






 , J. Mao 2,†, X. Wang 1, B. Yin 1, Z. Shen 3, C. Di 4, W. Gu 5, M. Wu 2
, J. Mao 2,†, X. Wang 1, B. Yin 1, Z. Shen 3, C. Di 4, W. Gu 5, M. Wu 21 Department of Medical Oncology, Jiangsu Cancer Hospital & Jiangsu Institute of Cancer Research & The Affiliated Cancer Hospital of Nanjing Medical University, Nanjing, Jiangsu Province, China
2 Jiangsu Collaborative Innovation Center of Traditional Chinese Medicine (TCM) Prevention and Treatment of Tumor, Nanjing University of Chinese Medicine, Nanjing, China
3 Medical Oncology of Zhangjiagang First People's Hospital, Zhangjiagang, China
4 Emergency Center, Affiliated Hospital of Xuzhou Medical University, Xuzhou, China
5 Department of Otorhinolaryngology, Zhangjiagang Hospital of Traditional Chinese Medicine, Zhangjiagang, China
† Contributed equally.
Abstract
Objective: The authors aimed to explore the apoptosis of triple-negative breast cancer (TNBC) MDA-MB-231 cells induced by ginsenoside Rh2 and the underlying mechanism. Materials and Methods: Changes in the viability of MDA-MB-231 cells after treatment with 20 (S)-Rh2 and 20 (R)-Rh2 for 48 hours were detected by MTT assay. Changes in the morphology of cell nuclei were observed by DAPI staining. The expressions of caspase-3, caspase-9, cytochrome c, Smac, Bak, and Bax related to the mitochondrial pathway were detected by Western blotting. Results: 20 (S)-Rh2 inhibited the proliferation of MDA-MB-231 cells, but 20 (R)-Rh2 failed to do so. After treatment with 20(S)-Rh2 for two hours under visible light, they shrank and had incomplete morphology, whereas the morphology of control group hardly changed. Under UV light, the nuclei stained with DAPI were blue. After treatment with 20 (S)-Rh2 for one hour, the nuclei shrank and ruptured, and nearly 90% ruptured at two hours. In contrast, the nuclei of PBS control group remained intact. The activity of caspase-9 in MDA-MB-231 cells treated with 7.5 µg/mL 20 (S)-Rh2 was increased at 30 minutes, and gradually increased over extended time. Under identical conditions, the activity of caspase-9 in control group did not change significantly. Cytochrome c and Smac were released from mitochondria to the cytoplasm at one hour after treatment with 7.5 µg/mL 20 (S)-Rh2. However, such release was not detected in the control group. Bax was translocated after 30 minutes of treatment with 7.5 µg/mL 20 (S)-Rh2, which then gradually accumulated in mitochondria over time and peaked at two hours. The Bax expression in the entire cell lysate remained unchanged. The translocation of Bax was not detected in control group. Conclusion: 20 (S)-Rh2 evidently inhibited the proliferation of TNBC cell line MDA-MB-231. It killed the cells by inducing apoptosis, probably by activating the mitochondrial pathway.
Keywords
- Ginsenoside Rh2
- Triple-negative breast cancer
- Mitochondrial pathway.
Breast cancer is one of the most common malignancies, and the second leading cause for women’s deaths. Over 1, 000, 000 women around the world suffer from breast cancer annually, and such patients are becoming younger on average. Therefore, its treatment has long plagued the medical field [1-3]. Currently, this cancer is mainly treated by surgical removal of mammary glands, which, however, is not applicable to all patients, which also devastates their physical and mental health [4]. Therefore, it is necessary to find an effective drug and to clarify the mechanism of action.
MDA-MB-231 is a triple-negative breast cancer (TNBC) cell line with extremely high malignancy, which is simultaneously deficient in estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2) [5]. As known, ER, PR, and HER2 are important targets for the clinical treatment of breast cancer. Due to the lack of these targets, TNBC patients cannot receive routine endocrine therapy or targeted therapy, resulting in poor prognosis as well as high recurrence, metastasis, and morality rates. Thus, the treatment of TNBC has been recently spotlighted [6].
