










1 Department of Third Clinical Medical College of Nanchang University, The First Hospital of Nanchang, 330008 Nanchang, Jiangxi, China
2 Department of Gynecologic Oncology, Jiangxi Cancer Hospital, 330029 Nanchang, Jiangxi, China
3 Department of Nanchang Key Laboratory for Precision Therapy of Gynecological Tumor, The First Hospital of Nanchang, 330008 Nanchang, Jiangxi, China
4 Department of Jiangxi Key Laboratory of Cancer Metastasis and Precision Treatment, The First Hospital of Nanchang, 330008 Nanchang, Jiangxi, China
5 Department of Clinical Medicine College, Jiangxi University of Chinese Medicine, 330001 Nanchang, Jiangxi, China
6 Department of the First Clinical Medicine, Gannan Medical University, 341000 Ganzhou, Jiangxi, China
Abstract
Background: Immune infiltration of the tumor microenvironment offers unlimited possibilities for therapeutic strategies in cervical cancer, where GRAP2 is an adaptor protein engaged in diverse signal activations. However, uncertainty exists regarding GRAP2’s prognostic significance and its relationship to immune infiltration. Methods: The data on cervical cancer cases were downloaded from The Cancer Genome Atlas (TCGA) database. The ESTIMATE computational technique was utilized to calculate the amount of immunological and stromal components, which helped us to identify the differential expression genes (DEGs). Among them, GRAP2 was considered to be related to overall survival based on a protein-protein interaction network and a univariate Cox regression analysis. Thus, based on the Gene Expression Omnibus (GEO) and TCGA databases, we evaluated GRAP2’s influence on clinical prognosis. Furthermore, GRAP2 expression was analyzed by Gene Set Enrichment Analysis (GSEA). Finally, we used CIBERSORTx analysis to assess the proportion of tumor-infiltrating immune cells (TICs) and the connection between GRAP2 and the tumor immune microenvironment. Results: ESTIMATEScore was associated with cervical cancer patient’s prognosis. There are 791 DEGs and 11 potential key genes were identified including GRAP2. In survival analyses with clinical information, We found that the GRAP2 high expression group exhibited a significantly longer overall survival (OS) than the low expression group and that the gene expression gradually declined as the Federation of International of Gynecologists and Obstetricians (FIGO) stage and M classification increased. GRAP2 was strongly linked with immunity and metabolism, according to GSEA. Finally, we discovered that 11 different TIC types and GRAP2 expressions were linked. Conclusions: GRAP2 may be a novel immune-related prognosis biomarker in cervical cancer.
Keywords
- cervical cancer
- GRAP2
- tumor microenvironment
- immunotherapy
- prognostic biomarker
The term cervical cancer is used to define the malignant neoplasm that arises from the uterine cervix, whereby it poses a threat to female health. Persistent high-risk human papillomavirus (HPV) infection is a primary etiological factor in cervical cancer. Although such an important mechanism has been identified (which makes the issue of cervical cancer one of the most preventable types of cancer), cervical cancer is persistently the fourth most common female cancer worldwide [1]. In the United States, which is the highest quality medical resource owner, cervical cancer took the lives of 4152 women in 2019, half of whom were under the age of 50 [2]. Furthermore, due to low prophylactic HPV vaccination rates, the number is larger in most low- to middle-income countries, wherein cervical cancer has a higher incidence and mortality [3, 4].
Although cervical cancer patients can be categorized according to the International Federation of Gynecology and Obstetrics (FIGO) to estimate their tumor burden and prognosis, the therapeutic strategy varies according to the tumor stage and the status of the patient. Chemotherapy and radiation therapy are the standard therapeutic strategies. For women with low-risk, early-stage disease, conservative, fertility-preserving surgical techniques are now considered the standard of care [5]. However, for advanced-stage patients with metastatic or recurrent disease, the overall prognosis remains poor [6]. Revolutionary immunotherapy has been able to provide hope for patients in this situation, and this therapy aims to recruit and “educate” immune cells to target cancer cells more effectively and specifically. The major strategy includes adoptive cell therapy (ACT) and reversing immunosuppression or effector T-cell suppression. Nevertheless, solid tumors have generally been less responsive to ACT [7], whereas only pembrolizumab (which is a PD1-blocking antibody) has been approved by the Food and Drug Administration (FDA) for PD-L1-positive metastatic or recurrent cervical cancer; moreover, even the response rate for these patients is only ~15% [8].
A sophisticated tumor microenvironment (TME) offers the foundation for immunotherapy, which consists of extracellular matrix, immune cells, inflammatory cells, fibroblasts, and surrounding blood vessels. Several special biomarkers in the TME have also been indicated to improve the prediction of several cancer prognoses. Approximately 1 decade ago, immune infiltration in the TME was observed as a prognostic factor that should not be ignored [9]. According to recent research, tumor-infiltrating immune cells (TICs) and stromal components play an important role in the development of breast cancer [10, 11], colorectal cancer [12], and cutaneous melanoma [13].
In this study, we screened differentially expressed genes (DEGs) related to the TME and clinical characteristics, through The Cancer Genome Atlas (TCGA) database and Gene Expression Omnibus (GEO), as well as several bioinformatic algorithms and tools, such as ESTIMATE algorithms and CIBERSORTx. ESTIMATE is a tool to predict tumor purity by using expression data to estimate stromal and immune cells in tumor; and it generates three scores: StromalScore (records the presence of stroma in the tumor tissue) ImmuneScore (represents the infiltration of immune cells in the tumor tissue) ESTIMATEScore (infers tumor purity) [14]. CIBERSORTx is a tool for the deconvolution of the expression matrix of human immune cell subtypes based on linear support vector regression [15]. Further analysis confirmed that GRAP2 may be a novel and potential immune-related prognosis biomarker for cervical cancer.
The transcriptome profiling data of patients with cervical cancer that we analyzed were retrieved from the TCGA database (https://portal.gdc.cancer.gov/) on May 30, 2022. The data included 309 RNA-seq files. The tumor-corresponding clinical characteristics of cervical cancer were also downloaded from TCGA, and the data included 304 cases of tumors. The GSE52903 cohort was downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/), which includes 55 cervical cancer RNA-seq files and corresponding clinical characteristics.
All of the transcriptome data were analyzed by using the “ESTIMATE” R package (https://www.r-project.org, v1.0.13) via Rstudio (https://www.rstudio.com, v2022.02.2+485). The calculated immune, stromal, and ESTIMATE scores were observed to display a component ratio in TME immune cell infiltration. ESTIMATES scores consisted of both ImmuneScores and StromalScores. Higher scores indicated a larger number of components in the TME.
The samples were divided into the “high scores” group and “low scores” group
according to the medians of the ImmuneScores, StromalScores, and ESTIMATEScores.
A Kaplan–Meier survival analysis was performed in two groups via the R package
“survival” (https://www.bioconductor.org/, v3.3-1) and “survminer” (https://www.bioconductor.org/, v0.4.9). The primary prognostic endpoint
was overall survival (OS). In addition, some clinical characteristic data were
permitted to assess the associations with the scores, which included age, stage
(defined by FIGO), histopathological grades, and Tumour, Nodes, and Metastasis
(TNM) stage. These comparisons were carried out using the Kruskal-Wallis rank-sum
test, with p
DEGs were distinguished by using the “high scores” group and “low scores”
group via the R package “limma” (https://www.bioconductor.org/, v3.48.3). A gene was only regarded as a DEG if
two conditions were met: (1) false discovery rate (FDR)
Gene Ontology (GO) analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis with 791 DEGs were performed by using the R package “clusterProfiler” (https://www.bioconductor.org/, v4.0.5) and visualized by using the website HIPLOT (https://hiplot.com.cn) and GOBubble Plot module (https://www.bioconductor.org/, v0.1.0).
