




 , Ahai Luvai 2, Paul Downie 1, Ayoob Ghani 1, Megan Maple 3, Wycliffe Mbagaya 1
, Ahai Luvai 2, Paul Downie 1, Ayoob Ghani 1, Megan Maple 3, Wycliffe Mbagaya 11 Department of Clinical Biochemistry, University Hospitals Bristol and Weston NHS Foundation Trust, BS2 8HW Bristol, UK
2 Department of Clinical Biochemistry, Newcastle upon Tyne Hospitals NHS Foundation Trust, NE1 4LP Newcastle upon Tyne, UK
3 Department of Haematology, UCL Cancer Institute, WC1E 6DD London, UK
Abstract
Despite accounting for a quarter of all UK deaths, atherosclerotic cardiovascular disease (ASCVD) is frequently undertreated in non-specialist hospital care due to the perceived complexity of modern lipid management. This review aims to bridge the gap between evolving guidelines and routine clinical practice. We examine the advances in lipid testing, including lipoprotein (a), apolipoproteins and genetic testing, which have allowed improved and personalised risk assessment. While statins remain the foundation of lipid-lowering therapy, the treatment options have expanded and we discuss the role of ezetimibe, proprotein convertase subtilisin/kexin type 9 (PCSK9) monoclonal antibodies, inclisiran, and bempedoic acid. We also look ahead to gene-editing strategies and lipoprotein (a) lowering therapies currently in clinical trials. By simplifying these recent advances, this review aims to provide hospital clinicians with a practical approach to identify high-risk patients, optimise their therapy and reduce cardiovascular disease burden.
Keywords
- lipids
- dyslipidemias
- cardiovascular diseases
- hypertriglyceridemia
- cholesterol
- low-density lipoprotein
- lipoprotein (a)
- atherosclerosis
- anticholesteremic agents
Atherosclerotic cardiovascular disease (ASCVD) remains one of the leading causes of morbidity and mortality in the UK, with an estimated 7.6 million people living with heart and circulatory disease [1]. Despite advances in care, ASCVD continues to account for 27% of all deaths in the UK, imposing a significant burden not only on public health but also on healthcare resources, with an estimated annual cost of $12 billion to the National Health Service (NHS). Addressing this challenge is central to the NHS long term plan, which hopes to improve the detection and management of patients with ASCVD to mitigate its impact on population health outcomes [2].
Effective management of blood lipids is a critical strategy both for reducing the initial risk of developing ASCVD and for lowering the likelihood of recurrent events in those already affected [3, 4]. In recent years, the field of lipidology has undergone significant advancements. Key developments include:
• Improved understanding of the genetic underpinnings of lipid disorders, alongside advances in genetic testing technologies that are increasingly accessible and informative [5].
• A growing recognition of the role of novel risk factors, such as elevated lipoprotein (a) [Lp(a)], in driving cardiovascular risk [6].
• The expansion of the pharmacological options available provides clinicians with the ability to tailor interventions to individuals.
While statins remain the first-line therapy for lipid-lowering, the novel and emerging oral and injectable therapies allow for further optimisation and address the needs of the statin-intolerant population. These recent advancements have been integrated into recent guideline updates, most notably those from the European Society of Cardiology in 2025 [3] and the National Institute for Health and Care Excellence (NICE) [7].
This review provides non-specialist clinicians with a concise UK-focused update on advancements in lipid management, assuming a foundational understanding of lipid metabolism and experience in managing ASCVD. It aims to update knowledge and provide a practical and actionable perspective on addressing lipid disorders and managing patients at elevated cardiovascular risk in routine hospital practice.
Lipids, including cholesterol and triglycerides, are insoluble in plasma and require transport by lipoproteins to circulate in the bloodstream. Lipoproteins are complex structures made up of cholesterol, phospholipids, triglycerides, and apolipoproteins (Fig. 1). Clinical laboratories measure key lipid parameters—total cholesterol, triglycerides, and high-density lipoprotein cholesterol (HDL-C), to assess lipoprotein levels and metabolism. From these, additional values like low-density lipoprotein cholesterol (LDL-C) and non-high-density lipoprotein cholesterol (non-HDL-C) are calculated. Together, these measured and derived values form the lipid profile, a fundamental tool for diagnosing and managing lipid disorders and ASCVD, summarised in Table 1 (Ref. [8, 9]) [10].
