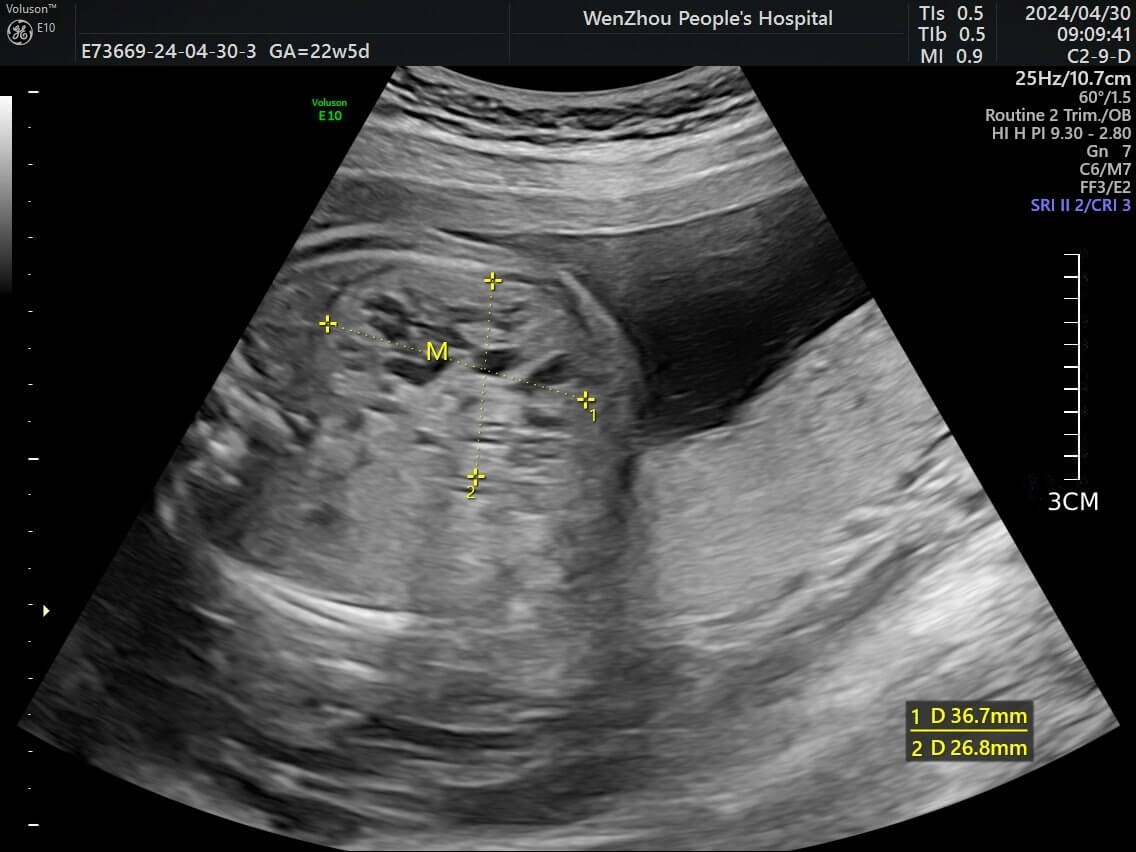 Fig. 1.
Fig. 1.
Fetal ultrasound image demonstrating a dendritic pattern at the intersection of yellow dashed lines within the right lobe of the liver. Image is de-identified.
Fetal MRI performed the same week (1.5-T Siemens; predominantly T2-weighted sequences) confirmed ill-defined hilar bile duct structures and diffuse intrahepatic ductal dilatation. The kidneys, bladder, and intestines were normal, with preserved corticomedullary differentiation and no nephromegaly (Fig. 2). The concordant imaging findings supported a ductal-plate malformation within the Caroli spectrum and prompted invasive genetic testing.