The protein and protein
The concerned gene GRAP2 was selected. Afterward, we summarized the
correlation between clinical information and GRAP2 expression of
patients in the TCGA database, Pearson’s Chi-squared test, Fisher’s exact test,
and Wilcoxon rank sum test were used to test the p-value. The expression
of each gene was matched to clinical features using “ggpubr” (https://www.bioconductor.org/, v0.4.0) and
“survival”. For these comparisons, a Wilcoxon rank-sum was employed, and
p
To further explore the role of GRAP2 in cervical cancer, GSEA, a
computational method that determines whether an a priori defined set of genes
shows statistically, was performed by using GSEA (
http://www.gsea-msigdb.org/gsea/index.jsp, v4.2.3) software to analyze the gene matrix of
GO and KEGG associated with the gene GRAP2 expression level. Moreover,
p
The website “CIBERSORx” (https://cibersortx.stanford.edu/) provides the CIBERSOR tool to evaluate the content of immune cells in tumor cases. The Wilcoxon rank-sum test and Pearson’s correlation were used to assess the proportions and gene expression. The results were visualized by using the “vioplot” (https://www.bioconductor.org/, v0.3.7), “ggplot2” (https://www.bioconductor.org/, v3.3.2), and “ggpubr” R packages.
The current study was performed by using the following analysis process (Fig. 1). First, we downloaded the RNA-seq profiles of 304 cervical cancer cases and the corresponding clinical information from the TCGA and GEO databases. Afterward, calculations of TIC ratios and immune and stromal proportions were carried out separately using the CIBERSORT and ESTIMATE algorithms. We identified DEGs between groups with low and high ImmuneScore and StromalScore. Simultaneously, clinical information and TME scores were integrated for univariate Cox regression analyses, and we found that 147 genes showed a significant association with prognosis (the Cox key genes). Subsequently, the DEGs were subjected to PPI network analysis and univariate Cox regression analyses, and 30 genes were selected by the cytoHubba plugin (the PPI key genes). Finally, an intersection analysis of Cox key genes and PPI key genes was used to obtain 12 prognosis key genes. The gene GRAP2 attracted our attention; therefore, GRAP2 was further analyzed, including expression, survival, and clinicopathological characteristics, as well as GSEA and correlations with TICs proportions.
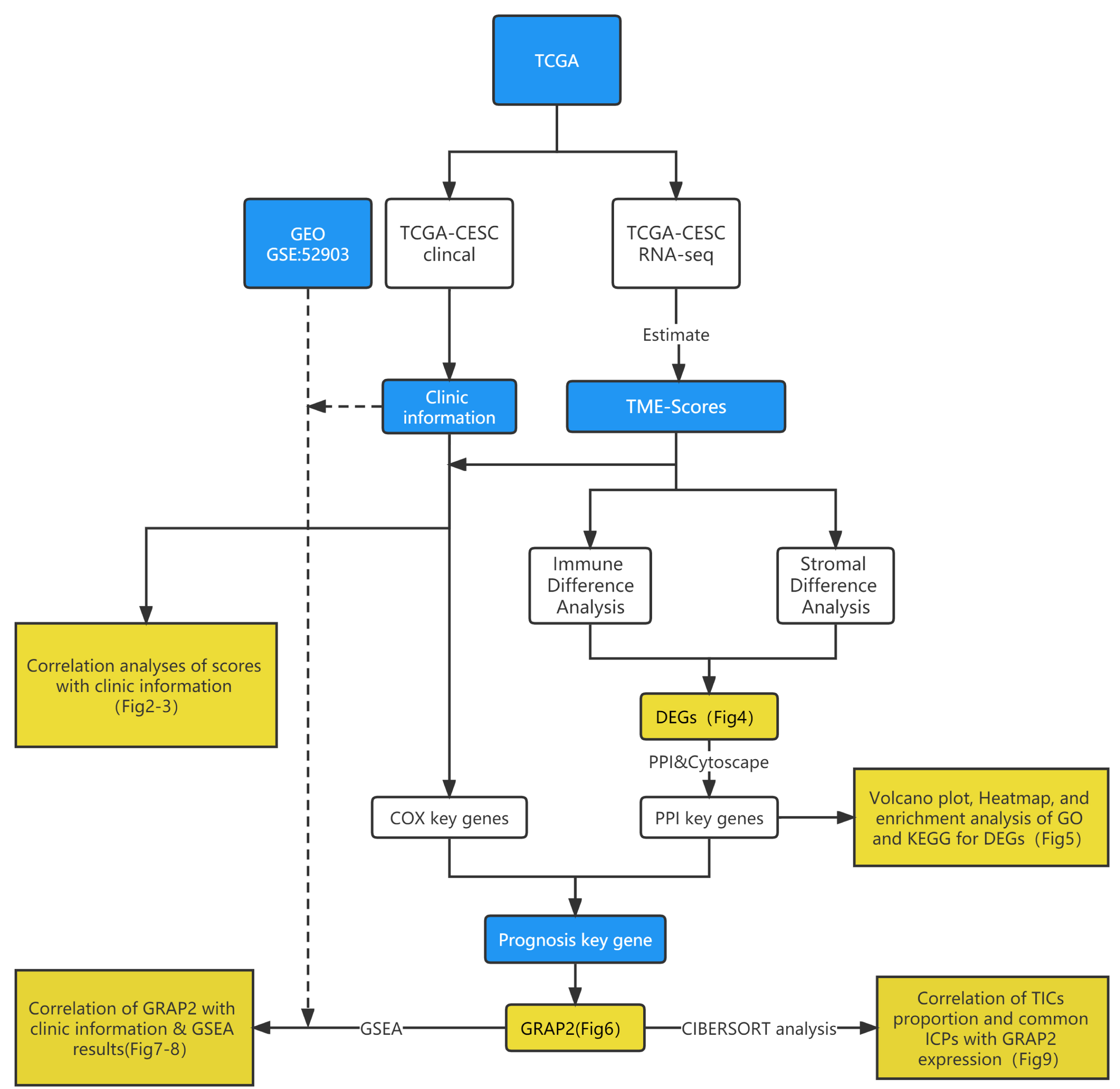 Fig. 1.
Fig. 1.Overview of this study’s analytical flowchart.
Cervical cancer patients’ immune infiltration scores were evaluated for prognoses (Fig. 2). For each of the TME scores (ImmuneScore, StromalScore, and ESTIMATEScore), half of the 304 tumor samples (with complete clinical data) were classified into the high score group (152 cases), and the other half of the samples were classified into the low score group (152 cases) based on each median score. Thereafter, Kaplan–Meier survival curves were constructed.
