


 , Nicholas J. Simmonds 1,2,*
, Nicholas J. Simmonds 1,2,*
1 Adult Cystic Fibrosis Centre, Royal Brompton and Harefield Hospitals, Guy’s and St Thomas’ NHS Foundation Trust, SW3 6NP London, UK
2 National Heart and Lung Institute, Imperial College London, SW3 6LY London, UK
Abstract
Cystic fibrosis (CF) is one of the most common life-shortening hereditary disorders, caused by a defect in the cystic fibrosis transmembrane conductance regulator (CFTR) protein. This defect causes multi-system disease, but primarily it affects the lungs and pancreas. Over 2000 CFTR gene variants have been identified; these can result in variable CFTR protein function and, consequently, a diverse clinical phenotype. CF is not only diagnosed in children but also in adults and is present in non-White populations. In adults, CF often presents with an atypical phenotype, usually due to residual CFTR protein function, which can be caused by rare CFTR variants. As a result, diagnosing CF in adults can be challenging. In this review, we discuss who should be considered for CF, how and where the diagnosis is made, and why a timely CF diagnosis is important for all patients, as well as their families.
Keywords
- cystic fibrosis
- CFTR-related disorder
- diagnosis
- sweat chloride concentration
- nasal potential difference
- intestinal current measurement
Cystic fibrosis (CF) is one of the most common autosomal recessive life-shortening diseases, affecting more than 11,000 people in the UK and over 160,000 people worldwide [1, 2]. It is caused by mutations on both alleles of the cystic fibrosis transmembrane conductance regulator (CFTR) gene, located on the long arm of chromosome 7, encoding an anion channel expressed on the apical surface of epithelial cells [3], playing a crucial role in regulating the fluidity of intraluminal secretions. CFTR dysfunction can affect many organ systems, with diverse clinical manifestations, including thick mucus production in the lungs, exo- and endocrine dysfunction of the pancreas, altered gut motility, chronic rhinosinusitis, infertility in men, and a risk of subfertility in women [3].
Historically, CF was considered a childhood disease with poor survival into adulthood due to severe suppurative lung disease. Over the decades, survival rates have significantly improved, for a variety of reasons, including high quality multi-disciplinary team (MDT) care and therapies, such as pancreatic enzymes. However, with the introduction of mutation-specific CFTR modulator therapies since 2011—and more specifically—since the widespread introduction of the triple combination Elexacaftor/Tezacaftor/Ivacaftor (ETI) therapy, for which ~90% of people with CF (pwCF) are eligible, survival rates and quality of life have dramatically improved further. Computational (‘microstimulation’) projective models suggest that with the early initiation of these treatments, pwCF may have a life expectancy approaching normal [4].
Before the widespread introduction of newborn screening (NBS) programs (e.g.,
2007 in the UK), CF was usually diagnosed in infants/children presenting with
serious conditions, such as meconium ileus, recurrent chest infections or failure
to thrive. However, with NBS, most diagnoses are now made soon after birth, often
before significant symptoms develop [5]. Nevertheless, a significant proportion
of pwCF are still diagnosed in adulthood—e.g., according to the UK CF Registry
2024 Annual Report, 12% of all pwCF diagnosed between 2020 and 2024 were aged
PwCF diagnosed in adulthood often present with a variable phenotype, which can sometimes be explained by residual CFTR protein function, highlighting the concept of variable CFTR protein function across a spectrum. Despite CF being a monogenetic condition, more than 2000 variants in the CFTR gene have been identified, many of which are exceedingly rare, making it challenging to determine their pathogenicity [9]. Additionally, some individuals with CFTR variants and/or borderline functional tests (e.g., sweat chloride; see below) do not exhibit classic CF features. Consequently, Bombieri et al. [10] introduced the concept of CFTR-related disorders (CFTR-RDs) to classify individuals whose genetic and functional profiles do not meet the strict diagnostic criteria for CF, but whose conditions are still linked to CFTR dysfunction. The diagnostic criteria and management of CFTR-RD have recently been updated by a series of papers from the European CF Society (ECFS) [11, 12, 13, 14].
In light of this greater understanding of the functional and genetic aspects of CF, combined with the therapeutic advancements, we provide an up-to-date review of the diagnostic process, suggest a pragmatic diagnostic algorithm and discuss some of the complexities and uncertainties that remain. This review is not only targeted at CF specialists, but also non-CF specialists in primary and secondary care across many specialities, as patients with conditions associated with CFTR protein dysfunction may present under their care, and will need to be appropriately investigated and diagnosed.