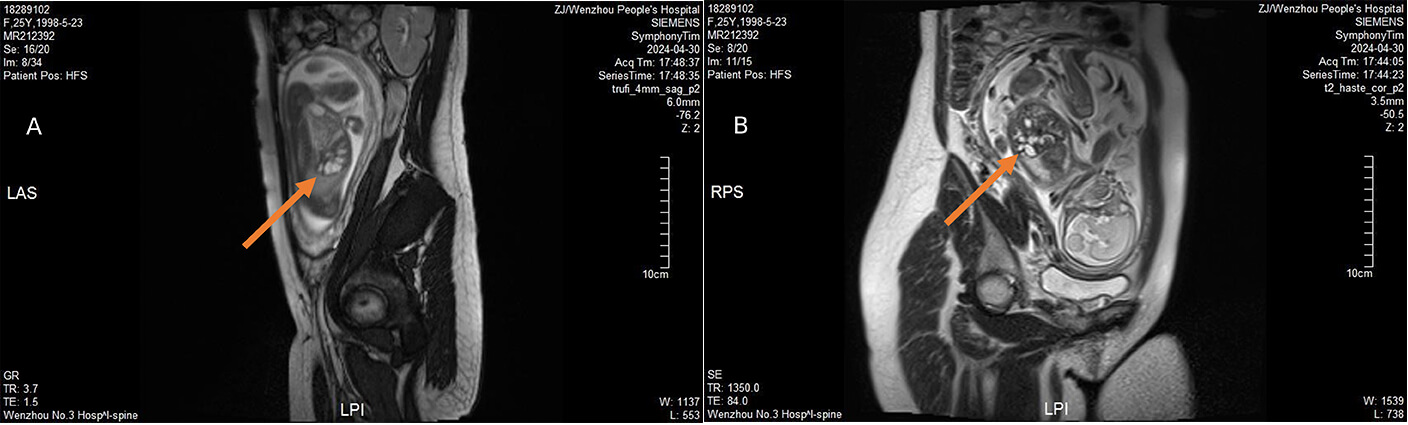 Fig. 2.
Fig. 2.
Fetal MRI findings. (A) MRI shows an ill-defined hilar bile duct (indicated by the orange arrow). (B) MRI reveals dilated intrahepatic bile ducts (indicated by the orange arrow), with no other significant abnormalities detected. Images are de-identified. MRI, magnetic resonance imaging.
Given the apparently isolated hepatobiliary finding and the couple’s adverse
obstetric history, amniocentesis (~35 mL) was performed for
genetic evaluation. Conventional karyotyping and chromosomal microarray analysis
(Affymetrix CytoScan 750K, Lot Number: 901859, Thermo Fisher Scientific, Waltham,
MA, US) showed no abnormalities. Trio-based WES-trio identified a homozygous
PKHD1 c.2507T
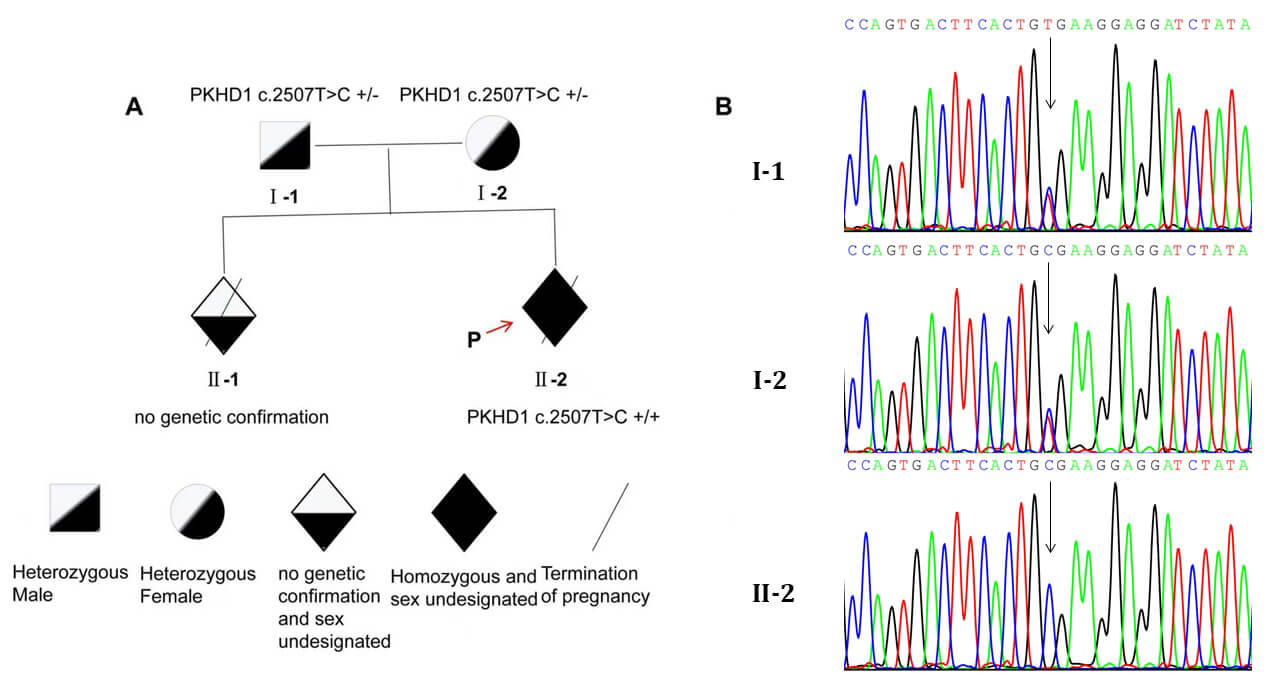 Fig. 3.