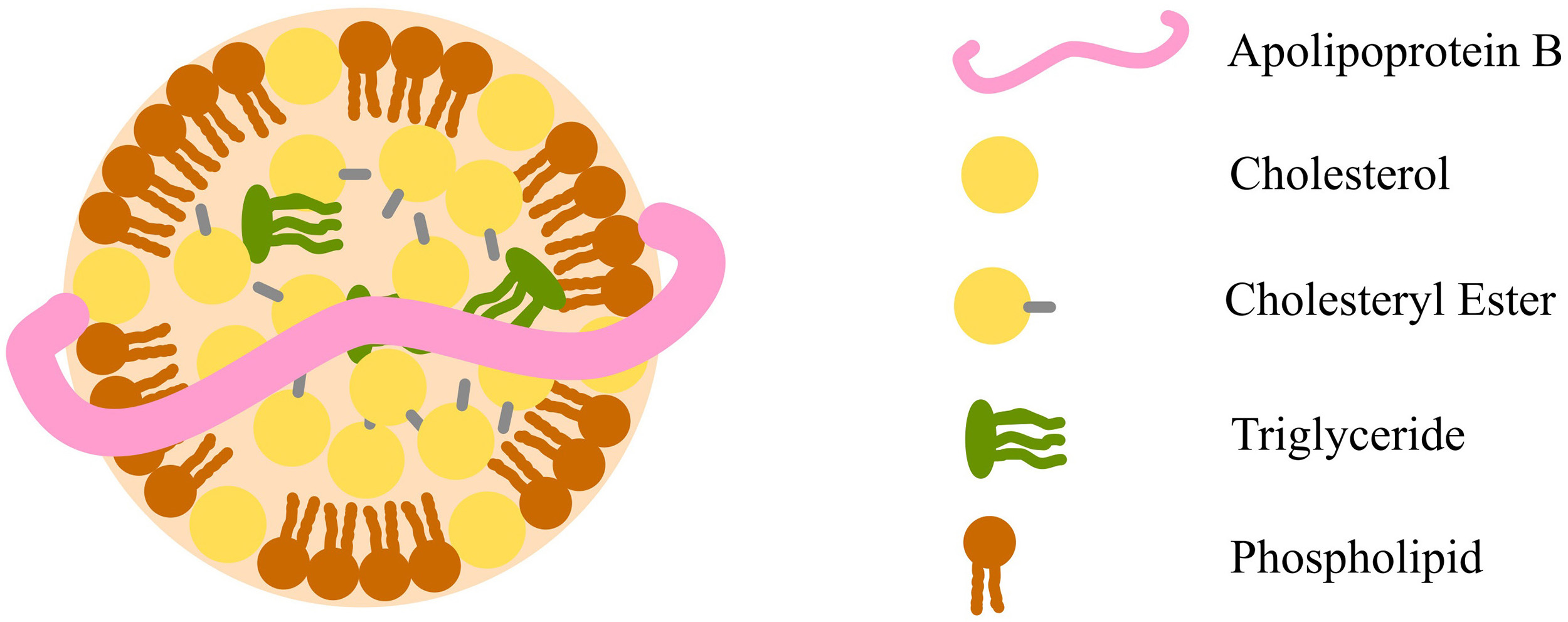 Fig. 1.
Fig. 1.
Diagram of a lipoprotein macromolecular structure, using low-density lipoprotein as an example. The illustration highlights a core composed of cholesteryl esters and triglycerides, encased by a surface layer of free cholesterol and phospholipids. A single apolipoprotein B molecule binds these components together. Fig. 1 was created using Inkscape (https://inkscape.org/).
| Constituent | Method/Calculation |
| Total cholesterol (mmol/L) | Measured using enzymatic assays |
| Triglycerides (mmol/L) | Measured using enzymatic assays |
| HDL-C (mmol/L) | Measured using enzymatic assays after precipitating apolipoprotein B particles |
| LDL-C (mmol/L) | Historically calculated using the Friedewald formula, more recently, the Sampson or Martin-Hopkins formula is being implemented for greater accuracy [8, 9] |
| Non-HDL-C (mmol/L) | Calculated by subtracting HDL-C from total cholesterol |
HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; non-HDL-C, non-high-density lipoprotein cholesterol. Note: Both non-HDL-C and LDL-C are valid treatment targets, particularly in patients with established atherosclerotic cardiovascular disease (ASCVD). However, LDL-C is more commonly used, as it is the primary focus of clinical trials and guidelines.
Cholesterol is predominantly carried in low-density lipoprotein (LDL) and high-density lipoprotein (HDL), whereas triglycerides are predominantly carried in chylomicrons, which carry dietary triglycerides, and very-low-density lipoproteins (VLDLs), which carry triglycerides derived from hepatic synthesis. VLDLs also contain a small proportion of cholesterol, and it is this proportion that necessitates the use of formulae to estimate LDL cholesterol. The older Friedewald equation had a significant limitation as it assumed a fixed proportion of cholesterol in VLDL, leading to an under-estimation of LDL cholesterol in cases of hypertriglyceridaemia. Modern equations, such as the Sampson and Martin-Hopkins equations, are more accurate, even with triglyceride levels of up to 9 mmol/L, leading to their widespread adoption [8, 9].
Direct LDL-C assays are available in some laboratories, but their accuracy varies between manufacturers and is often affected by high triglyceride levels [11]. Some specialised laboratories use advanced analytical techniques for LDL-C measurement, but these methods are labour-intensive, have longer turnaround times, and are mainly used for complex cases beyond this article’s scope.
Non-HDL-C represents the total atherogenic cholesterol carried by all circulating lipoproteins. While it primarily includes LDL-C, it also accounts for cholesterol within VLDL and its remnants.
Lipoprotein (a) [Lp(a)] is a unique lipoprotein consisting of an LDL molecule bound to apolipoprotein (a) [apo(a)] via a disulphide bond [12] (Fig. 2). Beyond carrying cholesterol, its oxidised phospholipids drive inflammation, while its plasminogen-like structure may reduce fibrinolysis, increasing thrombotic risk. Lp(a) is about six times more atherogenic than LDL, significantly increasing the risk of ASCVD and calcific aortic stenosis [13, 14].
