






 , Jianmei Gu 1, Zhen Yang 2, Ping Tang 1,*
, Jianmei Gu 1, Zhen Yang 2, Ping Tang 1,*
1 Department of Fetal Medicine, Jiaxing Maternity and Children Health Care Hospital (Afffliated Women and Children Hospital Jiaxing University), 314009 Jiaxing, Zhejiang, China
2 BGI Genomics, 201321 Shanghai, China
Abstract
Bartter syndrome (BS) is a rare autosomal recessive genetic disorder, typically characterized by hypokalemic metabolic alkalosis, and sometimes accompanied by hyponatremia and hypochloremia. BS is caused by mutations at multiple gene loci with strong genetic heterogeneity, making accurate genetic analysis crucial for diagnosis.
We collected peripheral blood samples from three family members: an 8-week pregnant woman, her husband, and their daughter, with BS symptoms. Whole-exome sequencing (WES) and whole-genome sequencing (WGS) were performed, with variants verified by Sanger sequencing. Minigene assays were used to investigate splicing effects, and in silico predictions were conducted to assess the impact of mutations on protein structure. A prenatal diagnosis was performed at 18 weeks of gestation via amniocentesis and Sanger sequencing.
This case highlights three critical values. First, clinically, WGS provides a feasible solution for diagnosing BS cases with negative WES results, which helps reduce hypokalemia-related complications. Second, this is the first report of the SLC12A1 c.2961-647T>G and c.1153G>A variants, and integrated validation confirms their pathogenicity, expanding the spectrum of pathogenic SLC12A1 variants. Third, our findings guide clinicians to consider deep intronic variants and WGS application in unexplained hereditary renal diseases, which is highly relevant to current prenatal diagnosis and genetic counseling practices. WGS can identify deep intronic variants in SLC12A1, and functional experiments can strengthen pathogenicity evidence, providing an effective prenatal diagnostic approach for families with a history of BS.
Keywords
- bartter syndrome
- genetic disease
- prenatal diagnosis
- whole-genome sequencing
- deep intron
Bartter syndrome (BS) is a rare hereditary renal disorder characterized by tubulopathy leading to electrolyte imbalances, including hypokalemia, metabolic alkalosis, and polyuria, which significantly affect quality of life [1, 2, 3]. BS is divided into five subtypes, with types I and II being the most severe, often presenting as an antenatal form of BS [4]. Type I BS is the most common, but the diverse clinical manifestations of this type and similarity to other kidney diseases make diagnosing type I BS challenging [5].
With the development of next-generation sequencing (NGS) technologies, an
increasing number of studies have shown that genetic mutations are closely
associated with the onset of BS [6, 7]. However, BS can be classified into five
types depending on the causative gene mutation, with each type associated with a
specific gene: solute carrier family 12 member 1 (SLC12A1) (MIM600839,
type I), inwardly rectifying subfamily J member 1 (KCNJ1)
(MIM600359, type II), chloride channel, voltage-sensitive Kb (CLCNKB)
(MIM602023, type III), Barttin (BSND) (MIM606412, type IVa),
CLCNKB, and chloride channel, voltage-sensitive Ka
(CLCNKA) (MIM602024, type IVb), or melanoma-associated
antigen D2 (MAGED2) (MIM 300155, type V). Currently, more than 100
mutations in SLC12A1 have been reported, of which 65 are associated with
type I BS and nine are found in the general population and are associated with
lower blood pressure levels [8, 9, 10]. Common mutations are located in exons and
can be detected using the current whole-exome sequencing (WES) techniques.
However, conventional WES—focusing on protein-coding
regions and flanking splice sites—often fails to detect deep intronic variants
(located
In this case, we used WGS to diagnose a child with suspected BS. Using WGS, we conducted a comprehensive high-throughput analysis of the genome, including both coding and non-coding regions. This allowed us to identify the gene variants associated with BS accurately. This not only fills the diagnostic gap but also provides clinically relevant evidence for the use of WGS in the prenatal diagnosis of BS.
This study was approved by the hospital Ethics Committee of Jiaxing Maternal and Child Health Care Hospital (2024-Y-107). Informed consent was obtained from all study participants, and their family members provided written informed consent. Parental consent was obtained for gathering prenatal fetal amniotic cell samples at 18 weeks of pregnancy. Peripheral blood samples were collected from the parents of the proband.