Ginsenoside Rh2 is an active ingredient of ginseng at trace amount, which can be classified into 20 (S)-Rh2 and 20 (R)-Rh2 according to optical activity. Ginsenoside Rh2 and its derivatives can inhibit the growth of a variety of tumors such as ovarian cancer, pancreatic cancer, and prostate cancer [7, 8]. It can furthermore inhibit cancer cell migration and invasion, and tumor growth in animal models.
The authors herein aimed to find effective drugs for TNBC treatment by studying whether ginsenoside Rh2 suppressed the growth of TNBC cell line MDA-MB231, and to clarify the molecular mechanism.
Cells were cultured in 96-well plates at a density of 104/100 µL, with three replicate wells for each drug concentration. After adherence, the cells were treated with ginsenoside Rh2 at specified concentrations (0, 0.5, 1, 3, 5, 7.5, and 10 mg/mL) which had been dissolved in serum-free DMEM. After incubation for four hours, 5 mg MTT powders were dissolved into 1 mL of PBS, and 20 µL of MTT solution (5 mg/ml) was added to 96-well plates and further cultured for four hours. After the medium was aspirated carefully with a syringe needle, the cells were dried for a period of time, added 150 µL/well of DMSO, and shaken rapidly for five minutes. After the purple crystals in 96-well plates were completely dissolved by DMSO, the absorbance at 570 nm was measured by a microplate reader. The survival inhibition rate was calculated. Inhibition rate = (ODcontrol-ODtest)/(ODcontrol-ODblank)×100%.
MDA-MB-231 cells in the logarithmic growth phase were collected and inoculated in 24-well plates at a density of 5×104/well. After adherence, the cells were treated with 7.5 µg/mL 20 (S)Rh2 for 0.5, 1, and 2 hours, respectively. The supernatant was then discarded, and the cells were rinsed with PBS and fixed in 75% ethanol (500 µL/well) at -20°C for five minutes. Afterwards, ethanol was removed, and 300 µL of DAPI (0.1 mg/ml) was added to each well. The cells were thereafter stained at room temperature for five minutes, and observed under a fluorescence microscope. Excited by UV light, the cell nuclei emitted blue fluorescence.
MDA-MB-231 cells in the logarithmic growth phase were collected and digested with 3 mL of trypsin for two minutes. After being counted by a hemocytometer, the cells were evenly inoculated in petri dishes at a density of 1.6×1106/10 mL. After adherence, the cells were added 7.5 µg/mL 20 (S)-Rh2 and treated for 0.5, 1, and 2 hours, respectively. After drug treatment, the cell culture medium was collected into a pre-cooled centrifuge tube, centrifuged at 3, 000 rpm for five minutes to discard the supernatant, then added 750 µL of pre-cooled PBS, washed twice, and centrifuged at 12, 000 rpm for one minute to remove the supernatant. The precipitate was added 9 µL of mitochondria isolation kit A solution (containing protease inhibitor PMSF), shaken by vortex at medium speed for 5 seconds, incubated on ice for two minutes, 10 µL of mitochondria isolation kit B solution (containing PMSF) added, shaken by vortex at high speed for five seconds, incubated on ice again for five minutes, 100 µL of mitochondria isolation kit C solution (containing PMSF) were added, mixed upside down carefully and slowly, and centrifuged at 4°C and 700 rpm for ten minutes to remove the precipitate. The supernatant was transferred to a new 1.5 mL pre-cooled EP tube, and centrifuged at 12, 000 rpm for 15 minutes at 4°C. The supernatant, which comprised mitochondria and cytoplasmic lysate, was transferred to another new pre-cooled EP tube to remove the precipitate, and the solution was centrifuged at 4°C and 10, 000 rpm for 15 minutes. The resulting supernatant was cytoplasmic protein, and the precipitate was mitochondria. Subsequently, the precipitate was added 60 µL of mitochondria isolation kit C solution (containing PMSF), washed, and centrifuged at 4°C and 12, 000 rpm for 15 minutes to discard the supernatant. The precipitate was thereafter added to 30 µL of cell lysis buffer RIPA, pipetted repeatedly, mixed well, and incubated on ice for one hour, while being mixed every 15 minutes. Then the mixture was centrifuged at 4°C and 12, 000 rpm for 15 minutes to discard the precipitate, and the supernatant was mitochondrial protein.