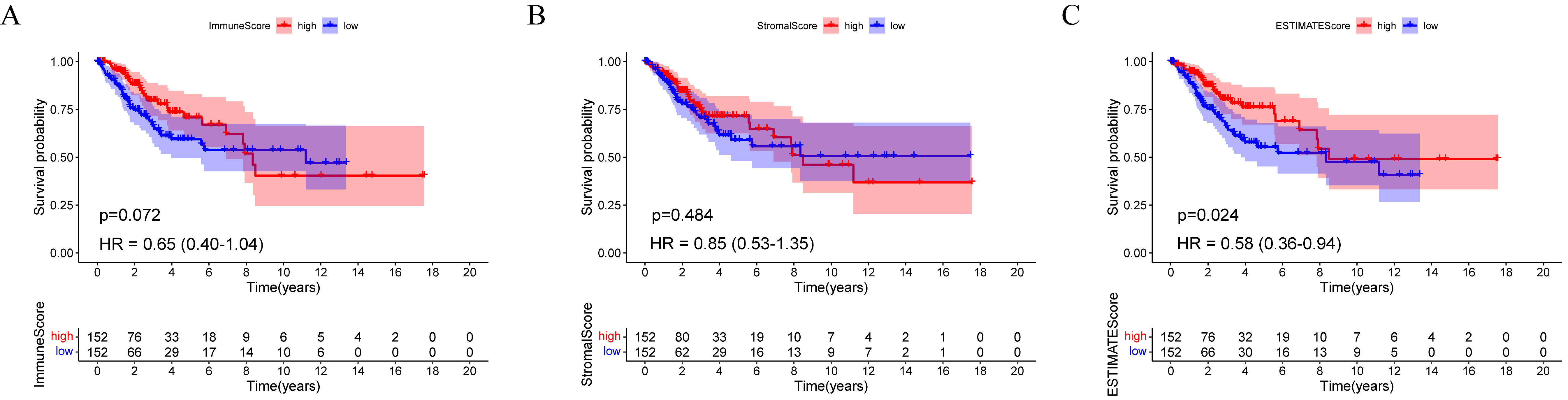 Fig. 2.
Fig. 2.A comparison of ImmuneScore, StromalScore, and ESTIMATEScore with survival among TCGA-CESC participants. (A) ImmuneScore (p = 0.072, HR = 0.65, 95% CI = 0.40–1.04), (B) StromalScore (p = 0.484, HR = 0.85, 95% CI = 0.53–1.35), and (C) ESTIMATEScore (p = 0.024, HR = 0.0.58, 95% CI = 0.36–0.94) survival analyses for cervical cancer patients with low and high scores. HR, hazard ratio; CI, confidence interval.
OS was better for patients with a high ESTIMATEScore (log-rank test p = 0.024, hazard ratio (HR) = 0.58) than the low scores (Fig. 2C). However, the StromalScore and ImmuneScore alone were not significantly associated with OS (Fig. 2A,B). According to these findings, the entire cervical TME might play an important role in cervical cancer prognosis.
We also analyzed the relationship between TME scores and clinical characteristics (Fig. 3). The latter parameter included age, FIGO stage, pathologic grade, and the TNM stage. Along with the progression of the M classification, the StromalScore and ESTIMATEScore notably declined (except for the ImmuneScore) (Fig. 3N,O). This result is different from that of a previous report [16] due to more samples being included and more cases being assessed by our team. These findings clarified that the TME of cervical cancer plays a crucial role in the invasion and metastasis of cancer.
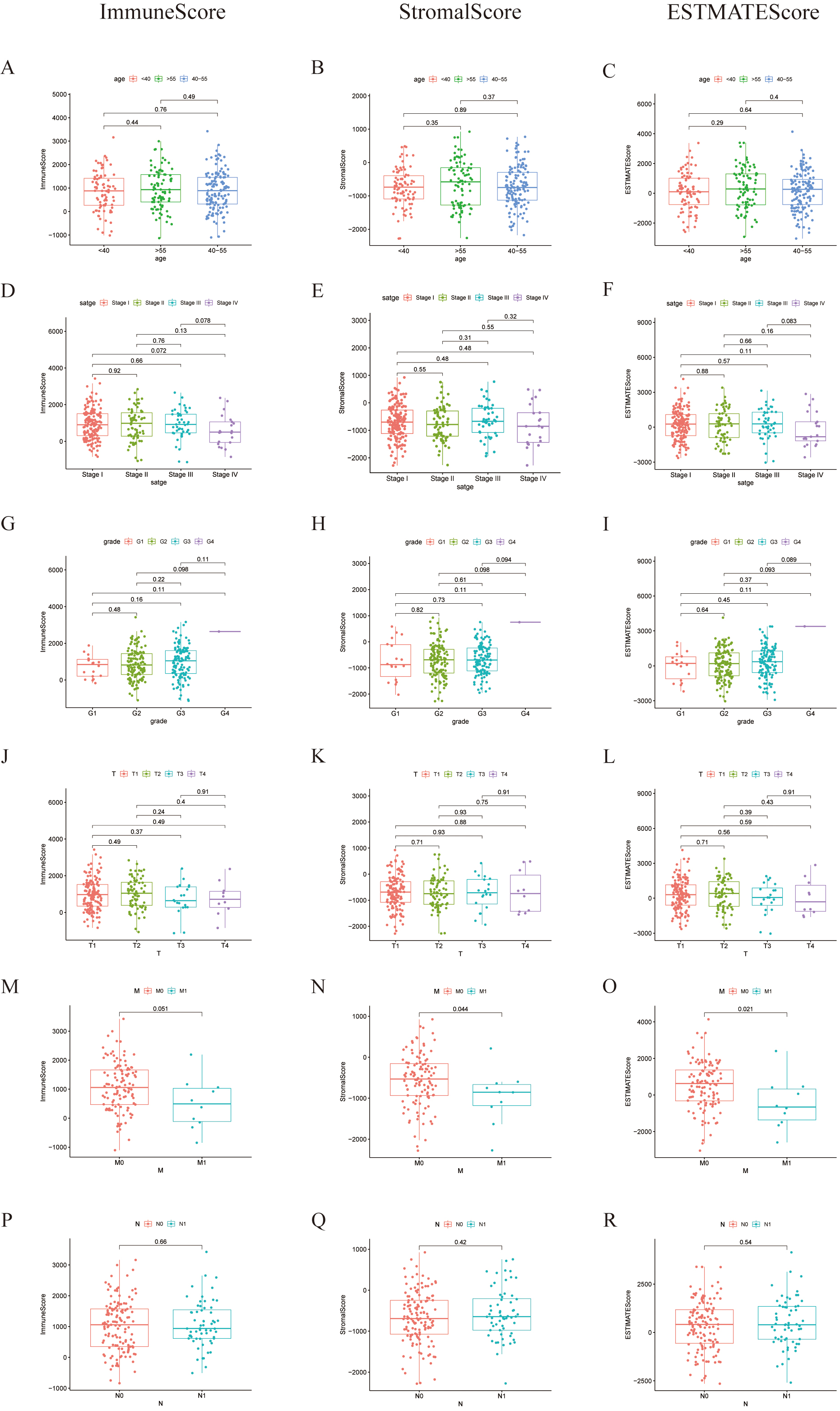 Fig. 3.
Fig. 3.Associations of ImmuneScore, StromalScore, and ESTIMATEScore with age, FIGO stage, grade, and TNM staging. (A–C) Kruskal-Wallis rank sum test of distribution of ImmuneScore, StromalScore, and ESTIMATEScore in the age classification, (D–F) in the FIGO stage, (G–I) in grades, (J–L) in T classifications, (M–O) in N classifications, (P–R) in M classifications.
The median TME score was used to divide patients into low and high TME groups, which can help us to identify a correlation between the TME and gene expression (Fig. 4). 1131 upregulated DEGs and 632 downregulated DEGs were acquired from ImmuneScore. 1299 upregulated DEGs and 51 downregulated DEGs were obtained from the StromalScore group. The intersection of the two groups, including upregulation and downregulation, was considered the final representation of the DEGs, which contained a total of 791 genes (749 upregulated genes and 42 downregulated genes). Moreover, these genes were used for the GO and KEGG enrichment analyses (Fig. 5). Based on the GO analysis, these DEGs are associated with immune functions, such as plasma membrane structure and leukocyte mediation (Fig. 5A–C). According to KEGG, DEGs are mainly involved in chemokine signaling pathways and cytokine–cytokine receptor interactions (Fig. 5D). It appears that immune factors play a pivotal role in cervical cancer TME.
