

Clinical and Experimental Obstetrics & Gynecology (CEOG) is published by IMR Press from Volume 47 Issue 1 (2020). Previous articles were published by another publisher on a subscription basis, and they are hosted by IMR Press on imrpress.com as a courtesy and upon agreement with S.O.G.
 , G. Ambrosini 2
, G. Ambrosini 21 Gynecologic and Obstetric Clinic, Department of Medical, Surgical and Experimental Sciences, University of Sassari, Sassari, Italy
2 Department of Women and Children’s Health, Unit of Gynecology and Obstetrics, University of Padua, Padua, Italy
3 University of Ferrara, Ferrara, Italy
Abstract
Congenital cystic adenomatoid malformation of the fetal lung (CCAMs) are rare malformations resulting from bronchial overgrowth with almost complete suppression of the alveolar development. Due to rarity of CCAMs, there is still a lack of evidence on the optimal management of this condition. Here the authors present a case of CCAM diagnosed during the second trimester of pregnancy and followed-up until delivery. This report shows that, in case of non-severe CCAMs and uncomplicated pregnancies, an optimal management of the patient can lead to spontaneous delivery of a healthy baby.
Keywords
- Congenital cystic adenomatoid malformation of the fetal lung (CCAMs)
- Diagnosis
- Management
Congenital cystic adenomatoid malformation of the fetal lung (CCAMs) are the most common malformation of the lower respiratory tract, with an incidence estimated in one in 11,000 to one in 35,000 live births [1]. CCAMs are hamartomatous lesions resulting from bronchial overgrowth, with almost complete suppression of the alveolar development. They are usually monolateral and unilobar, but can rarely involve multiple lobes of the same lungs or both the lungs [2, 3]. The macroscopic appearance of CCAMs varies from solid to cystic or mixed solid-cystic lesions.
In some cases CCAM is found in hybrid lesions with another lung malformation, namely broncopulmunary sequestration (BPS), suggesting their possible common pathogenetic origin. According to the latter hypothesis, both CCAM and BPS develop during the period of bronchial tissue proliferation (from the 7th to the 17th week of gestation), and become evident at ultrasound during the second trimester of pregnancy [1, 4-6].
During the last decades, different classification systems for CCAMs have been proposed. The most recent one by Di Stocker et al. [7] classifies five types of CCAM basing on the presumed site of its development: Type 0 is the rarest type, characterized by small cysts. It is usually lethal and arises from the tracheobronchial district. Type 1 is the most common type of CCAMs (from 40% to 70% of all malformations), marked by multiple macro-cysts (mean diameter greater than 20 mm). It arises from the distal bronchus or proximal bronchiole and has usually a good prognosis. Rarely, when the malformation is too large, it can lead to hydrops. Type 2 accounts for 15%-30% of CCAM and is characterized by small cysts (5-20 mm in diameter). It arises from terminal bronchioles and is often (up to 60% of cases) associated with other malformations. Thus, the prognosis mainly depends on the type of co-existing malformations. Type 3 CCAM accounts for 5-10% of the cases, arising from acinar-like tissue. It is composed of tiny cysts and resembles a solid lesion. Prognosis is mainly unfavourable due to the frequent mass effect of large lesions. Type 4 accounts for 10-15% of CCAMs, arising from the alveolar tissue. It is characterized by large cysts (even larger than 10 cm) and the prognosis is usually poor. Type 4 CCAM have been also associated with pleuropulmonary blastoma. At the ultrasound examination, types 1, 2, and 4 CCAMs have a cystic appearance, while type 3 CCAM appears as a well-circumscribed, echogenic mass. Statistically, 60% of CCAMs have a macrocystical aspect, while the remaining 40% are microcystic [6, 8].
In fetuses affected by CCAMs, the lung volume increases progressively during pregnancy, especially between the 20th and 26th week of gestation. As the residual pulmonary volume is the major predictor of neonatal prognosis, an accurate ultrasound study using volumetric probes (with 3D or 4D software) is of critical importance [6, 9].