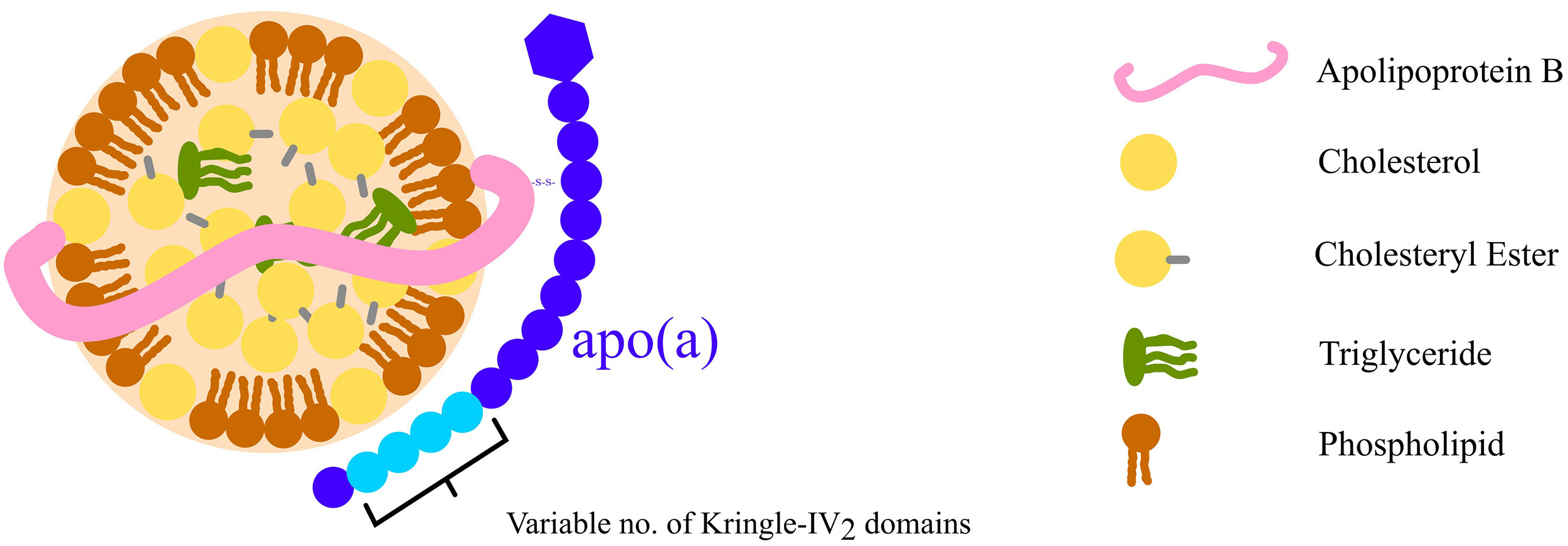 Fig. 2.
Fig. 2.
Diagram of Lp(a) structure illustrating an LDL molecule covalently linked to an apo(a) molecule via a disulphide bond. The number of KIV2 domains varies, influencing the size of apo(a) and, consequently, the overall Lp(a) structure. This variation is associated with differences in Lp(a) concentrations in plasma among individuals. KIV2, kringle-IV type-2; Lp(a), lipoprotein (a); apo(a), apolipoprotein (a). Fig. 2 was created using Inkscape (https://inkscape.org/)
Circulating levels of Lp(a) are largely genetically determined: the apo(a) component is encoded by the LPA gene, which follows an autosomal semi-dominant inheritance pattern [15]. Its size varies due to differences in the kringle-IV type-2 (KIV2) domain copy number [16], with larger isoforms associated with lower Lp(a) levels [17, 18]. Population levels of Lp(a) are positively skewed, meaning that most individuals have low levels with no associated increased risk, while a significant minority have very high levels corresponding to a far greater risk of ASCVD.
Guidelines recommend measuring Lp(a) at least once in a lifetime to refine cardiovascular risk assessment [19, 20]. While no targeted therapies are currently available, advanced clinical trials are underway. Meanwhile, aggressive management of traditional risk factors can help mitigate ASCVD risk. Notably, Lp(a) testing is inexpensive and accessible in routine laboratories [21].
Atherosclerosis progresses as cholesterol-rich lipoproteins containing apolipoprotein B (ApoB), primarily LDL, cross the arterial wall. Since each atherogenic particle carries one ApoB molecule, ApoB serves as a direct and reliable marker of atherogenic lipoprotein burden, offering critical insight into cardiovascular risk. In settings without advanced equipment, ApoB is the most robust marker for assessing atherogenic lipoproteins and helps characterise lipoprotein patterns when LDL-C is incalculable due to raised triglycerides. Like Lp(a), it is inexpensive and available in routine laboratories, though not yet standard nationwide in the UK [22].
In contrast, high-density lipoprotein (HDL) particles vary in apolipoprotein composition, often containing multiple apolipoprotein A1 (ApoA1) molecules. While the ApoB:ApoA1 ratio has been explored for cardiovascular risk assessment, it is rarely used in routine practice, and the clinical value of ApoA1 measurement remains uncertain [23].
Genomic testing for inherited lipid disorders is advancing, enabling better identification and management. Currently, 25 monogenic conditions affecting lipoprotein metabolism are recognised [24]. While not all are routinely tested in clinical practice, several key disorders can be assessed in the UK and should be considered when evaluating patients with suspected genetic lipid abnormalities.
For individuals presenting with isolated elevations of LDL cholesterol and a compatible family history, the primary lipid disorders to consider include [25]:
• Familial hypercholesterolaemia (FH): associated with variants in low-density lipoprotein receptor (LDLR), apolipoprotein B (APOB) and proprotein convertase subtilisin/kexin type 9 (PCSK9) genes.