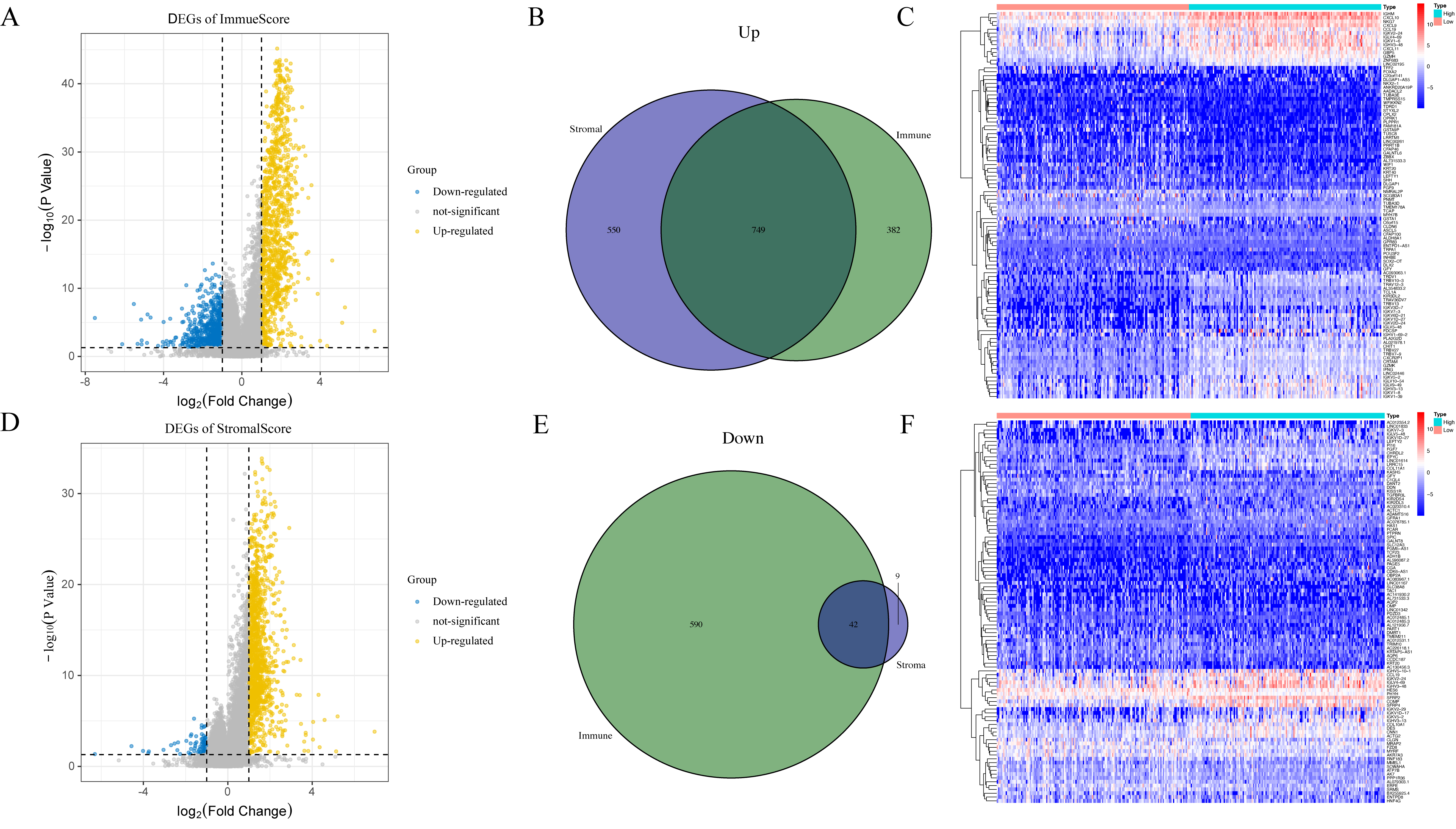 Fig. 4.
Fig. 4.Distribution of DEGs: Volcano plots, Venn diagrams, and Heatmap. Volcano plots showing the significance of DEGs between groups with high and low (A) ImmuneScore and (D) StromalScore. Venn diagrams showing the DEGs that were matched by the ImmuneScore and StromalScore assessments and were (B) upregulated and (E) downregulated. Heatmap for DEGs produced by contrasting the (C) ImmuneScore and (F) StromalScore groups with high and low scores. The gene name appears in the heatmap’s row, while the sample IDs that aren’t displayed in the plot appear in its column.
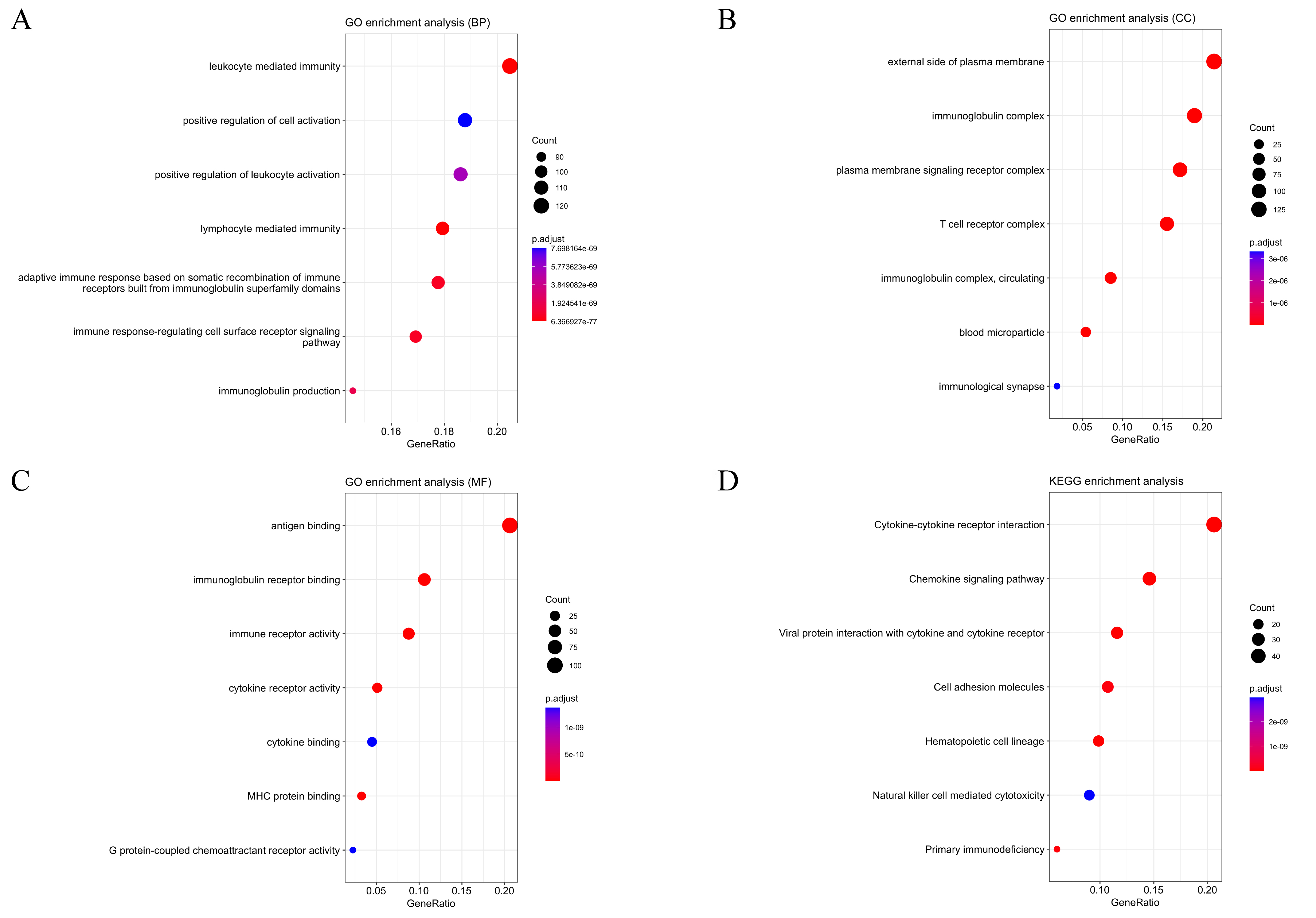 Fig. 5.