First, 50 µg of each sample was subjected to SDS-PAGE. Then proteins were transferred onto a nitrocellulose membrane that was blocked with 5% skimmed milk at room temperature for one hour and incubated with primary antibody at 4°C overnight. Afterwards, the membrane was washed, and incubated with secondary antibody at 37°C for one hour. Following ECL development and scanning, relative protein expressions were analyzed by Quantity-One software after internal reference correction.
Each experiment was performed three times independently. All data were analyzed by SPSS 22.0, and expressed as mean ± standard deviation. Comparisons between two groups were conducted by the t-test, and those among multiple groups were carried out by one-way analysis of variance. P < 0.05 was considered statistically significant.
MTT assay was used to detect the changes of viability of breast cancer cells MDA-MB-231 treated with 20(S)-Rh2 and 20(R)-Rh2, respectively. 20(S)-Rh2 had a strong inhibitory effect on the proliferation of MDA-MB-231 cells, and the inhibitory effect was in a dose-effect relationship with drug concentration. In contrast, 20(R)-Rh2 did not show tumor inhibitory effect at the same concentration (Figure 1). Therefore, only 20(S)-Rh2 was studied in subsequent experiments, using PBS as control.
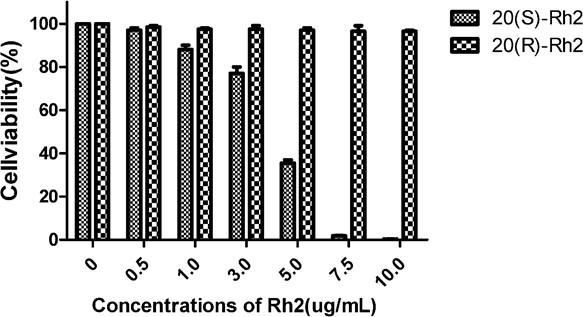 Figure 1.
Figure 1.— Viability changes of MDA-MB-231 cells after 48 hours of treatment with 20(S)-Rh2 and 20(R)-Rh2.
To investigate whether 20(S)-Rh2 can inhibit the proliferation of MD A-MB-231 cells by inducing apoptosis, the authors observed the morphological changes of the nuclei during drug treatment of cells by DAPI staining. Under visible light, after treatment with 20(S)-Rh2 for two hours, the morphology became incomplete and the cells shrank, but there was no obvious change in the cell morphology in the control group. Under UV light, the nuclei of DAPI were stained blue. After treatment with 20(S)-Rh2 for one hour, the nuclei began to shrink and rupture, and fragmentation was observed in nearly 90% of the nuclei in two hours. However, the nuclei remained intact in the control group (Figure 2). Accordingly, 20(S)-Rh2 inhibited MDA-MB231 cell proliferation by inducing apoptosis.
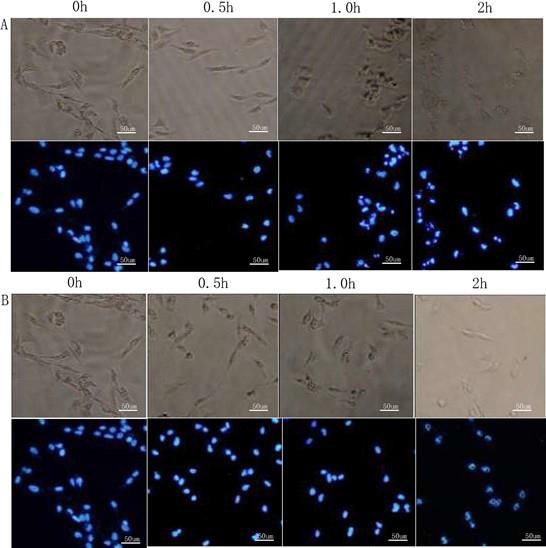 Figure 2.
Figure 2.— Morphological changes of MDA-MB-231 cells and nuclei after treatment with 7.5 µg/mL 20(S)-Rh2 for 0, 0.5, 1, and 2 hours. A: 20(S)-Rh2. B: PBS control.