The polymerase chain reaction (PCR) products were purified and sequenced using
BGI Genomics (Shenzhen, Guangdong, China). Candidate mutation sites were identified
following DNA extraction, quality inspection, genomic library construction,
target-region capture sequencing, and bioinformatic analysis. A total of 500 ng
genomic DNA was sonicated, yielding fragments ranging from 250 to 300 bp. After
adding a mixture of terminal repair buffer and repair enzyme, the fragments were
reacted on a PCR instrument. Then, the end repair system was added to the ligase,
buffer, and linker, and the reaction was incubated at 22 °C for 30 minutes. After
purification with magnetic beads, the sample mixture was added to the PCR
amplification reaction system for 8–10 cycles. After magnetic bead purification,
the library quality was assessed: the library was 25 µL, with a
concentration
The c2961-647T
The transmembrane region analysis (TMHMM) website was used to predict the effect
of the SLC12A1 gene c.1153G
Sanger sequencing validation of fetal variants was conducted based on whole-genome high-throughput sequencing results, and the PCR products were purified and sequenced on an ABI 3130. National Center for Biotechnology Information (NCBI) Basic Local Alignment Search Tool (BLAST) searches were performed against the human reference genome (hg19) and other reference genomes to identify mutation sites, and the pathogenicity of the variants was determined according to the American College of Medical Genetics and Genomics (ACMG) 2015 genetic variation classification criteria [11]. These variants are classified as pathogenic variants. The ClinGen Working Group on sequence variation Interpretation and the Association for Clinical Genomic Science (ACGS) were referred to for further interpretation of the guidelines.
A woman, who was eight weeks pregnant and had previously given birth to a daughter suspected of having BS, was included. In a previous pregnancy, ultrasonography at 28 weeks of gestation revealed an amniotic fluid index of 310 mm, consistent with polyhydramnios. The amniotic fluid level continued to increase progressively, resulting in preterm births at 30 weeks of gestation. The baby girl weighed 1300 g at birth. At the age of 2 years, the daughter developed polydipsia, with a daily water intake exceeding 2500 mL, accompanied by polyuria, with a daily urine volume of 2000 mL (normal range is about 400–600 milliliters/24 hours), as well as nausea and vomiting. Her 24-hour urinary calcium was 175.6 mg (equivalent to 13.5 mg/kg) (normal range is 150–600 milligrams per 24 hours, about 2.5–7.5 millimoles per 24 hours), and her blood potassium level was 2.5 mmol/L (serum potassium level is 3.5–5.5 millimoles per liter). Renal ultrasonography revealed petal-like medullary calcification. The patient is currently on long-term potassium supplementation.
The parents were not consanguineous, had no clinical symptoms, and had normal blood potassium levels. We reviewed the medical history, blood test results, and treatment plan of the 8-year-old daughter of the proband, as well as the history of polyhydramnios and preterm birth during the previous pregnancy. The Department of Pediatrics at our hospital confirmed a clinical diagnosis of BS (Fig. 1A).
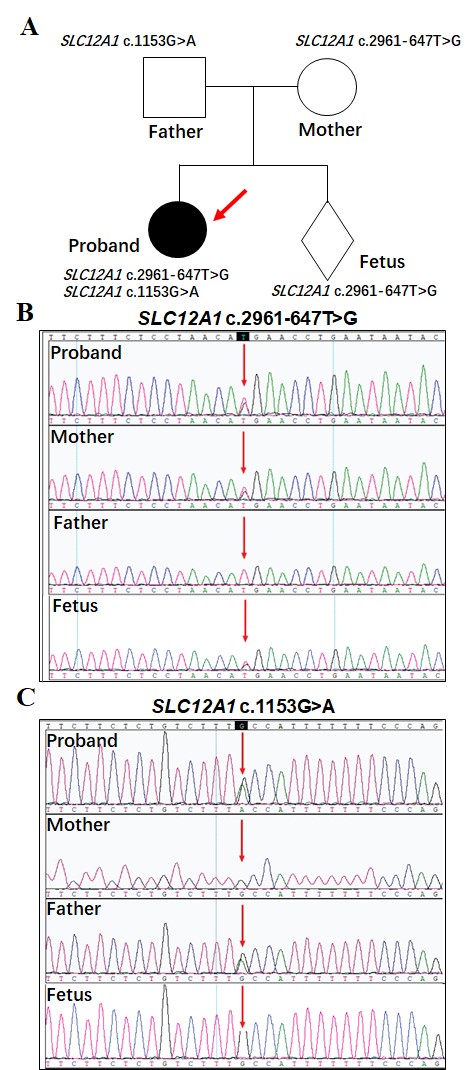 Fig. 1.