The 3D multislice ultrasound offers the possibility to evaluate the lesion on the three orthogonal planes. It allows the evaluation of the extent of the mass and its relationship with surrounding structures [10-11].CCAM is normally presented in an isolated form but can be rarely associated with renal abnormalities (agenesis, dysplasia), intestinal (atresia), heart, and skeletal. The association with chromosomal abnormalities is rare, in the order of 1.6%. Specifically, its association with trisomy 18 (1 case out of 48) [12], trisomy 13 [6, 8, 13], monosomy X [13], Prune Belly syndrome [14], and Fraser’s syndrome [15] have been previously reported [6, 8, 13]. After diagnosis of CCAM, a follow-up in utero should be conducted in order to estimate any possible change in lesions, pulmonary residual volume and the amount of healthy parenchyma that circumscribes the lesion, the relationship between CCAM/lung in toto and, lastly, the appearance of possible complications (hydrothorax, hydrops). In about 25% of cases, a mass regression is observed during the follow-up. In general, the larger the size of the lesion, the less likely a regression will occur [12]. About 10% of cases CCAMs are associated with hydrops, when large and/or bilateral lung lesions are present [12]. Sometimes it is possible to find a polyhydramnios due to the esophageal compression. In fetuses affected by hydrops, in utero surgery is the only life-saving option, even if the risk of intrauterine death is considerable. Differently, in uncomplicated CCAM, the delivery can be achieved at term by vaginal route. In 80% of cases, the CCAM is asymptomatic at birth, while in the remaining cases surgery is indicated as a life-saving procedure. In postnatal age, the asymptomatic CCAM should be subject to follow-up for the possible onset of complications such as infections, neoplastic degeneration, and hemorrhages, although they are all rare occurrences [14, 15]. Commonly, lesions are regressed within the third year of life. In case of non-regression after the third year of life, a surgical excision of the mass is indicated [16]. Here the authors report the case of CCAM diagnosed during the second trimester of pregnancy and followed-up until delivery at our Gynecologic and Obstetric University Hospital.
A 35-year-old, nulliparous woman had a normal antenatal course up to 21.6 weeks. She was in therapy with carbamazepine 900 mg daily due to epilepsy. Blood group was A Rhesus positive, with negative tests for glucose-6-phospate dehydrogenase and thalassemia. Serological tests for infectious diseases (HBV, HCV, HIV, and “TORCH complex”) were negative. The woman had a negative history for alcohol or drug use, environmental pollutants exposure and no I degree familiarity for genetics or chromosomal diseases. The pregnancy was achieved naturally, without use of assisted reproduction technology. The combined test of the first trimester resulted in a cumulative low risk of Down syndrome (1: 427), with an increased biochemical risk (1: 108) and normal nuchal fold thickness (1.4 mm).
At the second trimester ultrasound examination (at 21.6 weeks of gestation), a solid mass measuring 24 x 16 x 15 mm was identified at the bottom of the right lung (Figure 1). The mass was hyperechogenic and included multiple cystic lesions, the larger measured 10 mm in diameter. The mass was classified as macrocystic CCAM. No mediastinic shift was detected, nor other fetal malformations were identified. Fetal biometry was adequate for gestational age. A second ultrasound scan was performed five weeks later (26.5 weeks), confirming the presence of a mass suggestive for CCAM at the lower right pulmonary field. The size of the mass was unmodified from the previous measurement. Three subsequent ultrasound examinations were performed at 30, 32, and 38 weeks of gestation, showing neither size modifications of the CCAM, nor fetal complications. According to the classification reported by Stocker et al. [7], the picture was compatible with type 1 CCAM. Fetal echocardiography was performed at week 23 and repeated at week 35, showing no alterations in cardiac morphology and kinetics. The patient at 39 weeks and 1 day carried out a spontaneous delivery, without peri-partum complications.