The caspase-3 activity is one of the important indices for determining the occurrence of apoptosis [9]. To further determine the inhibitory effect of 20(S)-Rh2 on the proliferation of MDA-MB-231 cells by inducing apoptosis, the authors detected the activity of caspase-3 and found that caspase-3 could be activated after treatment with 7.5 µg/mL of 20(S)-Rh2 for one hour, and its activity was increased significantly with prolonged time. The activity of caspase3 at two hours was 6.5-fold that of the control group. In the control group, no significant changes were found in the caspase-3 activity within two hours (Figure 3). The results further confirmed that 20(S)-Rh2 allowed proliferation inhibition by inducing apoptosis of breast cancer cell line MDA-MB-231.
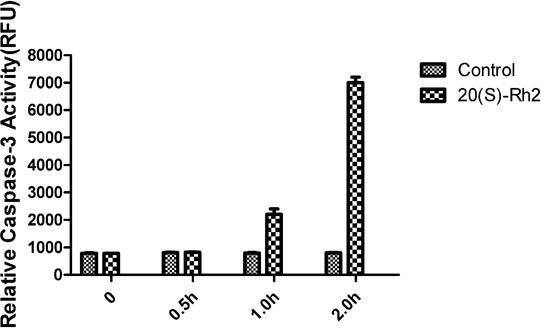 Figure 3.
Figure 3.— Caspase-3 activity changes of MDA-MB-231 cells after drug treatment.
Caspase-9 is a key enzyme in the mitochondrial pathway of apoptosis, and its activity is a crucial index in determining whether apoptosis is achieved via this pathway [10]. The activity of caspase-9 was increased 30 minutes after MDA-MB-231 cells were treated with 7.5 µg/mL 20(S)Rh2, further increasing gradually over extended time. After two hours, its activity peaked, 9.3-fold that of the control group. Under the same conditions, the caspase-9 activity hardly changed in the control group (Figure 4).
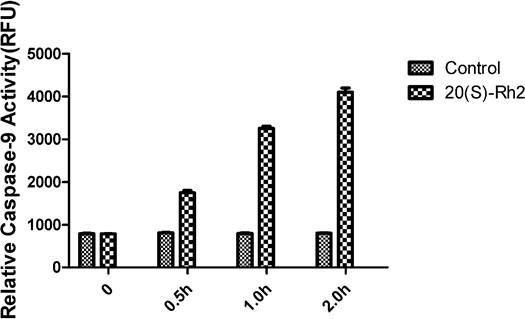 Figure 4.
Figure 4.— Caspase-9 activity changes of MDA-MB-231 cells after drug treatment.
Caspase-9 is activated mainly by the release of key pro-apoptotic factors cytochrome c and Smac through mitochondria to the cytoplasm. The cytochrome c and Smac expressions in the cytoplasm were detected by Western blotting. Cytochrome c and Smac began to be released from mitochondria to the cytoplasm at one hour after MDA-MB231 cells were treated with 7.5 µg/mL 20(S)-Rh2. Nevertheless, the release of cytochrome c and Smac was not detected in the cytoplasm in the control group (Figure 5). Thus, 20(S)-Rh2 induced apoptosis of MDA-MB-231 cells by activating the mitochondrial pathway.
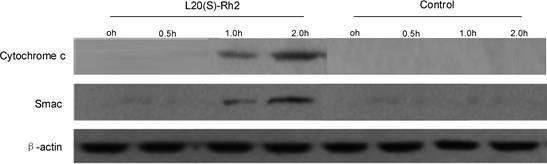 Figure 5.
Figure 5.— Release of cytochrome c and Smac from MDA-MB-231 cells after drug treatment.
In cells that are not normally stimulated by apoptotic signals, both the pro-apoptotic protein Bak on the mitochondrial membrane and the pro-apoptotic protein Bax in the cytoplasm exist in a non-active monomeric form. When the apoptotic signal stimulates the cells, Bax in the cytoplasm oligomerizes on the mitochondrial outer membrane to form a PT channel, thereby promoting the release of cytochrome c and Smac [11]. Therefore, the authors detected the expressions of Bax and Bak in whole cell lysate and mitochondria by Western blotting. Bax began to be translocated after MDA-MB-231 cells were treated with 7.5 µg/mL L20(S)-Rh2 for 30 minutes, and gradually accumulated in the mitochondria with elapsed time and peaked at two hours. However, the Bax expression in whole cell lysate did not change. Hence, the accumulation of Bax in mitochondria was not caused by the upregulation of overall expression, but the Bax translocation in the cytoplasm. In the control group, Bax translocation was not detected (Figure 6). Bax was translocated from the cytoplasm to mitochondria, cytochrome c and Smac were released from mitochondria to the cytoplasm, and caspase-9 was activated, all of which together demonstrated that 20(S)-Rh2 induced the apoptosis of MDA-MB cells by activating the mitochondrial pathway.
