


 , Chaofen Li 2,*
, Chaofen Li 2,*
1 Yunxian Township Health Center, 665003 Pu’er, Yunnan, China
2 Department of Pediatrics, Pu’er People’s Hospital, 665000 Pu’er, Yunnan, China
3 Department of Anesthesiology, Pu’er People’s Hospital, 665000 Pu’er, Yunnan, China
Abstract
Primary ciliary dyskinesia (PCD) is a rare autosomal recessive disorder characterised by structural or functional abnormalities of the cilia, which prevent proper oscillation. Patients typically present with symptoms such as chronic respiratory infections, bronchiectasis, and, in approximately half of the cases, inversion of internal organs. Due to its diverse clinical manifestations and lack of specificity, PCD is often misdiagnosed or underdiagnosed, and diagnosis in the neonatal period remains notably challenging. Therefore, this study aimed to analyse the clinical characteristics of the two cases, optimise PCD treatment, and improve the accuracy and timeliness of PCD diagnosis.
This article reports two cases of twin sisters at 34+3 weeks of gestation who were diagnosed with PCD during the neonatal period. The patients, twin girls conceived naturally, were transferred to Department of Pediatrics after cesarean delivery at Pu’er People’s Hospital on 26 September 2024, at 11:24, due to cyanosis and shortness of breath after birth. Prenatal examination at 13 weeks revealed “dextrocardia with abdominal situs inversus” in both fetuses. Delivery was by emergency cesarean section due to premature rupture of membranes. After birth, the twins presented with respiratory distress symptoms, including cyanosis and shortness of breath. Bedside chest radiography revealed hyaline membrane disease, dextrocardia, and abdominal situs inversus. Treatments such as non-invasive ventilator-assisted ventilation and anti-infective therapy were initiated after admission. However, the patients repeatedly experienced breathing difficulties, alternating atelectasis in both lungs, difficulty weaning from the ventilator, and episodes of desaturation.
Peripheral blood genetic testing confirmed a Dynein Axonemal Heavy Chain 5 (DNAH5) gene mutation as the cause of PCD. Following treatment with non-invasive ventilator-assisted ventilation, oxygen therapy in an incubator, nebulization (e.g., hypertonic saline), patting back and sputum aspiration, prone position ventilation, and postural drainage, both patients improved and were discharged.
In neonates with unexplained respiratory distress, especially those with situs inversus, early refinement of diagnostic tools, including genetic testing, is crucial to confirm PCD promptly, enable timely intervention, and prevent irreversible lung injury.
Keywords
- primary ciliary dyskinesia
- gene mutation
- neonate
- case report
Primary ciliary dyskinesia (PCD) is a rare, autosomal recessive inherited disorder or an X-linked biallelic mutation-related disorder characterised by ciliary motility dysfunction. It results from gene mutations that disrupt the structure and/or function of cilia, leading to impaired motility of the ciliated epithelium in various organs containing ciliary structures [1]. The estimated global prevalence of PCD ranges from 1 in 15,000 to 1 in 30,000 live births [2]. However, PCD has been infrequently reported in China. Guan et al. [3] analysed the genetic profile of Chinese children with PCD and identified Dynein Axonemal Heavy Chain 5 (DNAH5) as a common pathogenic gene. Approximately 40%–55% of patients exhibit lateral organ changes due to left-right asymmetric signalling deficits caused by abnormal ciliary movement during early embryonic development, resulting in visceral inversion, also known as Kartagener syndrome [4, 5]. About 5% of patients with PCD present with both visceral inversion and congenital heart disease. A smaller subset of Kartagener syndrome patients may also present with hydrocephalus, cleft palate, or anal atresia [4, 6].
The clinical manifestations of PCD are diverse and nonspecific. During the fetal period, it often presents as visceral inversion and ventricular dilation. In the neonatal period, it typically presents as unexplained respiratory distress syndrome, hypoxemia, and atelectasis. During childhood and adulthood, PCD commonly manifests as chronic sinusitis, chronic otitis media, bronchiectasis, chronic productive cough, and infertility [7, 8]. The diversity of clinical manifestations of this disease contributes to frequent misdiagnosis and underdiagnosis. Currently, there is no curative treatment for PCD. Management primarily focuses on preventing infections, strengthening airway clearance, and reducing lung injury.
At present, there is no universally accepted diagnostic criterion for PCD. Most clinicians refer to the diagnostic guidelines recommended by the American Thoracic Society (ATS) in 2018 [9]. Diagnostic methods include nasal exhaled nitric oxide measurements, high-speed video microscopy for ciliary morphology and function assessment, and genetic testing. However, each of these methods carries a risk of false negatives, and none can serve as a stand-alone diagnostic tool. Gene therapy holds promise as a potential future treatment for the condition. At present, increasing awareness of PCD and promoting early diagnosis and intervention are essential to improving the quality of life of affected children.