• FH phenocopy disorders: including variants in low-density lipoprotein receptor adaptor protein 1 (LDLRAP1), ATP-binding cassette subfamily G member 5 (ABCG5), ATP-binding cassette subfamily G member 8 (ABCG8), apolipoprotein E (APOE), and lipase A, lysosomal acid type (LIPA) genes.
FH is an autosomal semi-dominant disorder that disrupts the LDL receptor (LDLR) pathway, leading to lifelong LDL-C elevation and a high risk of premature ASCVD [25, 26]. In its most severe form, where individuals inherit biallelic (previously known as homozygous) variants, advanced atherosclerosis and vascular complications can develop in childhood [27]. Genomic testing typically follows phenotypic scoring to improve variant detection. In the UK, the Simon Broome and Dutch Lipid Clinics Network criteria guide eligibility for proband testing. Once a pathogenic variant is identified, cascade testing of family members is recommended for early intervention [28]. Except for the most severe cases, with timely lipid-lowering therapy, vascular complications are largely preventable.
For patients with mixed hyperlipidaemia [elevated triglyceride (TG) and non-HDL cholesterol (non-HDL-C)], the primary genetic lipid disorder to consider is:
• Dysbetalipoproteinaemia: associated with variants in the APOE gene [29].
Dysbetalipoproteinaemia is caused by pathogenic variants in the receptor-binding
region of APOE, impairing LDLR binding and leading to cholesterol-rich
remnant accumulation, significantly increasing vascular disease risk,
particularly occlusive peripheral arterial disease [30]. About 90% of cases
result from autosomal recessive APOE
Polygenic SNPs are increasingly being integrated into genomic testing panels for inherited lipid disorders. While some Genomic Laboratory Hubs include LDL-raising SNPs in their FH testing panels, these SNPs have not yet been formally incorporated into the UK Genomic Test Directory. Additionally, other polygenic risk scores, such as the cardiogenic risk score CardioinCode, are available through certain private providers [33]. However, concerns persist regarding the applicability and commutability of these polygenic risk scores across different populations.
Hypertriglyceridaemia (HTG) is a common biochemical abnormality, defined by
specific thresholds of plasma TG concentrations within the general population. A
fasting TG level below 1.7 mmol/L is generally considered normal [34, 35, 36]. Severe
HTG is classified as a fasting plasma TG level exceeding 10 mmol/L [34]. Severe
HTG can present with eruptive xanthomas, palmar crease xanthomas,
hepatosplenomegaly, lipaemia retinalis and most notably, acute pancreatitis,
which is associated with significant morbidity and mortality [37]. The risk of
pancreatitis is approximately 5% at plasma TG
Over 95% of dietary lipids are TGs, which are broken down into monoglycerides (MGs) and free fatty acids (FFAs) by gastric and pancreatic lipases [39]. MGs and free cholesterol are solubilised by bile acid micelles for intestinal absorption. Within enterocytes, MGs are reassembled into TGs, packaged into chylomicrons, and transported via the lymphatic system [40]. In the capillaries of adipose and muscle tissue, apolipoprotein C-II (ApoCII) on chylomicrons activates lipoprotein lipase (LPL), hydrolysing TGs into fatty acids and glycerol for storage or energy use [41]. The remaining cholesterol-rich chylomicron remnants are cleared by the liver mediated by apolipoprotein E (ApoE). This sequence of events constitutes the exogenous lipid pathway [42].
The liver synthesises VLDL, containing ApoB, and releases them into the circulation. These particles transport triglycerides and cholesterol to peripheral tissues. VLDL synthesis is stimulated by increased intrahepatic FFAs, which can result from a high-fat diet or excessive release of FFAs from adipose tissue into the bloodstream [43]. VLDL is metabolised into IDL, which is either cleared by the liver or converted by hepatic lipase into cholesterol-rich LDL, defining the endogenous lipid pathway [42]. Plasma TG levels reflect contributions from chylomicrons, VLDL, their remnants, and smaller amounts from LDL and HDL [44]. Dysregulation of the exogenous or endogenous lipid pathways can lead to hypertriglyceridaemia (HTG) [45].
Chylomicronaemia, the persistence of chylomicrons after fasting, is a key feature of lipid disorders such as familial dysbetalipoproteinaemia, multifactorial chylomicronaemia syndrome (MCS), and the rare familial chylomicronaemia syndrome (FCS). A key symptom of chylomicronaemia syndromes is severe hypertriglyceridaemia [46]. FCS is caused by biallelic loss-of-function variants in genes essential for triglyceride metabolism, primarily lipoprotein lipase (LPL) and its regulators, such as apolipoprotein C2 (APOC2), apolipoprotein A5 (APOA5), lipase maturation factor 1 (LMF1), and glycosylphosphatidylinositol-anchored HDL-binding protein 1 (GPIHBP1) [47, 48]. MCS results from a combination of heterozygous loss-of-function variants in key genes (as listed above for FCS), small-effect polygenic variants, and lifestyle factors, leading to a highly variable phenotype [49, 50, 51].