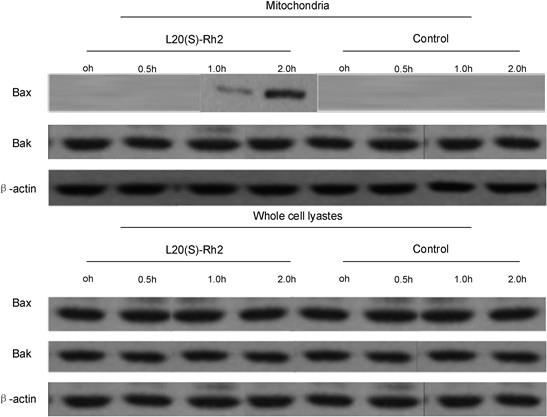 Figure 6.
Figure 6.— Bax and Bak expressions in mitochondria and whole cell lysate of MDA-MB-231 cells after drug treatment.
Apoptosis, also known as programmed cell death, is a concept discovered in a group of electron microscopy images and proposed by the three scientists of Kerr, Wyllie, and Currie in 1972. The process of apoptosis is accompanied by many changes in cell morphology, such as cell shrinkage, cell membrane and nuclear membrane foaming, organelle repositioning and constriction, and chromatin condensation eventually producing “apoptotic bodies” [12].
Complete mitochondrial function is the basis of cell survival. Mitochondria can integrate many pro-apoptotic signals. Among them, most of the proteins translocated onto the mitochondrial membrane upon stimulation by apoptotic signals are members of the Bcl-2 family like Bax. The Translocated Bax can change the mitochondrial outer membrane permeability (MOMP), and this process is also regulated by the BH3-only protein in the Bcl-2 family: BID can be hydrolyzed by caspase-8 and cleaved into tBID which can help Bax translocation, and in turn change MOMP. BAD can be translocated to mitochondria and interact with anti-apoptotic protein in the Bcl-2 family, and lift the inhibiting effect of Bcl-2 on Bak, thereby promoting apoptosis. Changes in the permeability of the mitochondrial outer membrane can promote cytochrome c, mitochondria and other apoptosis factors to activate Smac, Endo G, and apoptosis-inducing factor. Cytochrome c is undoubtedly one of the important factors in the process of apoptosis. It is usually associated with an oxidative respiratory chain, but it can cause very serious damage to cells once the integrity of mitochondria has been destroyed [10, 13]. Cytochrome c better allows the oligomerization of adapter molecule apoptosis-protease activating factor 1 (Apaf-1) in the presence of dATP, so as to form apoptosome. The apoptosome can recruit and dimerize seven procaspases-9 to activate caspase-9, which in turn activates downstream apoptotic pathways. The first four amino acids of mature Smac are Ala-Val-Pro-Ile, which can recognize the BIR domain of the IAPs family proteins and release the IAPs family’s inhibitory effect on caspase-9 and caspase-3, thus promoting apoptosis [14]. In this study, 20(S)-Rh2 effectively inhibited the proliferation of MDA-MB-231 cells. 20(S)-Rh2 exhibited its antitumor activity by inducing the apoptosis of these cells. Moreover, 20(S)-Rh2 initiated the endogenous pathway of apoptosis by inducing Bax translocation to the mitochondria and subsequent release of cytochrome c and Smac to activate caspase-9.
In summary, 20(S)-Rh2 inhibited the proliferation of breast cancer cell line MDA-MB-231 by activating the mitochondria-mediated apoptosis pathway. After MDA-MB-231 cells were treated with 20(S)-Rh2, pro-apoptotic protein Bax was translocated to mitochondria, which changed the permeability of the mitochondrial membrane. As a result, pro-apoptotic cytochrome c was released, which formed an apoptosome with Apaf-1 and precursor caspase-9, thus activating caspase-9, initiating the mitochondria-mediated apoptosis pathway and promoting apoptosis.