Fig. 1.
Identification and analysis of Bartter syndrome-related
mutations in a family. (A) Pedigree analysis shows that the proband carries both
a paternal missense mutation (SLC12A1 c.1153G
The patient was admitted to the Children’s Hospital for suspected BS; however,
prior WES performed at other hospitals did not identify pathogenic variants, and
a prenatal diagnosis was requested. Therefore, we performed WGS using the total
genomic DNA extracted from the proband and his parents. The proband carried a
paternal missense mutation (SLC12A1 c.1153G
To determine whether this variant causes abnormal splicing of SLC12A1
and affects the gene expression and function, we performed minigene splicing
assays in vitro. The software predicted that SLC12A1
c.2961-647T
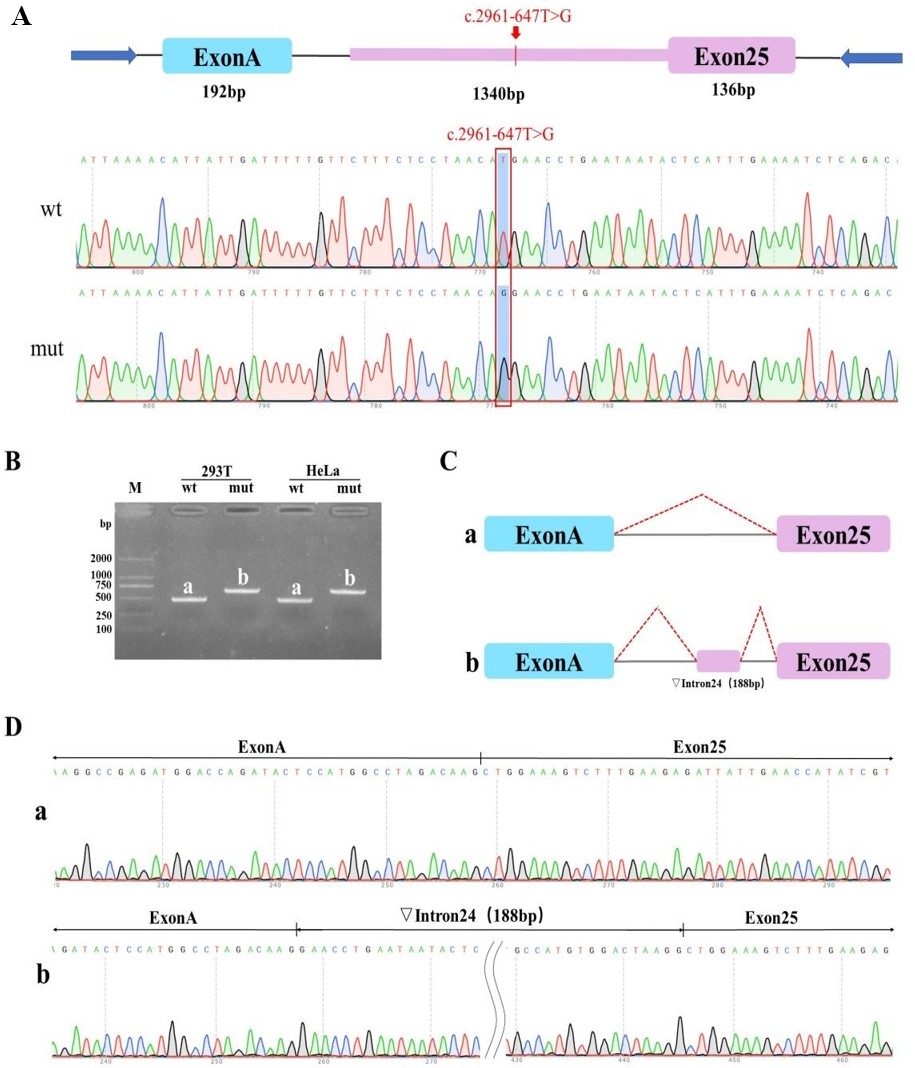 Fig. 2.
Fig. 2.
Analyses of abnormal splicing caused by the SLC12A1;
c.2961-647T
We compared the differences between wt SLC12A1 and mutant
SLC12A1 c.1153G
 Fig. 3.