Here, we report a case of twin infants with PCD and visceral inversion, documenting their clinical features, diagnostic process, and treatment approaches, aiming to provide a reference for improved management of PCD.
Case 1: The patient was a female infant, the elder of the twin siblings, aged 13 minutes at presentation. Chief complaint: premature delivery at 34+3 weeks with cyanosis and shortness of breath lasting 13 minutes after birth. Present medical history: The infant was delivered via cesarean section at Pu’er People’s Hospital due to maternal “premature rupture of fetal membranes and twin pregnancy”. Her birth weight was 1.96 kg, with clear amniotic fluid. Apgar scores [10] were 8 at 1 minute, 9 at 5 minutes, and 9 at 10 minutes. After birth, she presented with respiratory distress symptoms, including cyanosis and shortness of breath, and was admitted to Department of Pediatrics, Pu’er People’s Hospital on 26 September 2024, at 11:24. The parents of the infant and the elder brother were healthy, with no history of consanguinity or similar diseases in the family.
Physical examination at admission: Temperature (T): 35.3 °C (reference 36.5–37.7 °C), pulse (P): 145 times/min (reference 130–150 times/min), respiration (R): 56 times/min (reference 30–40 times/min), blood pressure (BP): 57/25 mmHg (reference: systolic 50–60 mmHg; diastolic 30–40 mmHg), peripheral capillary oxygen saturation (SPO2): 80% (reference 95%–100%), weight 1960 g, body length 44 cm.
Perioral and limb cyanosis was observed, with positive three-concave signs. Breath sounds in both lungs were coarse, without audible rales. Cardiac examination revealed dextrocardia with regular heart rhythm and strong heart sounds; the right cardiac border extended to the 4th intercostal space on the right, approximately 0.3 cm lateral to the right midclavicular line.
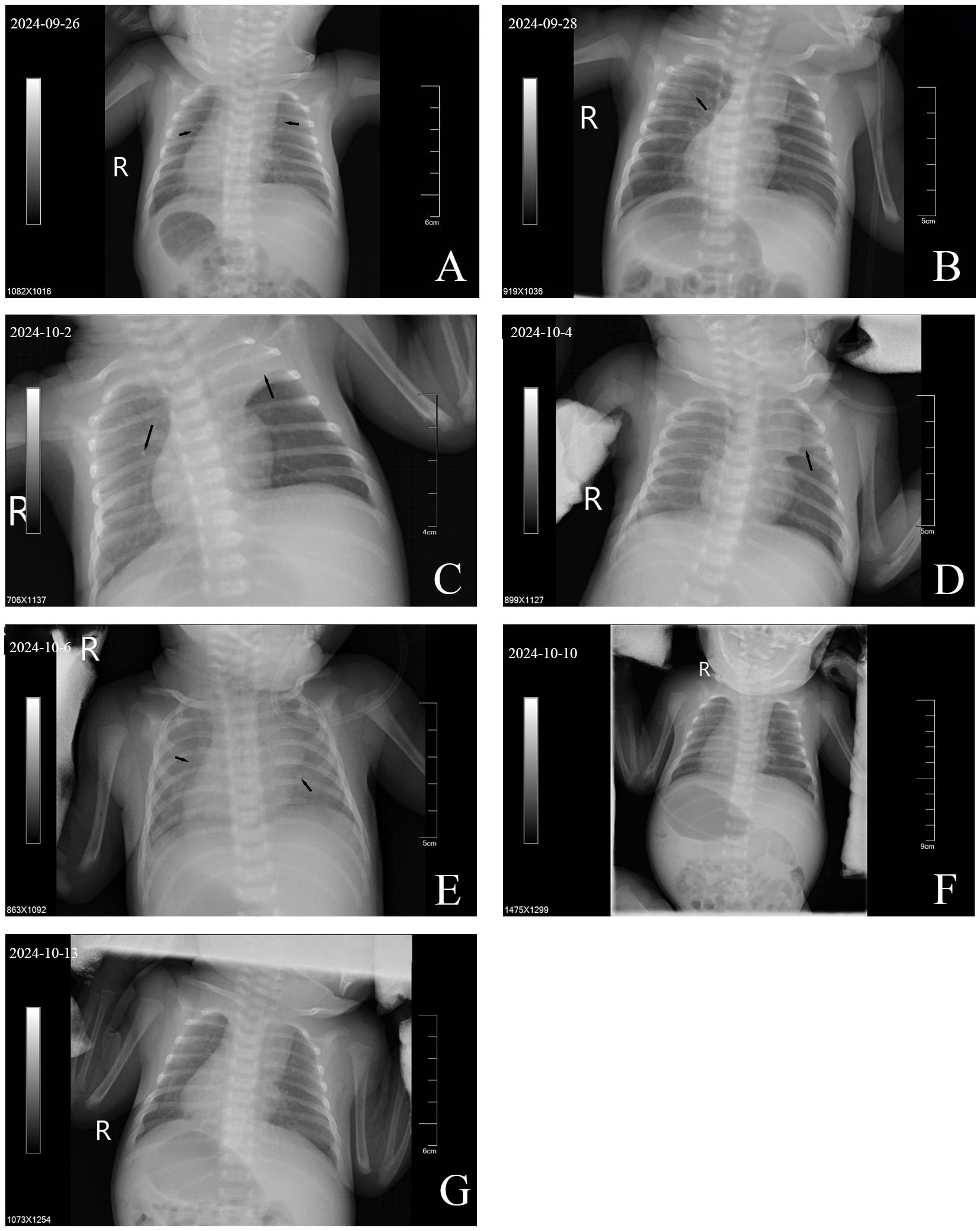
Imaging: The bedside chest radiograph is shown in Fig. 1A–G. In the early stage of admission, the patient exhibited decreased lung transparency and diffuse ground-glass opacities in both lungs. Although partial improvement was noted later, consolidation developed in the right lung and left upper lung. Following the subsequent diagnosis of PCD, targeted treatment was initiated, leading to reduced consolidation in the left upper lung and increased density with decreased transparency in both lungs.
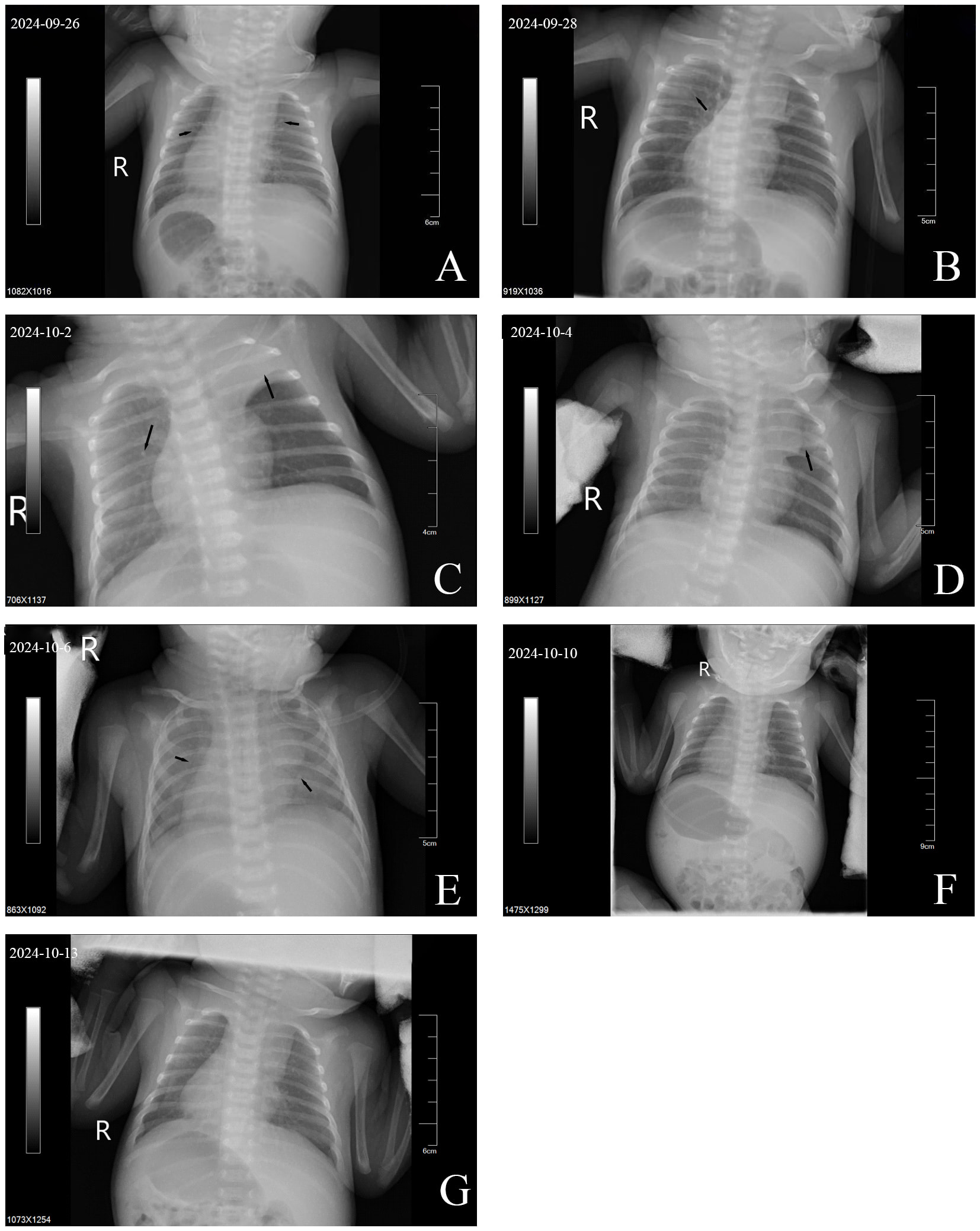 Fig. 1.