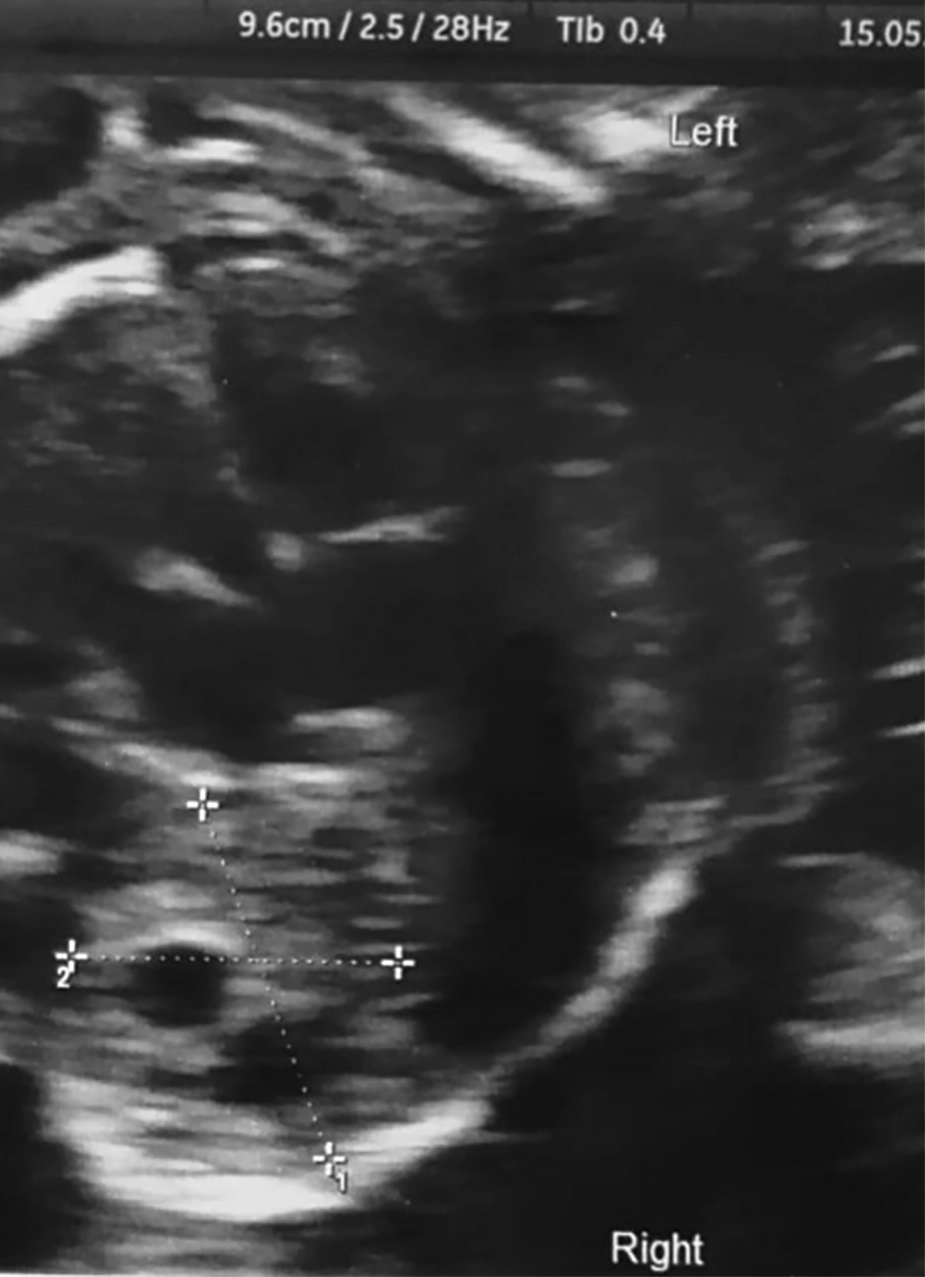 Figure 1.
Figure 1.— Ultrasound shows a solid mass measuring 24× 16×15 mm at the bottom of the right lung
A healthy, male baby weighted 2,770 grams was born. Venous pH of umbilical cord was regular (7.34, SaO2=94, base excess: -2). Apgar score was 9 at five and ten minutes. At the neonatological evaluation, the infant showed an excellent adaptation to extrauterine life without any kind of problem linked to the pathology. Neonatal thoracic radiogram and echocardiography were normal. Six months after birth, CT confirmed the presence of CCAM. In particular, a substantial reduction in the size of the right mass was shown (4.5 mm vs. 10 mm, > 50% reduction in size). Additionally, two small lesions (about 2 mm in diameter) with the same aspect were identified at the basal level of the left lung. After two years follow up, the baby is healthy, with a normal neurodevelopmental growth and without need for surgery.