Fig. 3.
Pedigree diagram and genetic analysis. (A) Parents are heterozygous carriers
(PKHD1 c.2507T
Upon receipt of the exome result, a multidisciplinary consultation involving maternal-fetal medicine, radiology, and clinical genetics was convened to integrate imaging and genomic data and guide counseling and management. The family received counseling regarding the diagnosis, potential perinatal course, and options for reproductive planning, including preimplantation genetic testing for monogenic disease (PGT-M). Interval surveillance was arranged to monitor lesion evolution and support shared decision-making.
At 25+5 weeks, follow-up ultrasound showed interval enlargement of the
right-lobe lesion to 47
In summary, the combination of arborizing intrahepatic ductal dilatation without
internal vascularity on ultrasound and diffuse intrahepatic involvement with
ill-defined hilar ducts on MRI most strongly supported the Caroli spectrum. A
choledochal cyst was unlikely, given the predominantly intrahepatic, branching
morphology and absence of a solitary extrahepatic cyst. Cystic biliary atresia
was less likely in mid-gestation with patent-appearing intrahepatic channels and
no ancillary sonographic signs. Hepatic hemangioma was disfavored by the lack of
internal Doppler flow and tubular rather than mass-like architecture. Taken
together with the homozygous PKHD1 c.2507T
Variants in the PKHD1 gene are the primary cause of ARPKD. The
PKHD1-encoded protein, fibrocystin/polyductin complex (FPC), interacts
with NEDD4 ubiquitin ligase-interacting protein 2 (NDFIP2), which interacts with
several members of the C2-WWW-HECT domain-containing E3 ubiquitin ligase family.
These interactions alter the subcellular localization and function of these
ligases, leading to increased activity of transforming growth factor
Caroli disease and ARPKD frequently co-occur, with a reported co-occurrence rate of up to 71% [10]. In addition, studies have reported cases of Caroli disease associated with PKHD1 gene variants in certain families, even in the absence of overt renal disease [11, 12]. Imaging is central to diagnosis. Ultrasonography may reveal cystic dilatation of the intrahepatic bile ducts, particularly in complex cases of Caroli syndrome associated with congenital hepatic fibrosis. In the prenatal setting, the spectrum of isolated congenital hepatobiliary malformations (ICHM) most commonly includes choledochal cysts and cystic biliary atresia, but also encompasses congenital hepatic cysts, gallbladder agenesis/non-visualization, and, less frequently, the Caroli spectrum [13]. A concise, literature-based overview of ICHM entities, prenatal imaging clues, and typical outcomes is provided (Table 1, Ref. [14, 15, 16, 17, 18]). Abdominal computed tomography (CT) may demonstrate cystic dilatation with the “central dot sign”, or communication between the bile duct and small ductules “tadpole sign” [19, 20, 21]. MRI and magnetic resonance cholangiopancreatography (MRCP) are key diagnostic modalities for Caroli syndrome, providing detailed information regarding lesion localization, size, and the presence of intrahepatic bile duct stones [22, 23, 24]. Fetal MRI complements ultrasound by delineating the cyst’s relationship to the biliary tree and the hepatoduodenal ligament, improving anatomic confidence for choledochal cyst and other ICHM [15]. For suspected CD/CS, prenatal MRI can reveal the central dot sign—the portal fibrovascular bundle within a dilated intrahepatic duct—thereby increasing diagnostic confidence and has been demonstrated in fetuses with ARPKD [14]. A recent prenatal series further reported a consistent central-dot sign across multiple fetuses diagnosed with Caroli disease, underscoring its in-utero utility [25].
