



1 Department of Pharmaceutical Sciences, Faculty of Pharmacy, Chiang Mai University, 50200 Chiang Mai, Thailand
2 Suchada IVF Center, Sriracha, Chon Buri, 20110, Thailand
3 Department of Ophthalmology, Faculty of Medicine, Chiang Mai University, Chiang Mai, 50200, Thailand
4 Department of Obstetrics and Gynaecology, Faculty of Medicine, Chiang Mai University, Chiang Mai, 50200, Thailand
†These authors contributed equally.
Academic Editor: Shigeki Matsubara
Abstract
Background: Oculocutaneous albinism type IA (OCA1) is the
most severe form of albinism, an autosomal recessive inherited deficit of the pigment
melanin causing distinctive alterations of skin, hair, and visual system. Pre-implantation genetic testing (PGT) is a
substitution for prenatal diagnosis. Methods: This study accomplished
SNP array with karyomapping for PGT of OCA1 and validated the results with
PCR-based PGT. Results: One family with a risk of having OCA1
c.819+3insATATGCC and c.896G
Keywords
- oculocutaneous albinism type IA (OCA1)
- embryo selection
- haplotyping
- karyomapping
- pre-implantation genetic testing for monogenic disease (PGT-M)
Albinism is a genetic complex class of hypopigmentation conditions that have a complete or partial inherited deficit of the pigment melanin and results in distinctive alterations in the skin, hair, and visual system (eyes and optic tracts) [1]. Melanin is synthesized and stored inside melanosomes, intracellular organelles that can be found in the stratum basale of the epidermis, hair bulbs, and intraocular epithelia [2]. The major form of albinism is oculocutaneous albinism (OCA) characterized by partial or complete deficiency of melanin pigmentation of the skin, hair, foveal hypoplasia, nystagmus, photophobia, and low visual acuity [3]. The prevalence of albinism varies and is estimated at 1:17,000. The prevalence of gene carrier is about 1.4% [4].
Oculocutaneous albinism type I (OCA1, MIM#203100), an autosomal recessive disorder, is the most severe form of albinism. OCA1 is caused by homozygous or compound heterozygous mutation in tyrosinase (TYR, MIM#606933) gene on chromosome 11q14.3 [5], leading to the deficiency of tyrosinase enzyme, i.e., tyrosinase-negative OCA. Over 60 different mutations have been addressed [6]. The parents of an affected child are obligate carriers and the chance of having another affected offspring is 25%. The offspring of an affected patient are obligate carriers. Carriers are asymptomatic. The life expectancy of OCA patients is not reduced. Development and intellectual capabilities are normal. Patients with OCA have normal fertility [1].
TYR consists of five exons spanning 65 kb and encoding a 529 amino acid protein [7]. Tyrosinase is a copper-containing enzyme that plays a role in several crucial steps in melanin biosynthesis [8]. The most important symptom of OCA1 is severely reduced vision. The lack of melanin pigment in eyes during embryogenesis causes developmental anomalies, including hypoplasia of macula, misrouting of optic nerve fibers to the visual cortex and translucent iris, and visibility of choroidal vessels in the pigment-lacking retina. These lead to nystagmus, decreased visual acuity, lack of stereoscopic vision, and severe photophobia. Refractive errors, i.e., myopia, hyperopia, astigmatism, and strabismus are commonly present. The second symptom is a consequence of sunlight on the skin. Frequent sunlight exposure leads to burn, degenerate changes, and an increased risk of skin cancer [1]. Severe visual handicap in OCA1 results in great daily living difficulties and limited professional options. Albinism is still incurable. Families at risk of having children with albinism will seek genetic counseling and prenatal diagnosis (PND).
PND of OCA by electron microscopy examination of fetal skin samples taken during fetoscopy at 20 weeks of gestation was first reported in 1983 [9]. A total of 12 cases of PND for OCA using fetal skin biopsies were reported in 2005 from the same group [10]. Thirty-one cases of PND by fetal scalp biopsies using fetoscopy with histological examination were reported in 1999 [11]. Fetal skins were then examined for the presence of absence of melanin in the epidermis and/or hair bulb melanocytes under light microscope. Subsequently, defining the stages of melanosomal differentiation to evaluate the course of melanogenesis was carried out using an electron microscope. Normal melanogenesis was revealed in 26 biopsies.