Fig. 3.
Effect of genetic variants on the secondary structure of the
SLC12A1 protein. This figure compares the secondary structures of the wild-type and mutant (c.1153G
We identified 385 mutations in the NCBI functional domain database, located in the 2a30 functional domain
(https://www.ncbi.nlm.nih.gov/). Using the TMHMM
website, the predicted SLC12A1 protein contained 12 transmembrane helices
(TMhelix) (Fig. 4), and the mutation at position 385 of the protein was predicted
to be located in the transmembrane helix (TMhelix). After the self-optimized
method with profile alignment (SOPMA) database analysis, the proportion of alpha
helices (Hh), extended strands (Ee), random coils (Cc), and other structures
changed, suggesting that the mutation affected the secondary structure. In
contrast, the proportions of the structures, such as the beta turn (Tt), 310
helix (Gg), Pi helix (Ii), beta bridge (Bb), bend region (Ss), and ambiguous
states, did not change. The c. 1153G
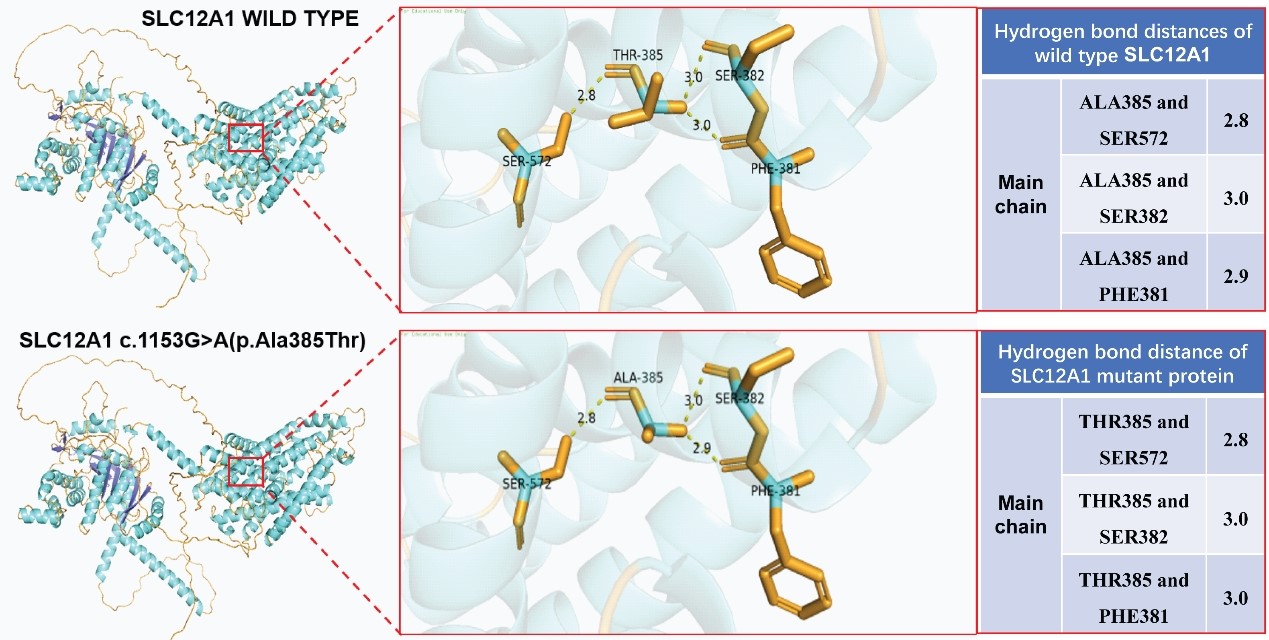 Fig. 4.
Fig. 4.
Effect of genetic variants on the tertiary structure of the
SLC12A1 protein. A comparison of the hydrogen-bond distances in wild-type and
mutant (c.1153G
We analyzed the structural and surface potential changes in both wild-type and mutant (p.Ala385Thr) SLC12A1 proteins. The wild-type SLC12A1 protein had a specific three-dimensional structure, and a particular surface potential distribution was observed around the locally magnified Ala385. The overall structure of the mutant SLC12A1 protein is similar to that of the wild-type protein. However, owing to the Ala385 to Thr385 mutation, the potential of the local surrounding surface changed significantly. This change in surface potential may affect interactions between the protein and other molecules, thereby influencing the biological functions and related physiological processes of the protein. Fig. 5 demonstrates the potential changes in surface potential of the SLC12A1 protein induced by mutations.