| Entity | Typical prenatal presentation (US) | Helpful imaging clues (US/MRI) | Postnatal confirmation/outcome | Representative ref. |
| Caroli disease/Caroli syndrome | Multiple tubular/branching intrahepatic anechoic channels; often an isolated liver finding | MRI “central dot sign”; assess concomitant ARPKD features | MRCP/CT confirms communication with the biliary tree; risk of cholangitis/portal HTN | [14] |
| Choledochal cyst | Solitary cyst at porta hepatis; may connect to hepatic/gallbladder ducts | Fetal MRI better defines cyst origin and course along the hepatoduodenal ligament | MRCP + surgery (cyst excision, biliary reconstruction) | [15] |
| Cystic biliary atresia | Small hilar cyst with non-visualized/small gallbladder | Cyst size threshold ( |
Early cholangiography; Kasai portoenterostomy | [16] |
| Congenital hepatic cyst | Isolated, unilocular intrahepatic cyst discovered in late gestation | No biliary communication on MRCP; often incidental | Many remain asymptomatic; selective resection if growth/pressure | [17] |
| Gallbladder agenesis/Non-visualization (NVFGB) | GB not seen or very small on US | Fetal MRI can re-identify a small/contracted GB and reduce false-positive NVFGB | Often benign if isolated; evaluate for BA/genetic causes if non-isolated | [18] |
ICHM, isolated congenital hepatobiliary malformations; MRI, magnetic resonance imaging; MRCP, magnetic resonance cholangiopancreatography; CT, computed tomography; HTN, hypertension; GB, gallbladder; ARPKD, autosomal recessive polycystic kidney disease; US, ultrasound; CBA, cystic biliary atresia; CC, choledochal cyst; BA, biliary atresia.
ICHM may be subtle in mild cases, and misclassification between choledochal cyst
(CC) and cystic biliary atresia (CBA) is common; in a systematic review of
prenatal “biliary cysts”, approximately 22% proved to be biliary atresia
postnatally [26]. Similarly, among perinatally detected subhepatic cysts, 15.4%
were ultimately CBA despite sonographic appearances overlapping with CC [27].
Simple biometric cues can assist—cyst width
The c.2507T
| Case | Age (years) | Gender | Genotype | Presentation at detection | Hepatic features | Renal features | Imaging highlights | Diagnosis (Caroli/ARPKD/Both) | Inheritance pattern | Ref. |
| 1 | 4 | M | c.2507T |
Early childhood ARPKD with polycystic kidneys and congenital hepatic fibrosis | Intrahepatic bile duct dilatation; CHF; liver size largely preserved | Bilaterally enlarged kidneys; increased medullary echoes; loss of corticomedullary differentiation; multiple renal cysts | US: enlarged echogenic kidneys with CMD loss; CT/MRI: multiple bilateral renal cysts with intrahepatic bile duct dilatation; spleen normal | Both | AR (compound het.) | Liu et al. [30] |
| 2 | 8 months | M | c.2507T |
Asymptomatic hepatomegaly and nephromegaly were found on routine check | Intrahepatic bile-duct dilatation; serum fibrosis markers elevated; extrahepatic ducts not dilated | Bilaterally enlarged kidneys with loss of corticomedullary differentiation; multiple microcysts; early renal injury (microalbuminuria) | US: enlarged echogenic kidneys with comet-tail; coarse, mesh-like hepatic echotexture; MRCP: mild saccular/branching intrahepatic ductal dilatation | Both | AR (compound het.) | Yang et al. [31] |
| 3 | 5 (Younger of twins) | M | c.2507T |
Splenomegaly by 1 y; at 5 y, presented with anorexia and upper abdominal mass, diagnosed as Caroli disease | Progressed to cirrhosis, portal hypertension with hypersplenism; severe anemia; mild jaundice during follow-up | Developed polycystic kidneys later in the course | US/CT/MRI: intrahepatic biliary dilatation; later bilateral renal cysts | Both | AR (compound het.) | Hao et al. [29] |
| 4 | 5 (Older of twins) | M | c.2507T |
Detected on family work-up; clinically quiescent at report | Intrahepatic bile-duct dilatation without symptoms | No overt renal disease at the time of report | Imaging showed intrahepatic ductal dilatation; no major extrahepatic findings | Caroli | AR (compound het.) | |
| 5 | 18 | F | c.2507T |
Adolescent evaluated for cystic kidney disease; hepatic abnormalities noted | Hepatic fibrosis; mild intrahepatic bile-duct dilatation reported in the cohort/literature | Bilateral renal cysts; renal function status not detailed in the source summary | US/MRI: renal cysts; intrahepatic ductal changes; no extrahepatic duct dilatation specified | Both | AR (compound het.) | Qiu et al. [32] |
| 6 | 17 | M | c.2507T |
Atypical Caroli syndrome with congenital hepatic fibrosis on biopsy; portal hypertension; bile-duct malformation/dilatation; cholestasis | Atypical Caroli syndrome with congenital hepatic fibrosis on biopsy; portal hypertension; bile-duct malformation/dilatation; cholestasis | No polycystic kidney disease; kidneys normal morphology on CT/US | US: dilated third-order intrahepatic bile ducts; CT: hepatosplenomegaly with giant spleen, no intrahepatic cysts; kidneys normal | Caroli | AR (compound het.) | Zhou et al. [34] |