PND using DNA analysis from fetal cells, i.e., chorionic villous sampling or amniocentesis, was also performed by direct mutation analysis (one case) and linkage analysis (two cases) in the same report [11]. PND of OCA by analysis of the fetal tyrosinase gene was reported in 1994 [12]. Following amniocentesis, PCR with dot-blot was successfully performed on fetal cells. In cases of unidentified mutation, an electron microscopic 3,4-dihydroxyphenylamin reaction test of fetal skin biopsy was employed for PND [13, 14]. A total of 55 PND of OCA using molecular genetic analysis in 37 families (32 families with OCA1) were reported from the same group in 2009 [15]. Half of the PND was performed by first trimester CVS and the other half by mid-trimester amniocentesis. Fetal samples underwent mutation analysis using either restriction fragment length polymorphism or direct sequencing and haplotyping analysis using 4–6 microsatellite linked markers.
DNA-based PND of a Korean family with the risk of having compound heterozygous OCA1 offspring was successfully performed in 1997 [16]. A combination of single-stranded conformation polymorphism/heteroduplex screening, restriction fragment length polymorphism, and allele-specific oligonucleotide hybridization analysis of DNA from chorionic villous sampling was employed. Two cases of molecular diagnosis using denaturing high-performance liquid chromatography and Sanger’s sequencing from fetal blood samples were successfully carried out in 2006 [17]. Ten PND using fluorescent sequencing were reported in 2014 [18]. It is noted that sampling techniques have moved from fetal skin biopsy to either chorionic villous sampling or amniocentesis and diagnostic techniques moved from electron microscope to molecular genetic analysis. However, PND of OCA1 is sometimes controversial.
PND provides fetal samples for genetic analysis. Selections of invasive PND include chorionic villous sampling in the first trimester and amniocentesis and fetal blood sampling in the second trimester. Negative results assure the couples that their baby is healthy. Nevertheless, positive results provide difficult decisions for the couples regarding dismissing or continuing the pregnancy and preparing for postnatal treatment [18]. Additionally, some pregnancies may abort following the PND procedures.
Pre-implantation genetic testing (PGT) [19] is a substitution for the conventional PND giving the couples an opportunity to start a pregnancy knowing that the baby will be unaffected. PGT for monogenic disorder (PGT-M) in ten families at risk of having OCA offspring was reported in 2018 [20]. A total of 28 PGT cycles were performed giving rise to six unaffected children. However, there are no details of molecular techniques for reproduction. A PGT-M of OCA was successfully carried out in a family for three clinical cycles [21]. One healthy infant resulted. Sequencing following whole genome amplification (WGA) was employed. However, molecular techniques were briefly described and analysis results were not demonstrated.
Karyomapping is a sophisticated molecular technique employing single nucleotide polymorphism array (aSNP) for simultaneous haplotyping and copy number variation (CNV) testing [22]. Haplotyping-based PGT-M cycles have been reported [23, 24]. However, clinical use of karyomapping is still uncommon. This study applied aSNP and karyomapping for PGT-M of OCA1 and PGT for aneuploidy (PGT-A) in one clinical PGT cycle and compared the results with those of PCR techniques.
One family with a risk of having OCA1 child entered the project after thoroughly
counseling and written, informed consent was obtained. The study was conducted in
accordance with the Declaration of Helsinki, and approved by the Research Ethics
Committee of the Faculty of Medicine, Chiang Mai University, Thailand (protocol
code OBG-2563-07801, December 30th, 2020). The proband was a 38 years old
housewife. She and her 39 years old husband were symptomless for OCA. Her
daughter was healthy. She experienced a spontaneous miscarriage in her second
pregnancy. Her third child had OCA type I and died soon after birth. Her fourth
and fifth pregnancies were terminated due to positive PND for OCA1. She and her
daughter carried c.819+3insATATGCC mutation (splicing) within Intron 1 of the TYR
gene. Her husband carried c.896G
The patient went through in vitro fertilization procedures. Ovarian stimulation was carried out. Intracytoplasmic sperm injection was done to eliminate the chance of sperm DNA contamination. Blastocysts were biopsied with a laser on day 5 post-fertilization. Five cells were biopsied for WGA and then molecular analysis, i.e., karyomapping and DNA analysis. Embryos vitrification was done following the biopsy.