Fig. 5.GO and KEGG enrichment analyses for 791 DEGs. (A–C) GO and (D)
KEGG enrichment analyses, terms with p and q
PPI networks, which were based on DEGs established in the STRING database and shown by Cytoscape software, were used to explore potential associations between these factors (Fig. 6). Subsequently, the top 25 key PPI genes (namely, PPI-key genes) were screened via the cytoHubba plugin in Cytoscape (Fig. 6A,B). 147 genes were significantly associated with the 791 DEGs using univariate Cox proportional hazard regression analysis (namely, COX-key genes). Thereafter, 11 genes existing in both groups of PPI- and COX-key genes were defined as being prognosis key genes and were visualized in the Venn diagram (Fig. 6C). Finally, we selected GRAP2, which was the best hazard ratio gene, for further analysis (Fig. 6D).
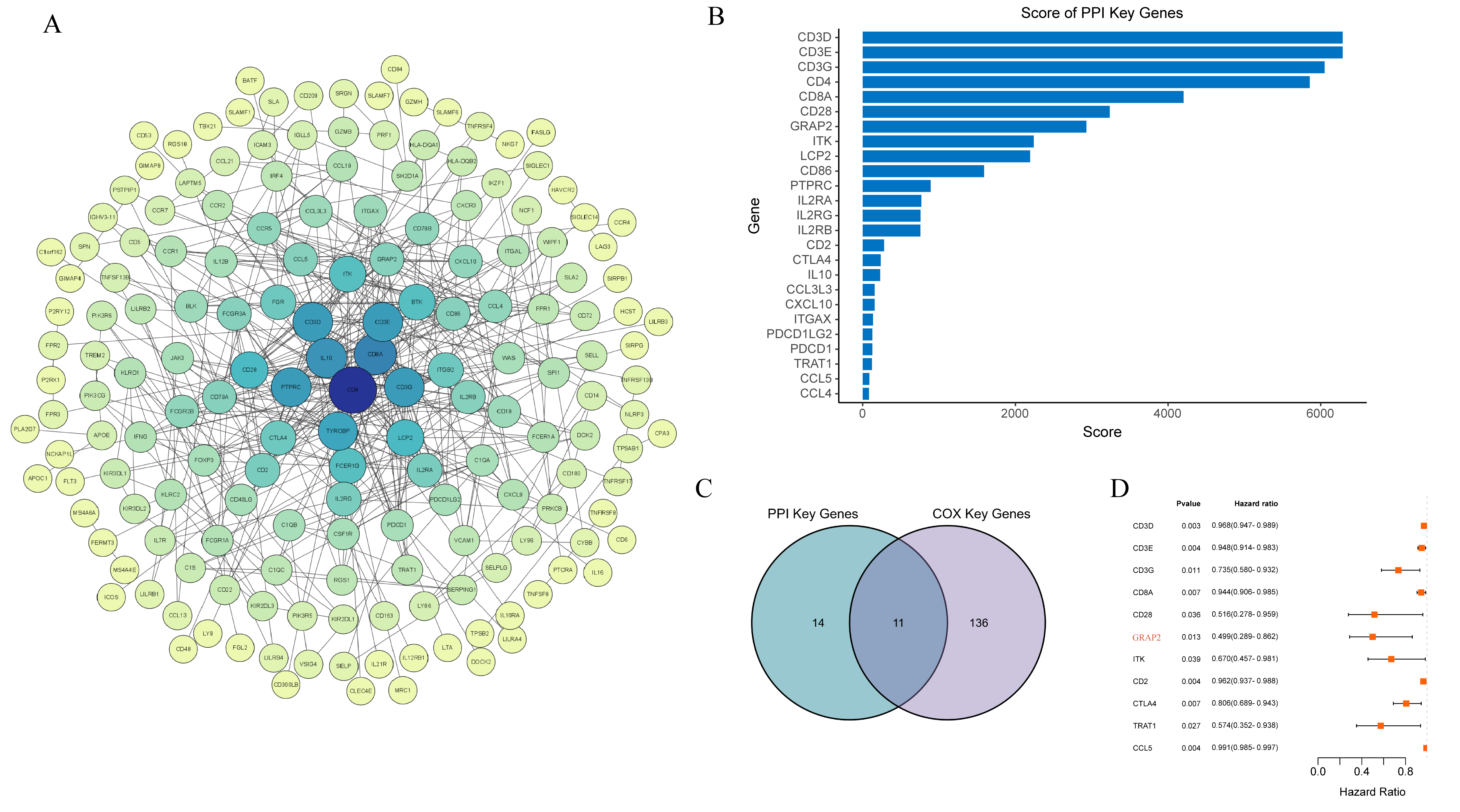 Fig. 6.
Fig. 6.PPI network visualization with univariate Cox regression analyses. (A) An experimental evidence type was selected to build the PPI network in the STRING database. The size and color of the node represent the degree value, the color of the edge represents the total score value, and the edge represents the interaction between the target gene. The degree value that corresponds to the color from yellow to blue increases with the size of the node. (B) Bar-plot diagrams of the top 25 genes identified by Cytoscape’s cytoHubba plugin. (C) A Venn diagram showing where the important COX and PPI genes overlap. (D) The GRAP2 gene is highlighted in red on the Forest plot, which shows the substantial deferentially altered genes between the two cohorts.
The GRAP2 gene encodes GRB2-related adapter protein 2, which is a 37 kDa protein in humans and is also known as a GRB2-related adaptor downstream of Shc (GADS). GADS is involved in Ras signaling [17]. The latest research suggests that GADS is recruited to phosphorylated CD28 to activate T cells [18]. Increasing evidence has demonstrated that GRAP2 seems to serve as a protective factor in lung cancer [19]. Furthermore, in this study, we outlined the relationship between GRAP2 expression and clinical information in the TCGA-CESC database (Table 1) and observed that GRAP2 expression is closely associated with survival time and M stage in cervical cancer patients. Besides, the GRAP2 high-expression group possessed a relatively longer OS than the low-expression group (Fig. 7A). To avoid biased results due to a single database source, we selected 55 samples from GEO (GSE:52903) to verify this result. Similarly, we found that high GRAP2 expression predicted better OS (Fig. 7B). After conducting an analysis of GRAP2 combined with clinicopathological characteristics, we found that GRAP2 expression gradually decreased following the advanced FIGO stage and M classification (Fig. 7C–H). It is widely recognized that GRAP2 expression is associated with improved prognosis, including survival rates and metastasis rates, in cervical cancer.