In the UK, studies indicate that approximately 50% of FCS cases are caused by pathogenic variants in LPL. More than 80 LPL variants have been identified, most of which significantly impair LPL function in the homozygous state, leading to the accumulation of triglyceride-rich lipoproteins [52]. However, many cases result from non-LPL variants affecting LPL activity, producing an indistinguishable clinical phenotype [48, 53, 54]. Because FCS is a rare disorder, with symptoms that overlap with more common causes of hypertriglyceridaemia, diagnosis is often challenging, leading to a delay in treatment. To combat this, Moulin et al. [55] have developed a clinical scoring system for evaluating the likelihood of a patient having FCS. This scoring system is a valuable tool for guiding genetic testing and specialist referrals for confirmation and management and is summarised in Table 2 [55].
| FCS score | Interpretation | Practical steps |
| FCS very likely | Prioritize genetic testing and seek specialist input | |
| FCS unlikely | Consider MCS and other secondary causes of HTG before FCS | |
| FCS very unlikely | Manage as a non-FCS HTG |
FCS, familial chylomicronaemia syndrome; MCS, multifactorial chylomicronaemia syndrome; HTG, hypertriglyceridaemia. Examples of secondary causes include alcohol, diabetes, hypothyroidism, metabolic syndrome, and glucocorticoid therapy.
Hypertriglyceridaemia is typically managed with lifestyle changes. These changes include abstinence from alcohol, and an increase in exercise. It is also recommended that HTG patients follow a low-fat diet, avoid saturated fats, and reduce their intake of rapidly metabolised carbohydrates. Diabetic patients should maintain good blood sugar control, while overweight patients will need to reach a healthy weight [56, 57].
In patients with severe HTG (
In the case of suspected or proven FCS, these should be referred to a clinician with specific expertise, as more specific interventions are likely to be required. FCS is a particularly severe condition with high morbidity and mortality due to acute pancreatitis [58]. Targeted therapy against FCS is available with volanesorsen, an ApoC3 inhibitor. This drug is licensed for patients with genetically confirmed FCS, at high risk of pancreatitis despite dietary and other triglyceride-lowering interventions [59]. While volanesorsen has demonstrated efficacy, regular monitoring is required for potential thrombocytopenia, a known side effect.
Epidemiological studies suggest that even mild elevations in TG levels are
associated with an increased risk of adverse cardiovascular outcomes [60, 61].
However, unlike LDL-C, proving a direct causal link to ASCVD is challenging due
to confounding factors such as the inverse TG to HDL-C relationship and
variations in other lipid particles. Hypertriglyceridaemia (TG levels of 2.2–5.6
mmol/L) independently raises the risk of non-fatal myocardial infarction (MI) and coronary
revascularisation in statin-treated patients with well-controlled LDL-C (1.0–2.6
mmol/L) [62]. Similarly, in a primary prevention cohort of patients with diabetes
mellitus and adequately treated hypercholesterolaemia (LDL-C
Despite these associations, the clinical benefit of lowering TG concentrations remains uncertain. Fibrates, despite improving lipid profiles, have shown inconsistent outcomes. The Triglyceride Lowering with Pemafibrate to Reduce Cardiovascular Risk (PROMINENT) trial found no significant ASCVD risk reduction with pemafibrate in statin-treated patients with elevated TGs and noted an increased risk of pulmonary embolism and venous thromboembolism [65]. In contrast, the Reduction of Cardiovascular Events With Icosapent Ethyl-Intervention Trial (REDUCE-IT) trial demonstrated substantial cardiovascular benefits with icosapent ethyl, a form of omega-3 fatty acid. While some benefits may be linked to TG reduction, other mechanisms likely contribute to the observed outcomes [66]. This medication has been approved by NICE for the reduction of cardiovascular risk in those with raised triglycerides for secondary prevention of cardiovascular disease.
In summary, while lowering triglycerides to prevent pancreatitis is clearly justified, the role of triglyceride reduction in ASCVD risk management remains less well-defined. Emerging evidence suggests that TG modulation could help address residual cardiovascular risk, but the effectiveness likely depends on the mechanism of reduction. Further research is essential to clarify these mechanisms and optimize treatment strategies.
ApoB-containing lipoproteins, particularly LDL and Lp(a), drive disease progression by penetrating the arterial intima, triggering oxidative modification, inflammation, and plaque instability, increasing the risk of acute events [67]. Meta-analyses from the Cholesterol Treatment Trialists (CTT) Collaboration show that lowering LDL-C by 1 mmol/L with statins reduces major cardiovascular events by 21% over five years [68, 69]. This benefit extends across various therapies, including ezetimibe and PCSK9 inhibitors, with cardiovascular risk reduction directly linked to the degree of LDL-C lowering [70, 71, 72, 73, 74].
Evidence from landmark studies confirms that reducing LDL-C significantly lowers the risk of cardiovascular events in those without prior events (primary prevention) and those with established ASCVD (secondary prevention) [75, 76, 77, 78, 79]. For primary prevention, lipid-lowering therapy is guided by comprehensive risk assessment and shared decision-making, considering lifetime cardiovascular disease (CVD) risk, comorbidities, and overall frailty. Risk stratification should be reassessed periodically or when clinical status changes, though the optimal reassessment interval remains undefined [80].
In the United Kingdom, risk stratification tools such as the Joint British Societies’ 3rd (JBS3) Iteration risk calculator and QRISK3/QRISK lifetime are commonly used, while the Systematic Coronary Risk Estimation (SCORE) tool is employed in Europe. These calculators provide slightly different perspectives: QRISK and JBS3 estimate both the 10-year risk of a CVD event and the lifetime risk, whereas SCORE focuses on the 10-year risk of fatal CVD and is tailored to specific countries. Despite their utility, all these tools have limitations and are known to underestimate risk in certain populations, such as those with high Lp(a) levels, FH, chronic inflammatory conditions that accelerate ASCVD and young individuals with significant elevations in individual CVD risk factors.