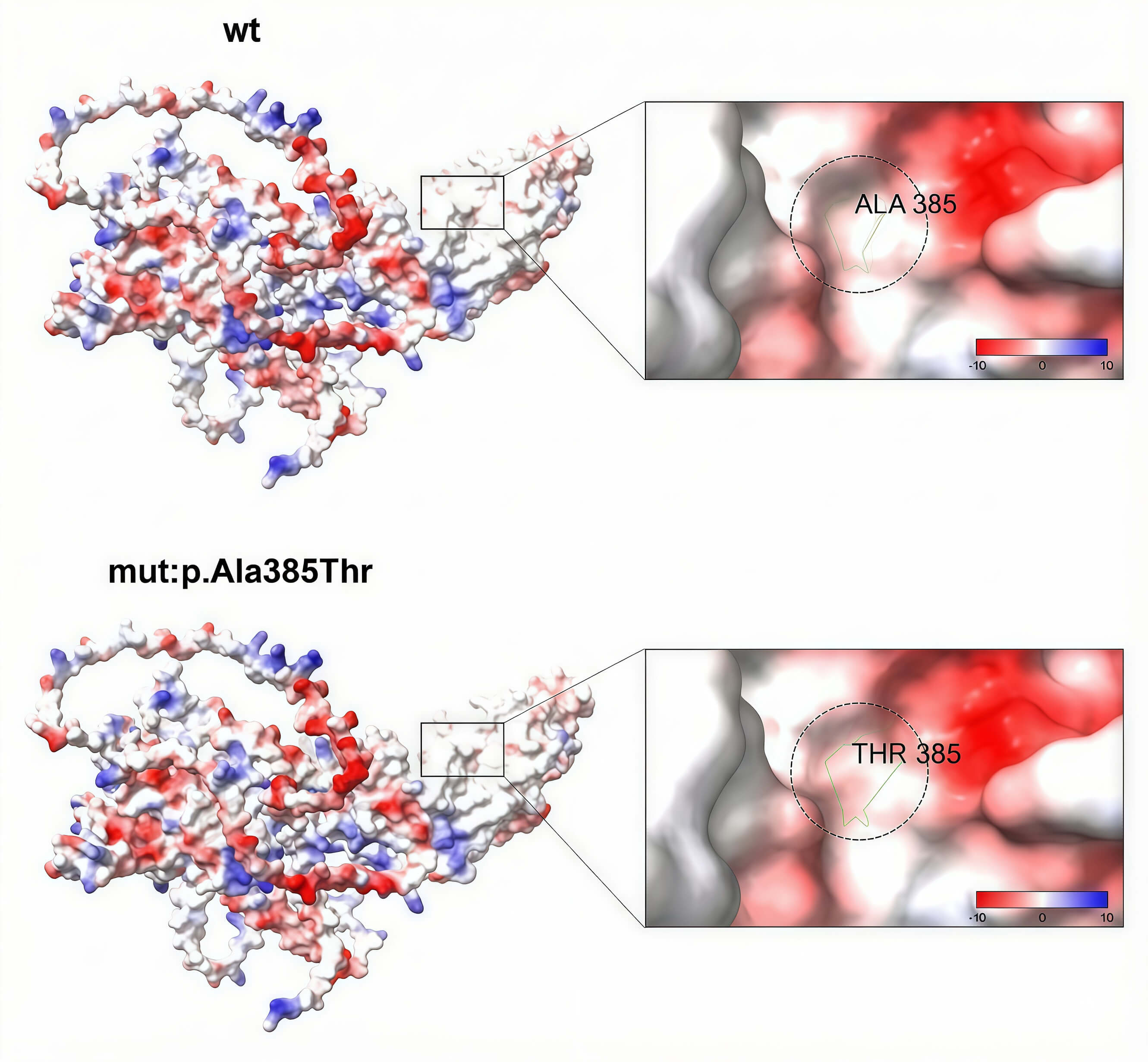 Fig. 5.
Fig. 5.