Fig. 1.
Bedside chest X-rays of case 1. (A) Compared with normal lungs, the transparency of both lung fields was generally decreased, with ground-glass opacities and scattered minute nodules. (B) Increased lung transparency compared with (A), though ground-glass opacities persisted in the upper middle field of the right lung. (C) Reduced right lung transparency compared with (B), with new consolidation in the left upper lung. (D) Continued decreased right lung transparency, with partial resolution of consolidation in the left upper lung compared with (C). (E) Diffuse ground-glass opacities in both lungs with a blurred cardiac shadow. (F) Further enhancement and blurring of lung markings, with slightly reduced bilateral lung transparency compared with (E). (G) Further increased lung markings, blurred textures of both lungs and decreased transparency of both lungs compared with (F). Notes: Arrows indicate key findings. R, right.
Cardiac ultrasound: The heart was located in the right thoracic cavity, with the apex pointing toward the right anterior side, consistent with dextrocardia. A patent foramen ovale measuring 1.6 mm was also observed.
Abdominal ultrasound: Findings included situs inversus, a moderate echogenic area in the splenic hilum suggestive of an accessory spleen, and dilation of both renal pelvises.
Cranial ultrasound: The examination revealed mildly widened bodies of the bilateral lateral ventricles (left: 0.40 cm; right: 0.42 cm), an unclosed cavum septum pellucidum, and increased resistance indices in the bilateral middle cerebral arteries.
Laboratory findings: No significant abnormalities were detected in blood gas analysis, complete blood count, liver and renal function tests, electrolyte levels, myocardial enzyme assays, coagulation profile, blood cultures, thyroid function tests, infection markers, G-test, or hearing screening. Spectral analysis of amino acids and acylcarnitines for inborn errors of metabolism also revealed no abnormalities.
The Care Checklist has been attached as Supplementary Material associated with this article.
The results of the peripheral blood gene detection conducted by KingMed
Diagnostics on 25 October 2024, revealed two mutation sites in the DNAH5 gene: c.11761G
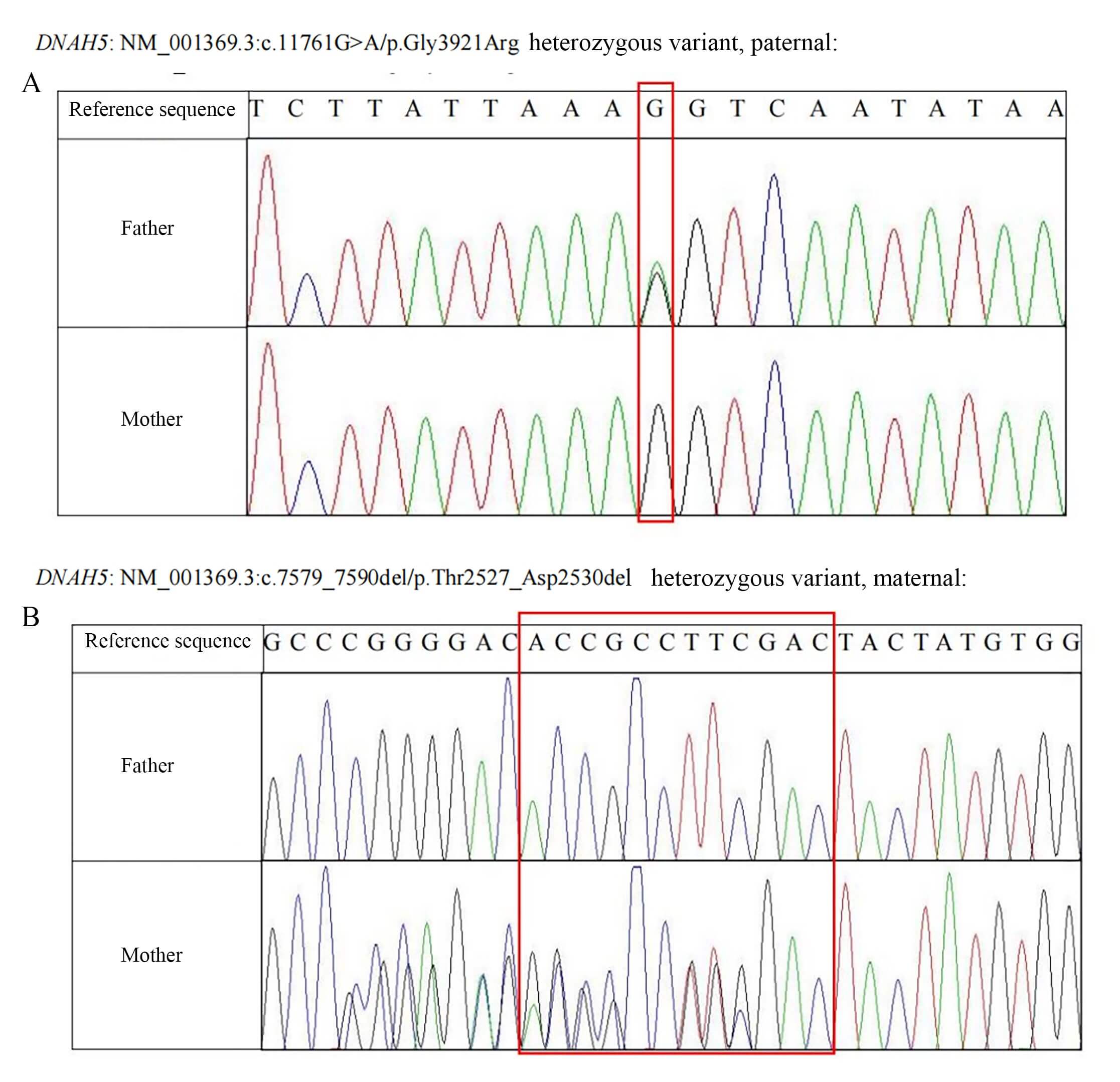 Fig. 2.
Fig. 2.