| 7 | 17 | M | c.2507T |
Evaluated for renal cysts with elevated creatinine; hyperuricemia | Not reported at diagnosis | Multiple bilateral renal cysts; creatinine ~276 µmol/L | US/CT: multiple renal cysts; no specific hepatic imaging findings noted | ARPKD | AR (compound het.) | Bi et al. [35] |
| 8 | 54 | M | c.2507T |
Adult-onset renal cysts; clinically stable | Incidental liver cyst | Multiple small ( |
Imaging: small bilateral renal cysts; additional incidental findings (prostate stones; colonic mucinous tubular adenoma; colon polyps) | ARPKD | AR (compound het.) |
CMD, corticomedullary differentiation; AR, autosomal recessive; M, male; F, female; *, stop codon.
The patient reported in this study is homozygous for the c.2507T
Regarding the prognosis of Caroli disease, some patients maintain good long-term health with appropriate medical or surgical management. For example, the literature describes a case in which the patient remained in good health for more than 21 years following surgical intervention [39]. Another study reported four familial cases, in which a male patient presented with acute cholangitis at 6 years of age, while the other three sisters were asymptomatic at diagnosis. All four patients remained alive and asymptomatic for over 22 years at the final follow-up [40]. These findings suggest that, in certain cases, the long-term prognosis for patients with Caroli disease can be favorable. However, Caroli disease can also lead to several serious complications. The most common clinical manifestation is recurrent cholangitis. Additionally, Caroli disease is associated with portal hypertension and cirrhosis, as intrahepatic bile duct dilatation may promote gallstone formation, which can subsequently cause biliary colic, jaundice, and acute pancreatitis [24]. There is also an increased risk of malignancy. A study has shown that approximately 7% of patients who are not candidates for radical surgery develop cancer [41].
In summary, patients with Caroli disease exhibit diverse clinical manifestations
and genetic variants. Although all cases involve PKHD1 gene mutations,
the phenotypic expression varies significantly depending on the specific variant
location. Despite our effort to collect all reported cases carrying the
c.2507T
Despite its limitations—including the lack of postmortem pathological data, which limits our understanding of hepatic fibrosis, and a relatively small sample size—further functional studies are warranted to confirm the pathogenicity of this homozygous variant. Our findings highlight the critical role of prenatal WES-trio combined with imaging in early diagnosis and provide essential support for PGT-M intervention in high-risk families. Future research should include functional experiments and long-term cohort studies to further elucidate the dynamic impact of this variant on bile duct development and its long-term clinical outcomes.
This study reports the first known prenatal diagnosis of isolated Caroli disease
caused by a homozygous PKHD1 c.2507T
The data that support the findings of this study are available from the corresponding author upon reasonable request. Due to ethical and privacy restrictions related to human genomic data, some raw sequencing data are not publicly available. The publicly accessible data have been deposited in Zenodo https://doi.org/10.5281/zenodo.18263668.
JZ and HW designed the case report study. HW, HC, ZZ, and HZ acquired and interpreted the clinical and imaging data. XZ and HP performed the genomic sequencing and interpreted the genetic results. YW performed the literature search and data synthesis. ZX interpreted the imaging findings and designed the figures. JZ critically revised the manuscript for important intellectual content. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
This study was carried out in accordance with the guidelines of the Declaration of Helsinki. This study was approved by the Ethics Review Committee of Wenzhou People’s Hospital (Approval No. KY-202411-009). The authors confirm that written consent for the submission and publication of this case report, including all associated images, clinical data, and other information contained in the manuscript, has been obtained from the patient.
We are grateful to the patient and her family for their participation and for generously consenting to the publication of this case report.
This study was funded by Wenzhou basic scientific research project (Y2023528). This study was funded by Wenzhou City Major Scientific and Technological Innovation Project (Research on New Technologies for Women, Children and Reproductive Diseases) (ZY2024023).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/CEOG45794.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.