Surface potential changes in the wild-type and mutant
(p.Ala385Thr) SLC12A1 proteins. The results of minigene splicing assays that
investigated the effect of the SLC12A1 c.2961-647T
Pregnant women with BS usually experience polyhydramnios between 18 and 30 weeks
of gestation, and their amniotic fluid index can reach 330–500 mm [12, 13]. In
cases of gestational polyhydramnios, the risk of fetal BS should be considered
after excluding pathological conditions such as gestational diabetes. Therefore,
this study provides valuable insights. In this case, the patient was a pregnant
woman with a history of adverse birth outcomes who required a prenatal diagnosis.
After the WES results were negative, WGS was performed to detect two variants of
uncertain significance in the SLC12A1 gene, which is associated with
type I BS and was found to be related to the phenotype of the subject. The
NM_000338.2: c.2961-647T
This variant was found to have enhanced pathogenicity through functional
verification. Specifically, the corresponding locus of the variant in the
SLC12A1 gene (NM_000338.2: c.1153G
Since WGS generates vast amounts of data, more sophisticated bioinformatics analyses are necessary, and genetic variants of uncertain significance have frequently emerged. Currently, the predominant classification criteria and guidelines for genetic variants published in 2015 are used [15]. For Mendelian diseases, a five-level conclusion based on 28 independent evidence judgments was proposed, and a classification system for sequence variants was established.
All compound heterozygous variants of SLC12A1 detected in this study were initially of uncertain significance and did not provide a basis for prenatal diagnosis. However, after meticulous consultation, functional testing was performed to validate and improve the pathogenicity rating.
SLC12A1 NM_000338.2: c.2961-647T
The supporting evidence for reclassification includes:
• PP3: Multiple in silico analyses (conservation prediction, evolutionary
analysis, and splice site impact assessment) consistently predicted harmful
effects on the gene or gene product. • PM2: The variant was absent in normal control populations (Exome Sequencing Project
(ESP), 1000 Genomes,
and Exome Aggregation Consortium (EXAC) databases) or presented as an extremely low-frequency locus in recessive
genetic diseases. • PS3: In vitro minigene assays confirmed that this variant affects
SLC12A1 expression and impairs protein function, consistent with
in silico predictions that this variant disrupts normal mRNA levels. • PP4: The phenotype and family history of the carrier were highly consistent with
a monogenic inherited disorder, supporting the potential pathogenicity of the
variant.
Collectively, the evidence (PM2+PS3+PP3+PP4) confirmed the pathogenicity of this intronic variant.
SLC12A1 NM_000338.2: c.1153G
• PP3: In silico predictions (conservation, evolutionary, and splice site
impact analyses) indicate harmful effects on the gene product. • PM2: The variant was not detected in normal control databases (ESP, 1000
Genomes, EXAC) or exhibited extremely low frequency in recessive disease cohorts. • PM3: In the context of recessive inheritance, the variant was identified in
trans with another pathogenic allele. • PP4: The phenotype and family history of the carrier were highly consistent with
a monogenic disorder, reinforcing the clinical relevance of the variant.
Functional assessments further demonstrated that the c.1153G
Given the stringency required for the prenatal diagnosis, clarifying the pathogenicity of these SLC12A1 variants through systematic validation provides a critical basis for accurate prenatal risk assessment.
Given the particularities of prenatal diagnosis, identifying the mutation sites
of pathogenic genes and their pathogenicity requirements remains necessary [17].
Therefore, we performed functional tests to clarify the significance of the
pathogenicity. Presently, mutations that affect splicing are often tested for
protein function, while protein structure is predicted using software that does
not affect splicing. However, the evidence items are frequently not accepted.
Similar to SLC12A1, NM_000338.2: c.2961-647T
As a single case report, although the novel deep intronic SLC12A1 variant identified here was validated by WGS (after WES failure) and supported by functional assays, the rarity of this variant and the uniqueness of the genetic background of the patient limit the generalizability of our findings. We cannot conclude whether this variant is a common pathogenic cause of BS in broader populations, nor can we confirm its genotype–phenotype correlation in other patients with similar clinical manifestations. Hence, future studies with larger cohorts (e.g., multi-center case series) are needed to verify the prevalence and clinical significance of this variant.
WGS successfully identified the deep intronic variant missed by WES. However, we did not systematically explore other non-coding regulatory regions that might also contribute to the pathogenesis of BS. Additionally, due to a lack of population frequency data for this specific deep intronic variant, we could not fully exclude the possibility of rare benign variation, despite the predicted pathogenicity of the variant by in silico tools and functional experiments.