Peripheral blood genetic testing results of cases 1 and 2. (A) Sanger sequencing results of the paternal DNAH5 gene (Red box: The missense variant site of the DNAH5 gene is marked. This site in the reference sequence is base G. The sequencing peak map of the father shows an overlapping peak of G and A here (suggesting heterozygous variation), while the peak map of the mother only shows a single peak of G, indicating that this variation originated from the father). (B) Sanger sequencing results of the maternal DNAH5 gene (Red box: The deletion variant sites of the DNAH5 gene are marked. This region in the reference sequence is “ACCGCCTTCGAC”. The sequencing peak map of the father is consistent with the reference sequence, while the peak map of the mother shows overlapping peaks with sequence disorder in this region (suggesting heterozygous deletion variation), indicating that this variation originated from the mother). DNAH5, Dynein Axonemal Heavy Chain 5.
Case 2: The patient was a female infant, the younger of the twin siblings, aged 12 minutes at presentation. Chief complaint: Premature delivery at 34+3 weeks with cyanosis and shortness of breath lasting 12 minutes after birth.
Present medical history: The infant was delivered via cesarean section at Pu’er People’s Hospital due to maternal “premature rupture of fetal membranes and twin pregnancy”. Her birth weight was 2220 g, with clear amniotic fluid. Apgar scores were 8 at 1 minute, 9 at 5 minutes, and 9 at 10 minutes. Following delivery, she presented with respiratory distress symptoms, including cyanosis and shortness of breath, and was admitted to Department of Pediatrics, Pu’er People’s Hospital on 26 September 2024, at 11:24.
Physical examination at admission: T: 35.4 °C, P: 140 times/min, R: 58 times/min, BP: 59/27 mmHg, SPO2: 81%, weight 2220 g, body length 44 cm.
Perioral and extremity skin cyanosis was observed with positive three-concave signs. Breath sounds in both lungs were coarse, with audible wet rales. Cardiac examination revealed dextrocardia with regular rhythm and strong heart sounds; the right cardiac border extended to the 4th intercostal space on the right, approximately 0.3 cm lateral to the right midclavicular line. Imaging: The bedside chest radiograph (Fig. 3A–H) initially demonstrated decreased transparency in both lungs with ground-glass opacities and small nodules, progressing to consolidation shadows in the upper lobes of both lungs. After modification of the treatment approach, the consolidation shadows showed alternating absorption and recurrence, eventually leading to a mild bilateral reduction in lung transparency without obvious consolidation shadows.