Biopsied trophectoderm were washed thoroughly in phosphate-buffered saline (Cell
Signaling Technology, Theera Trading Co. Ltd. Bangkok, Thailand) with 0.1%
polyvinyl alcohol (Sigma-Aldrich, Chiangmai VM Co., Ltd., Chiang Mai, Thailand)
before transferring to microcentrifuge tubes with a total volume of 4
Amplified MDA samples were tested with SNP array using Illumina HumanKaryomap-12 DNA Analysis Kit (Bio-Active Co. Ltd., Bangkok, Thailand) according to the manufacturers’ instruction [22, 26]. SNP genotyping information was analyzed using BlueFuse Multi software version 4.5 (Illumina, Inc. California, USA) for karyomapping analysis and molecular cytogenetics. Haplotyping analysis from SNP genotyping information of the couples together with an offspring or an informative relative as references reveals inheritance of unaffected or affected genes in the embryos. This allows the diagnosis of a monogenic disorder of the embryos. Additionally, SNP genotyping provides CNV details of every chromosome. The results were compared with PCR results.
Molecular mutation analysis using PCR was performed to confirm karyomapping
results. Aliquots of amplified WGA products were subjected to multiplex
fluorescent PCR and mini-sequencing analysis. 0.5
| Primers | Location on TYR gene | Sequences | Fragment length (bp) | Labeling | References | |
| TYR 819+3insATATGCC | Intron 1 | forward | 5′-CCA TGA AGC ACC AGC TTT TC-3′ | 292 | 6’FAM |
OMIM: AH003020.2 |
| reverse | 5′-CCC TGC CTG AAG AAG TGA TT-3′ | |||||
| TYR c.896G |
Exon 2 | forward | 5′-CCA ACA TTT CTG CCT TCT CC-3′ | 268 | HEX |
OMIM: AH003020.2 |
| reverse | 5′-GCA GCT TTA TCC ATG GAA CC-3′ | |||||
| mini-sequencing | 5′-CCG AGG GAC CTT TAC GGC-3′ | |||||
| HUMTH01 | forward | 5′-AGG GTA TCT GGG CTC TGG-3′ | 115–140 | NED |
[27] | |
| reverse | 5′-CTT CCG AGT GCA GGT CAC-3′ |
A mixture of 1
Mini-sequencing techniques were employed for mutation analysis of TYR
c.896G
A mixture of 1
One clinical PGT-M cycle for OCA was carried out in this study. Maternal (TYR
c.819+3insATATGCC carrier) and paternal (TYR c.896G
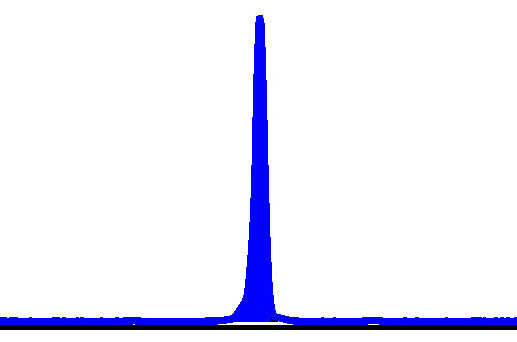
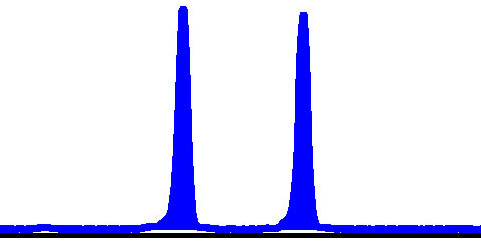
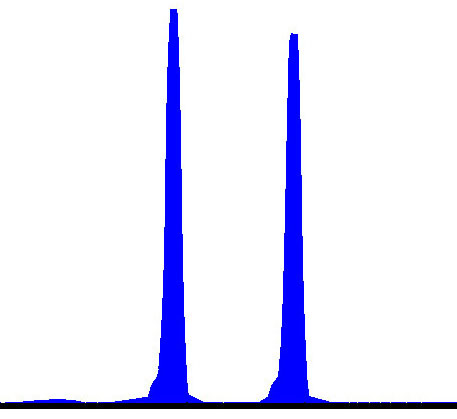
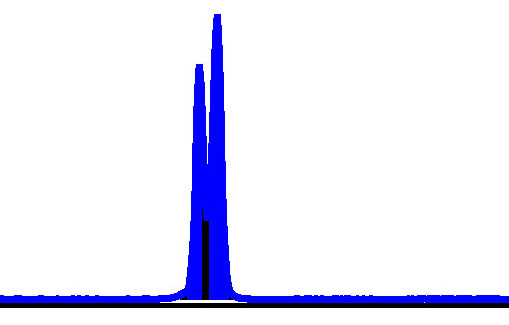
 Fig. 1.