Our functional assays (e.g., minigene experiments) confirmed the pathogenic effect of the variant on SLC12A1 function; however, these assays did not provide direct structural validation of the protein (e.g., via X-ray crystallography or cryo-EM). These limitations mean that the specific molecular mechanism through which the variant disrupts SLC12A1 function remains partially unclear.
To address the above limitations, future research should focus on two aspects: (1) Expanding the cohort to include more patients with BS who tested negative for WES, to explore the prevalence of deep intronic SLC12A1 variants and the associated genotype–phenotype correlations; (2) performing functional experiments in renal tubule-specific cell models to better simulate in vivo conditions.
Our case underscores one key clinical value of WGS-based diagnosis for BS: this study optimizes prenatal genetic counseling. Before the WGS results, the family faced uncertainty about the condition of the fetus (e.g., “whether the renal abnormality was genetic” or “whether future pregnancies would be at risk”). WGS can effectively identify deep intronic variants in SLC12A1, and functional experiments can provide critical evidence for the pathogenicity of the variant. The identification of the SLC12A1 variant allowed us to inform the family of the autosomal recessive inheritance pattern (both parents were found to be carriers via subsequent testing) and to offer an effective prenatal diagnostic approach for families with a history of BS.
Our findings are highly relevant to current clinical practice and research: For clinicians (especially neonatologists and prenatal diagnosticians), our case provides a “clinical algorithm” reference: when a patient presents with clinical features of BS (e.g., renal salt wasting, hypokalemia) but WES is negative, deep intronic variants should be considered, and WGS should be recommended as the next step. This directly addresses a common clinical dilemma and helps reduce misdiagnosis. For prenatal diagnosis programs, our case provides real-world evidence for incorporating WGS into the diagnostic workflow of suspected BS.
PM2, Pathogenic Moderate evidence 2; PM3, Pathogenic Moderate evidence 3; PP3, Pathogenic Supporting evidence 3; PP4, Pathogenic Supporting evidence 4; PS3, Pathogenic Strong evidence 3; BS, Bartter syndrome; WGS, whole-genome sequencing; VUS, variants of uncertain significance; NGS, next-generation sequencing; ACMG, American College of Medical Genetics and Genomics; ACGS, the Association for Clinical Genomic Science; PTC, premature stop codon; WES, Whole-exome sequencing; CNV, copy number variations; TMHMM, Transmembrane Protein Homology Model; TMhelix, transmembrane helices; Hh, alpha helices; Ee, extended strands; Cc, random coils; Tt, beta turn; Gg, 310 helix; Ii, Pi helix; Bb, beta bridge; Ss, bend region.
All data generated or analyzed during this study are included in the article. Additional raw/processed data supporting the conclusions of this article will be shared by the lead contact upon reasonable request. The charts presented in this manuscript are original works of the authors, have not been previously published, and do not involve third-party copyrights. The dataset used in the current research is available from the corresponding author upon reasonable request.
XW designed this study. XW, ZY and JG conducted research. PT provided critical guidance on the study design, offered in-depth suggestions on the research protocol optimization, and provided substantial editorial advice on the manuscript structure, logical framework, and academic expression during the writing process. XW analyzed these data. XW and JG jointly wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The study was carried out in accordance with the guidelines of the Declaration of Helsinki. Received approval from the Prenatal Diagnosis Ethics Committee at the Maternal and Child Health Care Hospital of Jiaxing, Zhejiang, China (Ethic Approval Number: 2024-Y-107), and all of the participants provided signed informed consent.
Special thanks to the staff of the Prenatal Diagnosis Laboratory at Jiaxing Maternal and Child Health Hospital for providing technical assistance in sample sequencing. We also appreciate the constructive comments from the reviewers, which greatly improved the quality of this manuscript. Finally, we would like to express our gratitude to all the research participants for their collaboration.
This study was also funded by Project of Science and Technology Bureau of Jiaxing, China (2022AY10028); Medical and Health Technology Project of Zhejiang, China (2023KY1220).
The authors declare that they have no affiliations with or involvement in any organization or entity with any financialinterest in the subject matter or materials discussed in this manuscript. As a third-party company, BGI Genomics did not participate in the preparation and publication of the manuscript.
During the preparation of this work the authors used ChatGpt-3.5 in order to check spell and grammar. After using this tool, the authors reviewed and edited the content as needed and takes full responsibility for the content of the publication.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/CEOG45631.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.