Fetal lung development is complex and is traditionally divided into different phases: the embryonic (4th to 6th week), pseudo-glandular (7th to 16th week), canalicular (17th to 23rd), saccular (24th to 36th week), and alveolar (36th week to the end) phases [6]. CCAM is considered secondary to embryonic insult occurring during the pseudo-glandular phase of lung development, which results in the development of hamartomatous tissue. The primum movens for CCAM may be an obstruction of the airways, tissue dysplasia/metaplasia or abnormal tissue proliferation. Nevertheless, the pathogenesis is still to be elucidated [1, 5, 8]. Furthermore, the physiological mechanisms underlying the regression of CCAMs (which can occur in uterus and/or in post-natal life) is unclear. It is possible that abnormal tissue growth leads to an insufficient maturation of the vascular supplement in CCAMs, resulting in atresia [2]. Alternatively, tissue atresia may be resultant from a persistent bronchial occlusion [3]. Last but not least, the dysregulation of the mechanisms of apoptosis [4] may cause the regression of CCAMs; nevertheless, further studies are needed to confirm these hypotheses [5]. Lung development follows well-structured phases induced and regulated by specific metabolic patterns. Various growth factors such as thyroid transcriptional factor-1 (TTT-1), hepatocyte nuclear factor (HNF) -3beta, HNF-3forkhead homologue-4 (HFH-4) are necessary for the proliferation and differentiation of alveolar epithelial cells [1, 5, 6]. The communication between the development of epithelial and mesenchymal tissue is regulated by the activity of the integrins, which also influence the adhesion activity of kinases and other adhesion molecules such as E-cadherin and numerous growth factors such as epidermal growth factor receptor (EGFR), fibroblast growth factor receptor (FGFR), vascular endothelial growth factor receptor (VEGF), and transforming growth factor beta (TGF-beta). Through these interactions associated with actin action in the cytoskeleton, integrins modulate migration, differentiation, and cell growth. In CCAMs, alterations of the cytoplasmic alpha2-integrin signals and of various cell adhesion molecules are demonstrated. All these factors indicate an alteration of the balance between the biochemical interactions of the epithelium and the mesenchyme in the initial phase of pulmonary morphogenesis as a cause of CCAM development [8]. From an pathologic point of view, the characteristic lesions of CCAM are constituted by an abnormal development of terminal bronchioles associated with the poorly differentiated tissue. The lesions consist of solid and cystic tissue (macro and microcyst); the latter is covered by respiratory epithelium [13, 14]. The cystic lesions communicate with the tracheobronchial tree. The diagnosis of certainty can be based on the histology of the lesions. Based on cytological characteristics, three main subtypes can be identified, CCAM I, II, and III which have a prevalent bronchiolar epithelium and the CCAM IV represented by an acinar-alveolar epithelium.
Recent advances in imaging techniques have radically changed the management of obstetric disorders [17-20]. In particular, ultrasound examination currently represent the gold standard for monitoring fetal well-being [19, 20] and the possible changes of uterine diseases (i.e. myomas) during pregnancy [17, 18] due to easy access, low costs, and lack of invasiveness. As previously discussed, CCAMs can be suspected only at fetal ultrasound examination during the second trimester of pregnancy (after the 20th week of gestation). Thereafter, the pathology requires a continuous ultrasound monitoring during pregnancy and a paediatric follow-up after birth, in addition to psychological support for the couple. A careful monitoring of CCAM features, the volumetric changes and the foetal well-being are mandatory for an adequate clinical management. In fact, complicated cases require more aggressive behaviour (interruption of pregnancy or intrauterine surgery).
In uncomplicated cases, a close ultrasound follow-up (with a frequency based on the severity of CCAMs) is recommended, potentially culminating in a spontaneous delivery. At this regard, it must be stressed that CCAM is not an indication for elective caesarean delivery, as demonstrated in the case reported. Nevertheless, we must stress the concept that an optimal management of CCAMs require the collaboration of a multi-disciplinary team including gynaecologist, paediatrician, surgeon, and psychologist [21, 22]. In conclusion, congenital lung lesions are detected during second trimester ultrasound scans and require a specialised, multi-disciplinary management. A careful evaluation of the fetus is required to determine the severity of the disease, the risk of complications, the need for surgery, and the optimal mode of delivery.
In case of non-severe CCAMs and uncomplicated pregnancies, an optimal management of the patient can facilitate a spontaneous delivery of a healthy baby.
Ph. Doctor School in Biomedical Sciences, Address in Gender Medicine, Men, Woman and Child, Sassari University, Italy, supported the study.