| Variable | Overall, N = 304 |
High, N = 152 |
Low, N = 152 |
p-value | |
|---|---|---|---|---|---|
| Age | 0.2 | ||||
| 82 (27%) | 41 (27%) | 41 (27%) | |||
| 83 (27%) | 36 (24%) | 47 (31%) | |||
| 40–55 | 137 (45%) | 75 (49%) | 62 (41%) | ||
| unkonwn | 2 (0.7%) | 0 (0%) | 2 (1.3%) | ||
| Grade | 0.6 | ||||
| G1 | 18 (5.9%) | 8 (5.3%) | 10 (6.6%) | ||
| G2 | 135 (44%) | 62 (41%) | 73 (48%) | ||
| G3 | 119 (39%) | 66 (43%) | 53 (35%) | ||
| G4 | 1 (0.3%) | 1 (0.7%) | 0 (0%) | ||
| unknown | 7 (2.3%) | 4 (2.6%) | 3 (2.0%) | ||
| unkonwn | 24 (7.9%) | 11 (7.2%) | 13 (8.6%) | ||
| FIGO stage | 0.2 | ||||
| Stage I | 162 (53%) | 83 (55%) | 79 (52%) | ||
| Stage II | 70 (23%) | 35 (23%) | 35 (23%) | ||
| Stage III | 45 (15%) | 26 (17%) | 19 (12%) | ||
| Stage IV | 21 (6.9%) | 6 (3.9%) | 15 (9.9%) | ||
| unkonwn | 6 (2.0%) | 2 (1.3%) | 4 (2.6%) | ||
| T stage | 0.5 | ||||
| T1 | 140 (46%) | 74 (49%) | 66 (43%) | ||
| T2 | 72 (24%) | 39 (26%) | 33 (22%) | ||
| T3 | 20 (6.6%) | 9 (5.9%) | 11 (7.2%) | ||
| T4 | 10 (3.3%) | 4 (2.6%) | 6 (3.9%) | ||
| unkonwn | 62 (20%) | 26 (17%) | 36 (24%) | ||
| N stage | 0.057 | ||||
| N0 | 134 (44%) | 76 (50%) | 58 (38%) | ||
| N1 | 60 (20%) | 31 (20%) | 29 (19%) | ||
| unknown | 110 (36%) | 45 (30%) | 65 (43%) | ||
| M stage | 0.039 | ||||
| M0 | 116 (38%) | 68 (45%) | 48 (32%) | ||
| M1 | 10 (3.3%) | 3 (2.0%) | 7 (4.6%) | ||
| unknown | 178 (59%) | 81 (53%) | 97 (64%) | ||
| Time (Days) | 640 (370, 1250) | 782 (440, 1398) | 584 (278, 1068) | 0.024 | |
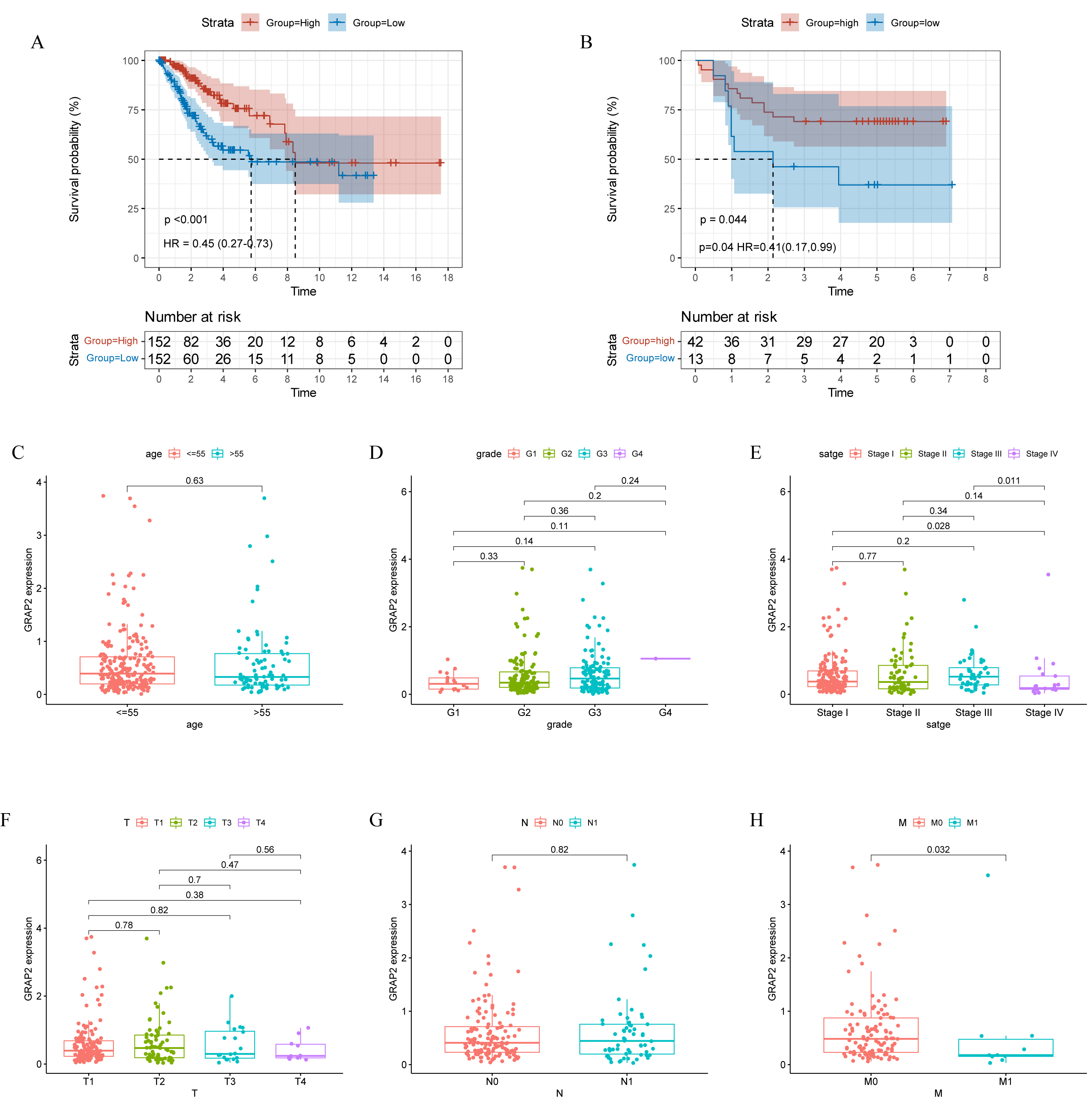 Fig. 7.
Fig. 7.Relationship between GRAP2 expression and clinical
information. Low and high GRAP2 expression with (A) TCGA-CESC patients
survival analyses (p
According to the abovementioned results, we believe that GRAP2expression may play a role in the malignant progression of cervical cancer. To further verify our theory, GSEA was used to explore the enrichment of GRAP2 expression in function and pathway. Genes within the GRAP2 high-expression tended to be associated with immune processes, such as cell activation involved in immune responses, cytokine-mediated signaling pathways, and mononuclear migration (Fig. 8A). Moreover, in the KEGG pathway analysis, the group mainly included immune-related pathways, including cytokine receptor interaction, chemokine signaling pathway, antigen processing and presentation, and T-cell receptor signaling pathway (Fig. 8C). Synchronously, the GRAP2 low-expression cohort was enriched in several different biological processes, including binding of sperm to the zona pellucida, establishment of mitochondrion localization, isoprenoid biosynthetic process, and negative regulation of nuclear division (Fig. 8B). Similarly, a wide variety of metabolic function pathways were enriched in low expression, which included the TCA cycle, glycerolipid metabolism, GPI anchor biosynthesis, and glyoxylate and dicarboxylate metabolism (Fig. 8D). In this figure, only the top ten items of pathways are shown in each section. All of the results illustrated that GRAP2 may serve as a promising indicator of immune and metabolic statuses in cervical cancer.