Fig. 1.Haploblock chart of TYR from karyomapping
(BlueFuse Multi software) using SNP array data (Illumina HumanKaryomap-12 DNA
Analysis Kit) and mutation analysis using multiplex fluorescent PCR (F-PCR) and
mini-sequencing for TYR c.819+3insATATGCC and TYR c.896G
Novel multiplex fluorescent PCR with mini-sequencing protocols specifically for
TYR c.819+3insATATGCC and TYR c.896G
| Embryo No. | F-PCR (TYR c.819+3insATATGCC) and Mini-Sequencing (TYR c.896G¿A) Analysis | HUMTH01 STR (bp) | PCR Results Conclusion | Karyomapping Analysis | Karyomapping Conclusion | Concordant Results | Chromosome Analysis | |||||
| c.819+3insATATGCC | antisense c.896C¿T | Alleles | ||||||||||
| Normal | Mutant | Normal | Mutant | Paternal | Maternal | Paternal | Maternal | |||||
| Father | Y | N | Y | Y | 131/131 | Ht | P1/P2 | |||||
 |
 |
|||||||||||
| Mother | Y | Y | Y | N | 127/134 | Ht | M1/M2 | |||||
 |
 |
|||||||||||
| Daughter | Y | Y | Y | N | 131 | 134 | Ht | P1 | M1 | |||
 |
 |
|||||||||||
| Embryo 1 | Y | N | Y | N | 131 | 127 | Normal | P1 | M2 | Normal | Yes | 47,XY +14,‑7q |
 |
 |
|||||||||||
| Embryo 2 | Y | N | Y | N | 131 | 134 | Normal | P1 | M2 | Normal | Yes | 47,XY +22 |
 |
 |
|||||||||||
| *Y, present; N, absent, Ht, heterozygous; Normal, homozygous normal. | ||||||||||||
In addition to haplotyping results, high-resolution SNP microarray in this study provided CNV information with parental origins information of chromosomal gain or loss. aSNP analysis exhibited an additional chromosome 14 (maternal gain) and segmental loss of 7q (paternal loss) in embryo No. 1, i.e., 47,XY+14,-7q (Fig. 2a) and an additional chromosome 22 (maternal gain) in embryo No. 2, i.e., 47,XY+22 (Fig. 2b). Therefore, both embryos were unsuitable for transfer. The patient will return for the next PGT-M cycle.