Guidance on risk assessment and treatment thresholds varies across different recommendations. NICE guidance focuses on stratifying patients into primary or secondary prevention, whereas JBS recommends the same treatment target for all patients on lipid-lowering therapy. In contrast, the latest European Society of Cardiology (ESC) guidelines have removed this arbitrary classification and instead defined multiple risk thresholds defined as a combination of SCORE and patient-specific factors (such as diabetes, chronic kidney disease (CKD) and the presence of ASCVD). Table 3 (Ref. [3, 7, 80]) summarises and contrasts the three guidelines.
| Guideline | Risk threshold | Definition | Treatment target |
| NICE 2023 guideline [7] | Primary prevention | QRISK |
|
| Secondary prevention | Established ASCVD | LDL-C | |
| JBS3 2014 [80] | All patients in whom lipid lowering is indicated | JBS3 risk calculator |
Non-HDL-C |
| ESC 2025 update [3] | Low risk | SCORE2 |
LDL-C |
| Moderate risk | SCORE2 |
LDL-C | |
| High risk | SCORE2 |
LDL-C | |
| Very high risk | Established ASCVD; SCORE2 |
LDL-C | |
| Extreme risk | Recurrent ASCVD events while taking maximal statin-based therapy; Polyvascular arterial disease | LDL-C |
ASCVD, atherosclerotic cardiovascular disease; CVD, cardiovascular disease; CKD, chronic kidney disease; ESC, European Society of Cardiology; NICE, National Institute for Health and Care Excellence; JBS3, Joint British Societies’ 3rd; SCORE2, Systematic Coronary Risk Estimation 2.
Computed tomography coronary angiography (CTCA) or carotid ultrasound are not
routinely employed for risk assessment in the general population. However, in
patients with very high-risk dyslipidaemias such as FH, particularly when
combined with other risk factors and/or when lipid-lowering treatment is delayed,
CTCA may be considered to assess coronary atheroma burden. As an example,
it is the authors’ view that if a patient has confirmed FH and is untreated
The Pravastatin or Atorvastatin Evaluation and Infection Therapy (PROVE-IT) trial demonstrated that high-intensity statin therapy provides superior cardiovascular protection compared to moderate-intensity treatment [81]. This was reinforced by the Intensive Lipid Lowering with Atorvastatin in Patients with Stable Coronary Disease (TNT) trial, which showed greater benefit with 80 mg versus 10 mg of atorvastatin [82]. Subsequent studies evaluating non-statin therapies, including ezetimibe and PCSK9 inhibitors, confirmed additional reductions in cardiovascular events and the safety of achieving very low LDL-C levels [70, 73, 74]. These findings have shaped European guidelines, with the adoption of lower LDL-C thresholds. Treatment targets recommended by NICE remain influenced by the balance between clinical benefit and the cost-effectiveness of newer therapies.
Statins have been the cornerstone of lipid-lowering therapy for over 30 years, but high-intensity statins, which can reduce LDL-C by up to 50%, may be insufficient for some patients to reach target levels [83]. Additionally, adherence to high-intensity statin therapy is frequently limited by side effects, as documented in the literature [84]. These challenges have driven the development of recent therapeutic agents, including inclisiran, bempedoic acid, PCSK9 monoclonal antibodies (mAbs) and icosapent ethyl. A suggested practical approach to utilising these new therapies is summarised in Fig. 3, which is adapted from the local guidelines in Bristol.
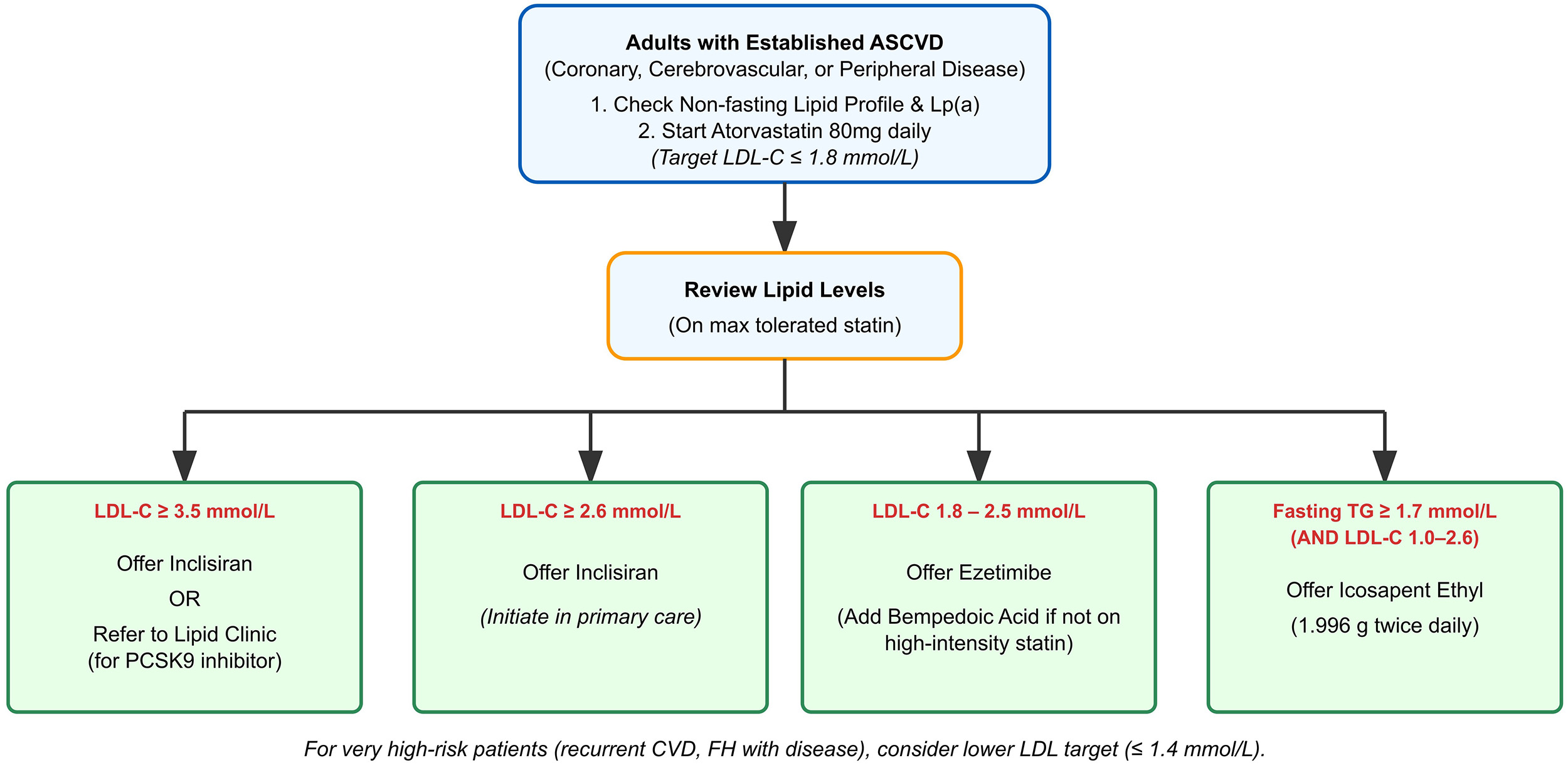 Fig. 3.
Fig. 3.
A practical approach to achieve a target LDL-C of
Inclisiran is administered via subcutaneous injection, starting with an initial
dose followed by a second dose at three months, and subsequent doses every six
months. This dosing regime is a notable improvement on previous monoclonal
antibody-based PCSK9 inhibitors. NICE recommends inclisiran for patients with CVD
and LDL-C