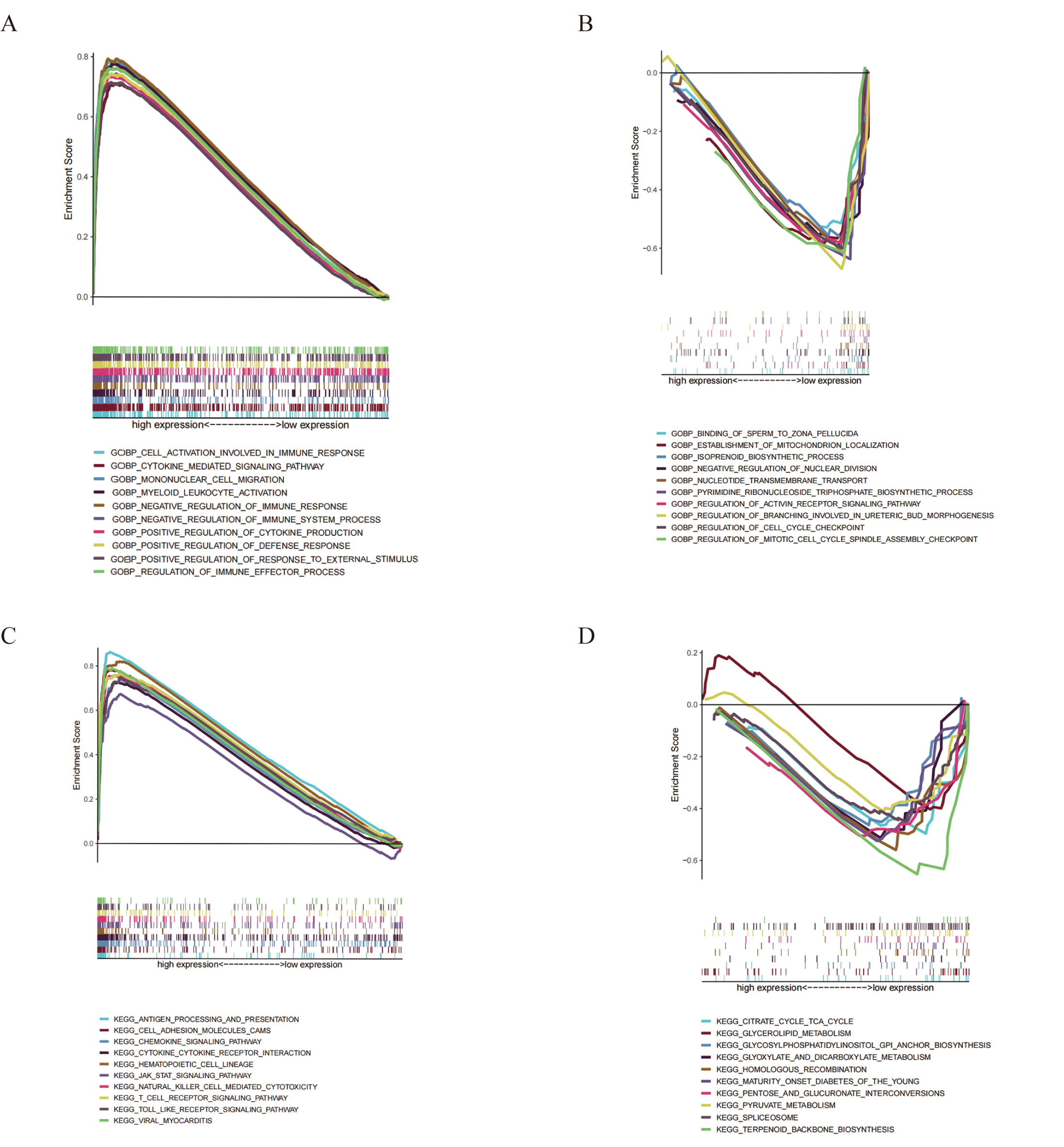 Fig. 8.
Fig. 8.GSEA for GRAP2 expression. Up-regulated genes are
situated on the left, which approaches the origin of the coordinates; in
contrast, down-regulated genes are located on the right of the x-axis. Each line
represents one specific gene set with a distinct color. The only gene sets that
were deemed significant have NOM p
The CIBERSORT algorithm was applied to further investigate the interplay between GRAP2 expression and TICs. First, Among the 22 immune cell types, 11 TICs had significant differences between the GRAP2 expression groups (Fig. 9A). Coincidentally, 11 TICs were strongly associated with GRAP2 expression from the correlation and difference analyses (Fig. 9B). Surprisingly, the intersection of the analyses showed a perfect coincidence of the two analyses (Fig. 9C). These TICs contain CD8 T cells, resting memory CD4 T cells, activated memory CD4 T cells, follicular helper T cells, M0 macrophages, M1 macrophages, activated dendritic cells, resting dendritic cells, resting mast cells, activated mast cells, eosinophils. According to these results, GRAP2 is essential to the immune TME of cervical cancer.
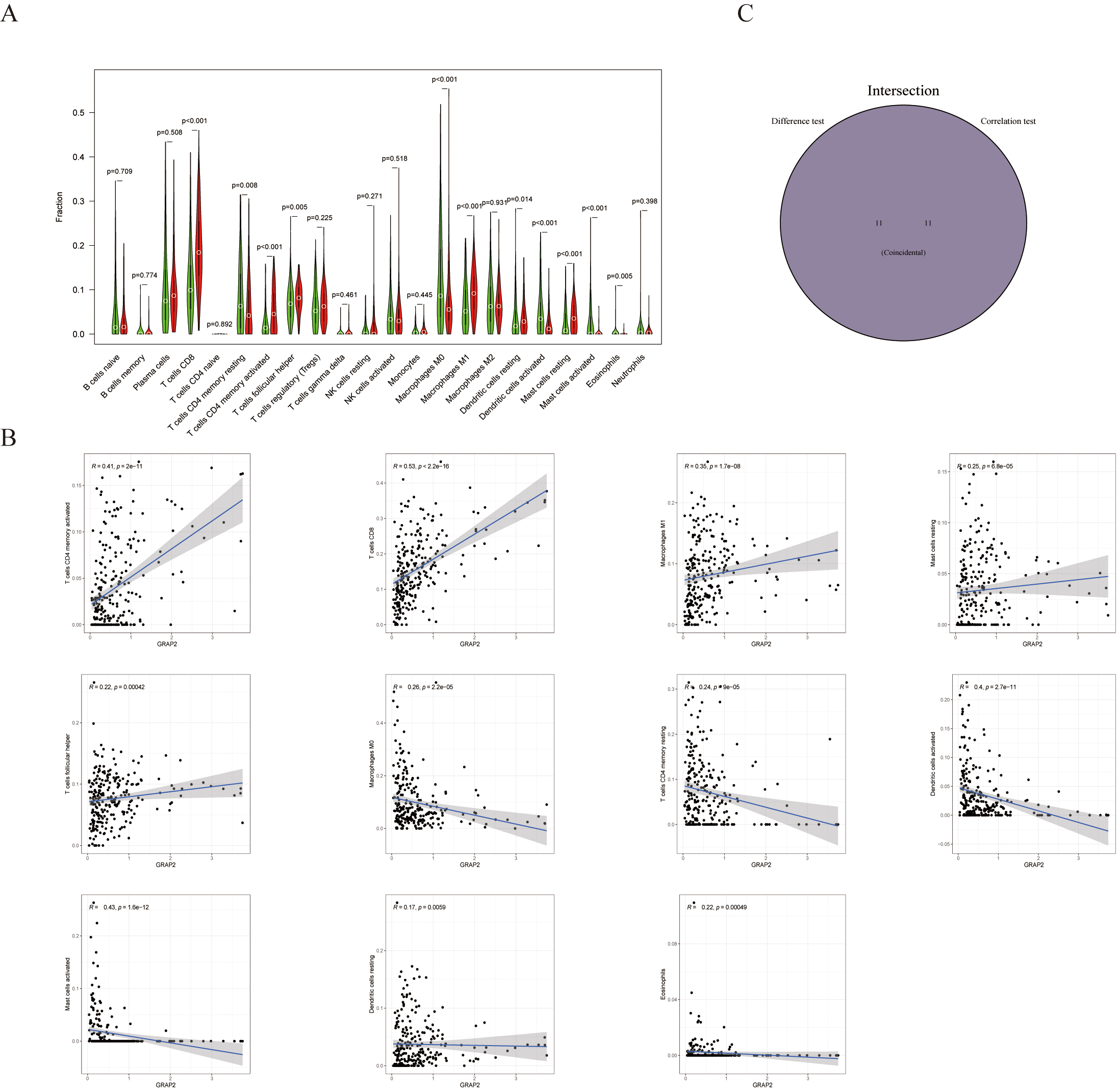 Fig. 9.