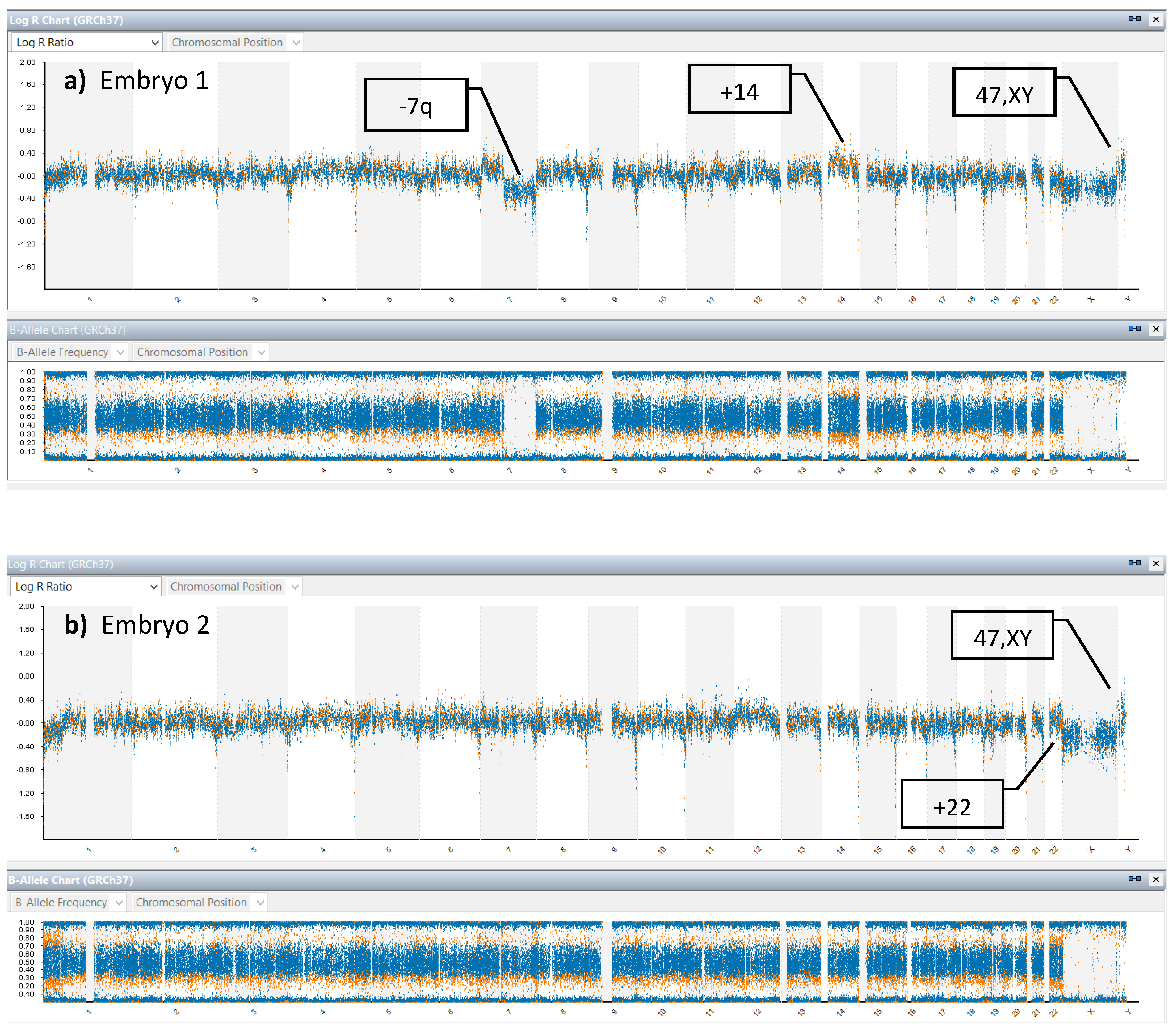 Fig. 2.
Fig. 2.Chromosome balance analysis from aSNP results. (a) shows copy number variation (CNV) of embryo 1, 47,XY+14 (maternal gain),-7q (paternal loss). (b) shows CNV of embryo 2, 47, XY+22 (maternal gain).
Homozygous or compound heterozygous mutations within the tyrosinase (TYR) gene on chromosome 11 causes OCA1 which is inherited in an autosomal recessive manner [5]. Severely reduced vision is the most crucial disability to the patients. There are a wide variety of different mutations, over 60 mutations have been reported [6]. Compound heterozygous subjects are common. PND of OCA in the early age employed electron microscopy examination of fetal skin biopsy in utero [9] and later using DNA analysis techniques [11]. Even with a lot of recent advanced molecular techniques, the nature of a wide variety of mutations causing OCA1 among the population makes molecular diagnosis complicated. Developing specific molecular analysis protocols for each OCA1 family is labor-intensive and time-consuming. The success of PGT-M of OCA attempts has been reported but the details of the techniques and the results were untold [20, 21]. Identification of specific OCA mutations and the development of PCR protocol for PGT-M for each family is labor-intensive and challenging. This study is the first to perform PGT-M for OCA1 employing advanced aSNP with karyomapping algorithm for haplotyping and simultaneous CNV analysis. Additionally, molecular analysis using fluorescent PCR and mini-sequencing was done alongside to validate karyomapping results. The details of PCR protocol and results analysis were also exhibited.