The slightly older PCSK9 mAbs (alirocumab and evolocumab) are administered by 2-weekly subcutaneous injection by a pre-filled self-administered injection pen. They are highly effective and achieve an approximate 60% reduction in LDL cholesterol, with proven efficacy for reducing cardiovascular end points, but in the UK are only prescribable in secondary care [73, 74].
Bempedoic acid, an adenosine triphosphate citrate lyase inhibitor, is an oral, once-daily lipid-lowering medication. It is administered as a prodrug and is activated in the liver to its active form, reducing LDL-C by up to 18% (38% with ezetimibe) while minimising muscle-related side effects [86]. It is particularly useful for statin-intolerant patients but may increase uric acid levels and gout risk. The Cholesterol Lowering via Bempedoic Acid, an ACL-Inhibiting Regimen (CLEAR) outcomes trial confirmed its cardiovascular benefits, and it is approved for patients with elevated LDL-C despite ezetimibe therapy [87].
Icosapent ethyl, a high-dose purified form of eicosapentaenoic acid, has been shown to reduce ASCVD risk in patients with established CVD and elevated triglycerides. Although it lowers triglyceride levels, the observed reduction in ASCVD risk in the REDUCE-IT trial was not directly correlated with the degree of triglyceride reduction. The precise mechanism by which icosapent ethyl reduces cardiovascular risk remains incompletely understood. Its benefits are believed to be multifactorial, likely involving antithrombotic effects, plaque stabilisation, and anti-inflammatory properties.
New developments in lipid-lowering therapies may enable further tailoring of the treatment to the specific circumstances of the patient, reducing their risk of ASCVD. For example, therapies which target alternative risk factors may help patients who still retain a high risk of ASCVD following LDL-lowering treatment. Gene therapies aim to achieve lifelong lipid-lowering from a single infusion. This section will explore some of these newer therapies and the potential impact that they could have on reducing ASCVD risk.
Lp(a) is an important, inherited risk factor for ASCVD. Currently, there are no specific treatments to lower Lp(a), so management has focused on reducing other cardiovascular risk factors. However, new therapies directly targeting Lp(a) are showing promise and are now being tested in large clinical trials.
Pelacarsen is a drug which blocks the production of apo(a) by binding to the
messenger RNA (mRNA) of the LPA gene and inhibiting the translation of apo(a)
[88]. In early studies, it reduced Lp(a) levels by 66–92% [89]. It is now being
tested in a major outcomes trial to see if it reduces the risk of heart attacks
and strokes in people with ASCVD and high Lp(a) levels (
Olpasiran and lepodisiran are another class of drugs that silence Lp(a) production in hepatocytes by disrupting LPA expression [90, 91]. In a recent trial, olpasiran lowered Lp(a) by nearly 100% at high doses in a placebo-controlled study, with minimal side effects apart from mild injection-site reactions [92]. Both drugs are now in large phase 3 trials to determine whether they prevent cardiovascular events in people with ASCVD and those at high risk for their first event [93, 94].
These therapies are most likely to benefit patients with very high Lp(a) levels
(
Cholesteryl ester transfer protein (CETP) plays a key role in lipid metabolism by transferring cholesteryl esters from HDL to more atherogenic lipoproteins like VLDL in exchange for triglycerides [96]. CETP deficiency leads to higher HDL cholesterol and lower LDL cholesterol, which is generally considered beneficial for cardiovascular health [97]. However, studies on the relationship between CETP deficiency and ASCVD have produced mixed results due to various confounding factors [98]. Despite this, CETP inhibition has emerged as a potential strategy for reducing ASCVD risk [98].