Fig. 9.The proportions of TICs in groups with high and low GRAP2 expression and their correlation with GRAP2 expression. (A) By using the Wilcoxon rank sum test, 22 immune cell types in tumor tissues with low (green) or high (red) GRAP2 expression were compared. (B) An illustration of Pearson’s correlation between the number of TICs and GRAP2 expression, with blue lines denoting the best fit linear models. (C) An intersection between analyses in (A,B) shows 11 TICs shared by both.
In this study, we found there is a close correlation between the microenvironment and the prognosis of cervical cancer patients. We also identified several immune-related prognostic genes from the TCGA database. Furthermore, an important role for GRAP2 was found in immune-related biological processes.
An essential element of tumor biology is the immune resistance of the tumor
microenvironment, which refers to the mutual resistance of malignant tumors and
tumor cells. [20]. Herein, we used the ESTIMATE algorithm to estimate the purity
of the tumor microenvironment’s components based on gene expression to calculate
stromal scores, immune scores, and estimate scores, thereby aiding to identify
candidate immune TME-related biomarkers. The results suggested that a high
EstimateScore led to better clinical outcomes, including survival rate and M
classification. Specifically, some TME-related genes were potentially able to
predict prognosis. The same conclusions could be drawn from other studies
[10, 21]. Immediately thereafter, a total of 749 upregulated and 42 downregulated
genes were detected between groups with high and low ImmuneScores and
StromalScores, respectively. GO and KEGG analyses suggested that the vast
majority of these DEGs were involved in immune-related processes, such as
leukocyte-mediated immunity and the chemokine signaling pathway. To further
investigate TME-related genes associated with prognosis, we constructed a PPI
network by using Cytoscape based on the DEGs, and we selected the top 25 key PPI
genes via cytoHubba plugin scores. Thereafter, 11 genes were defined as key
prognostic genes in both groups of PPI- and COX-key genes. In the survival
analyses, all 11 genes indicated a favorable prognosis. Interestingly,
GRAP2 (log-rank p
The GRAP2 gene encodes the GRB2-related adaptor protein 2, a protein involved in leukocyte-specific signaling by protein-tyrosine kinases [22]. This protein can interact with other proteins, such as GRB2-associated binding protein 1 (GAB1) and the SLP-76 leukocyte protein (LCP2), to activate T cells via multiple signaling pathways [23]. However, uncertainty surrounds the process of GRAP2 in cervical cancer. In this study, we found that patients with cervical cancer who expressed a high level of GRAP2 had an earlier FIGO and M stage, and a better survival ending. This suggests that GRAP2 may be used as a new prognostic biomarker to guide clinical treatment strategies. Similar findings were confirmed in another study [19], wherein GRAP2 was shown to predict survival status in lung adenocarcinoma patients. Furthermore, it has been claimed that GRAP2 is linked to metastasis in cervical cancer. Unfortunately, there is no research in this field to support this idea.
In the current study, we analyzed the components associated with GRAP2 in the cervical cancer microenvironment. Both GO and KEGG analyses, as well as GSEA, showed that GRAP2 was associated with immune function. This is consistent with the known function of GRAP2. For instance, GRAP2’s carboxy-terminal SH3 structural domain constitutively binds SLP-76, whereas the SH2 structural domain of GRAP2 can bind to the tyrosine-phosphorylated linker for activation of T lymphocytes (LAT). A heterotrimeric complex made up of LAT, GRAP2, and SLP-76 mediates TCR signaling, thus linking proximal membrane signals to downstream signaling pathways and activating T cells as a result [23, 24]. Interestingly, we also found that the low expression group of GRAP2 significantly enriched several metabolism-related pathways in GSEA, such as glycerolipid metabolism. It is not difficult to understand this phenomenon, as immune cells are also key to the microenvironment and tumor metabolic reprogramming [25, 26]. Although there is little proof that GRAP2 is linked to tumor metabolic reprogramming, this could be a fruitful area of future investigation. This characteristic of GRAP2 provides new insights into immunotherapy for cervical cancer. There is a critical clinical need for new medicines or therapeutic approaches for advanced cervical cancer, notwithstanding the significant contribution of HPV vaccinations to cervical cancer prevention. Regardless of the second-line drug that is employed, the outcomes are quite dismal for patients who relapse after receiving first-line platinum-based chemotherapy [27]. Immunotherapy is an attractive treatment strategy for cervical cancer, which is closely associated with human papillomavirus infection and is often accompanied by high PD-L1 expression and T-cell infiltration. Nevertheless, response rates with immune monotherapy remain under 15%, despite mounting evidence in favor of immunotherapy for cervical cancer [28]. The TME is a critical factor limiting the efficacy of immunotherapy in cervical cancer [20]. Another reason for the suboptimal therapeutic efficacy of adoptive T-cell therapies (ACT) in cervical cancer is the influence of “cold tumors”, which are cancers that have poor immune activation and lymphocyte infiltration, thus making it challenging to kill tumor cells by activating immune effector cells to accomplish this feat [29]. Herein, we found that GRAP2 plays an essential role in cervical cancer, particularly in the immunological microenvironment. We have reason to believe that GRAP2 has a promising future as an immunotherapy target and prognostic biomarker in cervical cancer.
GRAP2 demonstrates immunity-related activity in the TME of cervical cancer, thus suggesting that it may influence the biological behavior of cervical cancer, particularly in regard to metastasis. As a result, GRAP2 may be a unique molecular type of indicator, and it can be combined with the FIGO stage to enhance the prediction of clinical outcomes. An improvement in the identification of immunotherapy-sensitive cervical cancer patients will aid in the advancement of precision medicine.
The datasets generated and analyzed during this study are available in the TCGA database (https://gdc.cancer.gov/) and GEO database (https://www.ncbi.nlm.nih.gov/geo/).
SL, JG, and JZ conceptualized the study. SL and XM wrote the manuscript and constructed the figures. GL, YG, and DY performed the data analysis. JG and JZ reviewed and edited the manuscript. All authors contributed to the article and approved the final version.
Not applicable.
We thank the TCGA, HIPLOT, and GSEA working groups for generously sharing their data and technology. We also thank the Rstudio team for affording the free software.
This work was supported by the National Natural Science Foundation of China (grant no. 82060477), Jiangxi Provincial Natural Science Foundation (grant no. 2020BAB206054), Science and Technology Project of Health Commission of Jiangxi (grant no. 202120073), Nanchang Science and Technology Support Plan Key Project (grant no. Hongkezi2019-258-3), Nanchang Key Laboratory for Precision Therapy of Gynecological Tumors (grant no. 2020NCZDSY-003), 2021 Nanchang High-level Science and Technology Innovation Talents “Double Hundred Plan” Project (grant no. Hongkezi 2021-156-29), Nanchang Advantage Science and Technology Innovation Team Project (grant no. 2019-15-3).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.