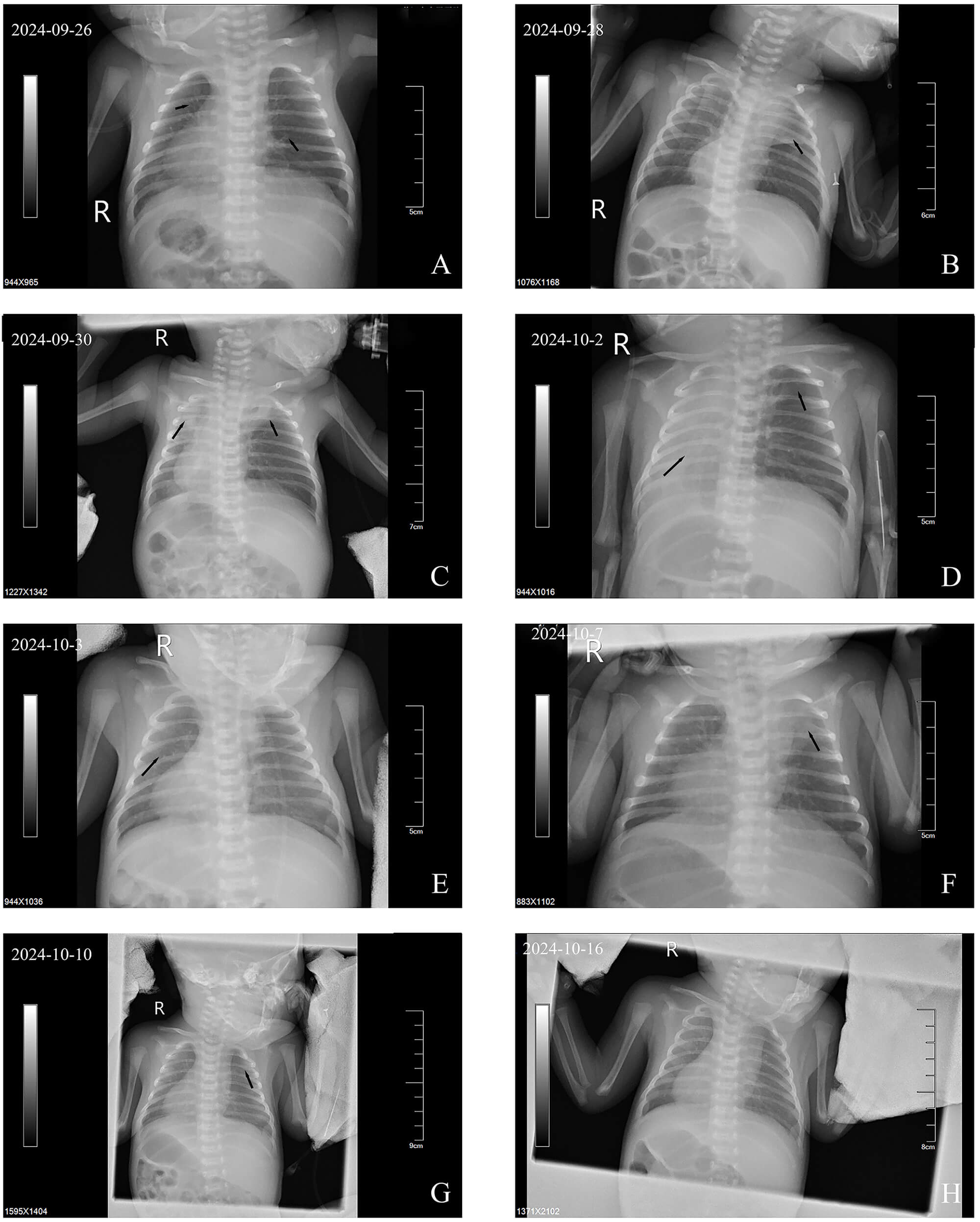 Fig. 3.
Fig. 3.
Bedside chest X-rays of Case 2. (A) Compared with normal lungs, the transparency of both lung fields was generally decreased, with ground-glass opacities and scattered minute nodules. (B) Ground-glass opacities observed in the upper left lung. (C) Markedly reduced right lung transparency compared with (B), with consolidations in the right and left upper lobes. (D) Diffuse consolidations in the right lung field with complete resolution of the left upper lobe consolidation. (E) Partial resolution of right lung consolidation with persistently reduced bilateral lung transparency. (F) New consolidation shadows in the left upper lung with slightly reduced bilateral lung transparency compared with (E). (G) Absorption of the left upper lobe consolidation, with persistently reduced bilateral lung transparency compared with (F). (H) Compared with (G), bilateral transparency was slightly reduced, with no new consolidations observed. Notes: Arrows indicate key findings. R, right.