Several CETP inhibitors have been tested since the early 2000s, but many have had to be discontinued due to safety concerns or ineffectiveness [99, 100, 101]. Obicetrapib has demonstrated the most promising results to date. It achieves near-complete CETP inhibition, has been well-tolerated, and has not shown significant off-target effects [102]. In patients on high-intensity statins, obicetrapib (5 or 10 mg) reduced LDL cholesterol and apolipoprotein B levels by up to 51% and 30%, respectively, compared to placebo over eight weeks [103]. It also significantly lowered lipoprotein (a) levels. Ongoing phase III trials in patients with ASCVD will provide further insights into its clinical benefits [104]. If successful, obicetrapib could offer a new treatment option for patients with high residual cardiovascular risk despite optimal statin therapy.
Gene disruption at the DNA level offers a promising approach to achieving lasting changes in protein expression [105]. VERVE-101, developed by Verve Therapeutics Inc., is an investigational therapy using Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) base-editing technology to inactivate the PCSK9 gene in liver cells. This is done by modifying a single DNA base to introduce a premature stop codon, effectively silencing the gene [106, 107]. Early phase 1b trial results showed that a 0.45 mg/kg dose of VERVE-101 reduced LDL cholesterol by up to 73%.
Building on this, Verve has developed VERVE-102, which uses the same base-editing system as VERVE-101 but features a new delivery mechanism. This updated approach has demonstrated good tolerability in other clinical trials, suggesting potential improvements in safety and effectiveness [108]. Although still in early development, gene therapy is gaining recognition. The UK Medicines and Healthcare products Regulatory Agency (MHRA) recently approved a CRISPR-based gene therapy for sickle cell anaemia and beta thalassaemia, highlighting the potential of such treatments for targeted interventions in high-risk patients [109].
Inflammation is increasingly recognised as a contributor to atherosclerosis and an independent cardiovascular risk factor [110, 111, 112, 113]. Advances in clinical assessment include high-sensitivity c-reactive protein measurements and imaging techniques like perivascular fat analysis to evaluate coronary inflammation [114]. Several anti-inflammatory therapies have been investigated for cardiovascular risk reduction, with mixed results. Colchicine and canakinumab have shown potential benefits, whereas methotrexate has not demonstrated efficacy [115, 116, 117]. Among these, colchicine stands out due to its low cost and positive early trial outcomes. It is the most extensively studied agent in this field and has already received Food and Drug Administration (FDA) approval for secondary prevention of coronary artery disease.
Given its potential for widespread clinical application, we focus here on colchicine’s trial outcomes and the future directions for its use. Several trials support colchicine’s role in cardiovascular prevention. A study of 532 patients with chronic coronary disease showed that 0.5 mg daily reduced cardiovascular events over three years compared to placebo [115]. A larger trial with 5522 patients confirmed these benefits [118]. In the post-myocardial infarction (MI) setting, the Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction (COLCOT) trial (4745 patients) demonstrated event reduction over nearly two years [119]. These findings suggest colchicine could be beneficial in both chronic and post-MI settings.
However, some studies have raised concerns. The Colchicine in Patients With Acute Coronary Syndrome (COPS) trial (795 post-acute coronary syndrome patients) found no significant cardiovascular benefit and reported a higher mortality rate in the colchicine group, raising safety concerns [120]. Additionally, a recent large-scale trial following patients with acute MI over five years did not show significant reductions in major cardiovascular events [121]. These mixed results highlight the complexity of targeting inflammation in cardiovascular disease. While colchicine shows promise, current evidence is insufficient for broad clinical adoption. Ongoing research is needed to determine its role and identify patients who may benefit most from its use.
This article provides an overview of current strategies for managing dyslipidaemia and reducing ASCVD risk, highlighting significant advancements in the range of available therapeutic options. These innovations enable greater personalisation of care and the achievement of much lower LDL-C targets than were feasible in previous decades. Promising therapies in development, particularly those targeting residual ASCVD risk factors like elevated Lp(a) levels, hold the potential to further enhance clinical outcomes.
• The older Friedewald equation for estimating LDL-C should be replaced with modern equations (Sampson and Martin-Hopkins) to allow for more accurate calculation of LDL-C, especially in patients with high triglyceride levels.
• Lp(a) is a genetically determined risk factor for ASCVD, and as such, should be measured once in a patient’s lifetime to refine cardiovascular disease risk assessment.
• Risk stratification to determine treatment goals for managing cardiovascular disease risk requires an individualized approach and the ESC guidelines offer the most nuanced targets for managing dyslipidaemia to reduce ASCVD risk.
• Genomic testing should be integrated into routine lipid care in order to identify inherited disorders such as familial hypercholesterolemia, enabling family cascade screening and early intervention.
• While statins remain the first-line lipid-lowering therapy, a significant proportion of patients require combination therapy to reach lower modern LDL-C targets, with a multitude of highly effective additional treatment options now available.
• The primary goal of treating severe hypertriglyceridaemia
(
Not applicable.
KA, AL, PD, AG, MM and WM designed the work. KA, AL, PD, AG, MM and WM contributed equally to the writing of the first draft of the manuscript. All authors contributed to the important editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
Figs. 1 and 2 were created using Inkscape (https://inkscape.org/). The authors declare no financial or personal relationship with Inkscape, and the use of this software does not imply any endorsement. The authors declare no conflicts of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.