Karyomapping in this clinical PGT-M cycle exhibited both embryos to be absent of
both paternal and maternal mutations by haplotyping based analysis from aSNP
information. Novel PCR protocols using multiplex fluorescent PCR with
mini-sequencing techniques were developed and applied for PGT-M of OCA1 (TYR
c.819+3insATATGCC and TYR c.896G
Additional advantage of karyomapping which is demonstrated in this study is the CNV and parental origins information of the embryos. aSNP reveals chromosomal unbalance and parental origins in both embryos, i.e., 47, XY,-7q (paternal loss),+14 (maternal gain) in Embryos 1 and 47, XY,+22 (maternal gain) in Embryos 2. Both embryos were not suitable for transfer due to chromosomal imbalance. Due to the COVID-19 pandemic, the couples will return for the next PGT-M cycle later. It was shown that karyomapping can be used as a precise, time-saving for protocol development, widely applicable PGT-M protocol for single gene disorders of different natures of mutations (i.e., small deletion and single base substitution mutations) and provides the additional CNV and parental origin details which are common during pre-implantation development of human embryos.
Before performing aSNP there is a need to have a high precision 0.5
Most PGT laboratory already has all the necessary equipment and consumables for PCR-based PGT which are a lot cheaper than those the microarray-based. Designing primers and PCR protocols for new monogenic diseases or new mutations is difficult and labor-intensive. However, the additional expenses of the PCR-based PGT are just the cost of the additional primers for each new mutation which is insignificant. For the regions with monogenic diseases of common mutations, i.e., alpha- and beta-thalassemias, the running cost of PCR-based PGT for the laboratory is not expensive at all because one stock of primers can be used for several cases. Therefore, microarray-based PGT may be suitable for those laboratories with monogenic diseases of various mutations, i.e., OCA, Marfan syndrome, etc., and with adequate funding.
This study presents a clinical PGT-M cycle using karyomapping for a family at risk of having a compound heterozygous OCA1 offspring. The novel PCR protocols using fluorescent PCR and mini-sequencing were described, tested and confirmed karyomapping results. Both embryos in this clinical PGT-M cycle were absent of both paternal and maternal mutations. However, aSNP revealed chromosomal unbalance in both embryos and excluded them from transfer. This study demonstrates that karyomapping can be used as a precise, time-saving for protocol development, widely applicable PGT-M protocol for single gene disorders of different natures of mutations (i.e., small insertion and single base substitution) and provides the additional CNV and parental origin details which are common during pre-implantation development of human embryos.
No datasets were generated or analyzed during the current study.
This study presents pre-implantation genetic testing of monogenic disorders
(PGT-M) for oculocutaneous albinism type IA (OCA1) including insertion
c.819+3insATATGCC and point mutation c.896G
This study presents pre-implantation genetic testing (PGT) for oculocutaneous albinism type IA (OCA1) employing aSNP with karyomapping for haplotyping and copy number variation (CNV) analysis. In addition, PCR-based PGT-M was performed to validate karyomapping.
ADO, allele drop-out; aSNP, single nucleotide polymorphism microarray; CNV, copy number variation; ICSI, intracytoplasmic sperm injection; IVF, in-vitro fertilization; MDA, multiple displacement amplification; OCA1, oculocutaneous albinism type I; PCR, polymerase chain reaction; PGT-A, pre-implantation genetic testing for aneuploidy; PGT-M, pre-implantation genetic testing for monogenic disorders; PND, prenatal diagnosis; WGA, whole genome amplification.
Study conception and design were performed by SP, SM and WP. Ovarian stimulation, oocytes collection, and embryology laboratory were performed by TP and SM. aSNP and karyomapping analysis were performed by RS, SM, SP and WP. PCR analysis and mini-sequencing were performed by WS, SP and WP. Data collection and analysis were performed by SP, SM and WP. WC took care of clinical diagnosis and assessment. Prenatal and postnatal diagnoses were performed by TT and WP. SM and SP contributed equally to this work. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
The study was approved by the Research Ethics Committee of the Faculty of Medicine, Chiang Mai University, Thailand (protocol code OBG-2563-07801, December 30th, 2020).
The authors thank Ruth Leatherman for the proofreading.
This research was funded by the National Research Council of Thailand and Chiang Mai University Research Fund, grant number CMU-2563.
The authors declare no conflict of interest.