Cardiac ultrasound: The heart was located in the right thoracic cavity, with the apex pointing toward the right anteriorly, consistent with dextrocardia. A patent foramen ovale measuring 2.0 mm was also observed.
Abdominal ultrasound: Findings included situs inversus and dilation of both renal pelvises.
Cranial ultrasound: Mild dilation of the bodies of lateral ventricles (left: 0.42 cm; right: 0.41 cm), an unclosed cavum septum pellucidum, and increased resistance indices in the bilateral middle cerebral arteries were observed.
Laboratory findings: No significant abnormalities were found in blood gas analysis, complete blood count, liver and renal function tests, electrolyte assays, myocardial enzymes, coagulation profile, blood cultures, thyroid function tests, infection markers, G-test, or hearing screening. Spectral analysis of amino acids and acylcarnitines for inborn errors of metabolism also revealed no abnormalities.
Consistent with case 1, peripheral blood genetic testing by KingMed Diagnostics
on 25 October 2024, identified two mutation sites in the DNAH5 gene:
c.11761G
Non-invasive ventilator support (non-invasive continuous positive airway pressure mode: Fraction of inspired oxygen (FiO2) 25%–35%, positive end-expiratory pressure (PEEP) 6 cmH2O, combined with ceftazidime and ampicillin for infection prevention was administered from 26 September 2024 to 1 October 2024. Supportive treatments included Vitamin K1 to prevent hemorrhagic disease of the newborn, intravenous nutrition, and formula milk feeding. Despite these interventions, the patients continued to experience paroxysmal breathing difficulties, blood oxygen desaturation, and difficulty weaning from ventilator support. Dynamic chest radiographs indicated alternating atelectasis in both lungs, particularly pronounced in the younger twin. Airway secretions in both infants were abnormally abundant and viscous. Based on the literature review, ciliary motility disorder was considered.
Therapeutic adjustments included oral azithromycin suspension for infection prevention starting 1 October 2024, intravenous ambroxol, nebulization with hypertonic saline and acetylcysteine for sputum dilution, patting back and sputum aspiration, postural drainage (lying on the healthy side), and prone position ventilation. These measures led to reduced airway secretions, gradual improvement in paroxysmal breathing difficulties, and decreased oxygen requirements. Both infants were successfully weaned off non-invasive ventilation on 4 October 2024, transitioned to oxygen therapy in an incubator, and were fully weaned off supplemental oxygen on 13 October and 17 October 2024, respectively.
Both patients improved and were discharged at 18 days (14 October 2024) and 21 days (17 October 2024) after birth, respectively. They were successfully weaned from oxygen. Repeat blood tests, including complete blood count and biochemistry, were normal at discharge, and follow-up chest radiographs showed resolution of atelectasis. Families were advised to avoid respiratory tract infections, use a pulse oximeter, oxygen concentrator, as well as a suction device at home, and perform sputum clearance as needed to prevent airway obstruction.
Currently, more than 40 gene mutations have been confirmed to be associated with the pathogenesis of PCD, among which DNAH5 is the most significant. In this case, prenatal examination of the mother at 13 weeks of pregnancy revealed that both twins had dextrocardia and abdominal situs inversus. No cardiovascular or other severe malformations were detected in subsequent routine prenatal examinations. After birth, the infants presented with respiratory distress symptoms, including cyanosis and shortness of breath. Based on gestational age and clinical presentation, hyaline membrane disease was initially considered. However, routine treatment, including non-invasive ventilator support and infection prevention, had a limited effect, while the patients continued to experience recurrent breathing difficulties, atelectasis, excessive airway secretions, and difficulties in weaning from ventilation. Furthermore, infection indicators and pathogen testing did not reveal evidence of infection.
Treatment was adjusted to include comprehensive airway clearance strategies: Nebulised hypertonic saline and acetylcysteine for sputum dilution, patting back for sputum aspiration, and postural drainage. Gradual reduction in airway secretions was observed, atelectasis resolved without recurrence, and both patients were successfully weaned from oxygen support. Subsequent genetic testing confirmed a diagnosis of PCD type 3 with heterotaxy caused by DNAH5 gene mutations. These findings highlight that in neonates with unexplained respiratory distress and ventilator dependence, especially those with dextrocardia and abdominal situs inversus, early genetic testing is crucial to confirm the diagnosis and guide timely intervention.
PCD typically manifests in childhood. However, due to the non-specificity of its clinical symptoms and limited awareness among clinicians, diagnosis is often delayed for many years, sometimes until adulthood. In particular, patients without organ inversion often present repeatedly with sinusitis, recurrent pneumonia, atelectasis and bronchiectasis, leading to progressive impairment of lung function [11, 12]. In China, the commonly adopted clinical diagnostic criteria are as follows: ① Presence of at least two of the following PCD clinical features: unexplained neonatal respiratory distress syndrome in full-term infants, onset of persistent cough before 6 months of age, onset of persistent nasal congestion before 6 months of age, or visceral inversion. ② Transmission electron microscopy (TEM) reveals characteristic ciliary ultrastructural defects (e.g., outer dynein arm deletion, combined outer and inner dynein arm deletion, inner dynein arm deletion with microtubule disorganisation, or central pair deletion). ③ Genetic testing showing biallelic pathogenic mutations in PCD-related genes.
PCD can be confirmed when criteria ①+② or ①+③ are met [5].
In this case, the twins were late-preterm infants. Prenatal ultrasound
identified organ inversion early in gestation, and after birth, they developed
acute respiratory distress with clinical features inconsistent with the imaging
findings. As high-speed video microscopy for ciliary function analysis was not
available at our hospital, and nasal nitric oxide testing was not feasible for
newborns, these examinations were not conducted. However, comprehensive genetic
testing identified two pathogenic DNAH5 mutations: c.11761G
At present, there remains a lack of specific treatment protocols for PCD syndrome, and management is primarily symptomatic. Numerous studies have explored therapeutic options for this disease, both domestically and internationally. Some studies suggest that deoxyribonuclease (dornase alfa), mRNA therapy, and uridine 5′-triphosphate (UTP) may be beneficial in treating PCD [13, 14, 15]. Ringshausen et al. [16] reported that nebulised inhalation of the epithelial sodium channel blocker idrevloride combined with hypertonic saline significantly improved percent predicted forced expiratory volume (PPFEV) and overall lung function compared to hypertonic saline alone. In adolescents and adults with primary ciliary dyskinesia, this combination improved mucus hydration, enhanced mucus clearance, reduced airway obstruction, and improved lung function [16]. However, these treatment methods still lack large-scale clinical evidence.
An increasing number of scholars believe that gene therapy may become an effective curative option for this disease. Gene therapy has already been successfully applied in various mammalian models of ciliopathies [17, 18]. However, PCD treatment mainly emphasises airway management, infection control, and supportive care to delay disease progression, preserve or improve lung function, and prevent chronic lung injury. In our case, once PCD was suspected, the conditions of the infants improved following the addition of hypertonic saline, acetylcysteine for sputum dilution, enhanced chest physiotherapy with percussion, and postural management. After discharge, family education focused on preventing respiratory tract infections, optimising airway clearance, and reducing lung injury risk.
Although these cases provide valuable insights into the clinical manifestations and management of PCD caused by DNAH5 gene mutations, this study has several limitations. First, PCD diagnostic methods were incomplete, as histological examinations to assess ciliary ultrastructural defects were not performed. Second, the follow-up period was relatively short, and long-term outcomes such as growth, development, and lung function changes were not evaluated. Therefore, larger-scale and higher-quality clinical studies are needed in the future to guide the diagnosis, treatment, and long-term management of PCD.
PCD is often misdiagnosed as a common respiratory disorder during the neonatal period, especially in premature infants. In clinical practice, paediatricians’ awareness of PCD should be enhanced. Early diagnosis and timely intervention are crucial to prevent irreversible lung injury and improve the quality of life in children with PCD.
• PCD should be highly suspected in neonatal patients presenting with unexplained respiratory distress and difficulty weaning from the ventilator support, especially when accompanied by dextrocardia and situs inversus.
• Detailed diagnostic approaches, such as genetic testing, facilitate early intervention and can prevent irreversible lung injury.
• Strengthening airway management and implementing anti-infection strategies can delay disease progression and improve or maintain lung function.
• Long-term management of PCD should prioritise preventing respiratory tract infections, optimising airway care and minimising lung injury.
All data included in this study are available from the corresponding authors upon reasonable request.
RM, WYW and CFL designed the research study and wrote the first draft. SYY, LZ and DJP performed the research. ZL, WYW and RM contributed to data acquisition and literature review. All authors contributed to the important editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
This study was conducted in accordance with the basic principles of the Declaration of Helsinki. Informed consent for publication of the case was obtained from the patient’s family. This study has been approved by the Ethics Review Committee of Pu’er People’s Hospital (Approval No. 2025-014-01).
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/BJHM54134.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.