CFTR protein is an adenosine-triphosphate (ATP)-binding ion channel which transports negatively charged ions (mostly chloride and bicarbonate) across apical epithelial cell membranes, regulating the viscosity and pH of epithelial secretions. It is expressed variably in multiple organs, including the upper and lower airway epithelium, sweat glands, pancreatic, biliary, and intestinal epithelia, as well as the female and male reproductive tracts [3, 15, 16, 17]. Additionally, CFTR regulates other ion channels, particularly the epithelial sodium channel (ENaC) which plays a crucial role in salt reabsorption [3].
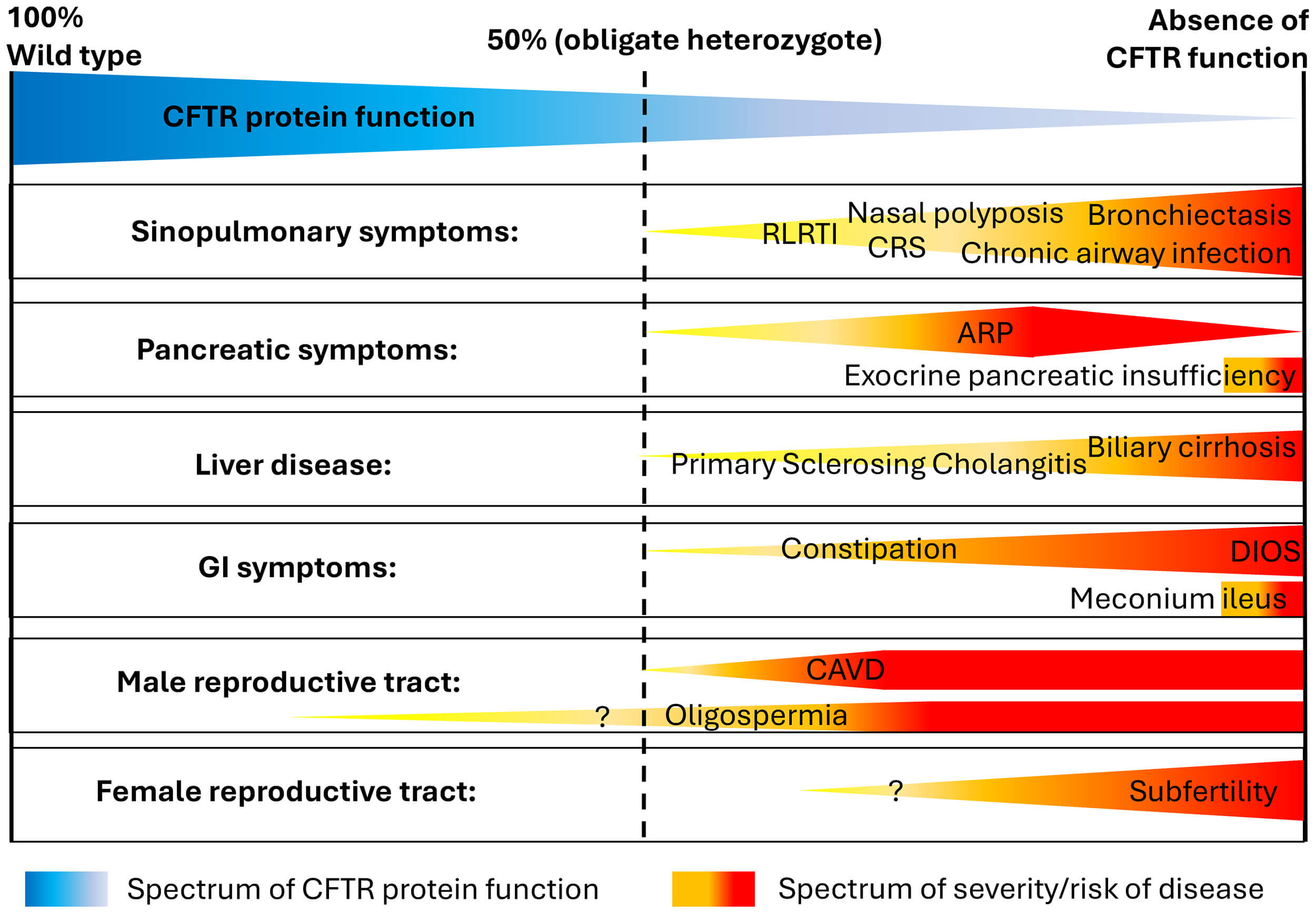
CFTR protein dysfunction can therefore result in a broad spectrum of symptoms which typically lead to multisystem disease. Clinically, some individuals develop severe multi-organ disease from birth, whereas others present in adulthood with mild or even mono-organ conditions. This variability supports the hypothesis that CFTR protein function exists in a continuum, where each individual’s combination of CFTR gene variants determines their CFTR function, ranging from “wild type” (100% function) to complete absence of function (Fig. 1, Ref. [18]). Evidence supporting this hypothesis stems mostly from data based on in vitro studies of different CFTR gene variants, investigating transepithelial chloride transport [19, 20].
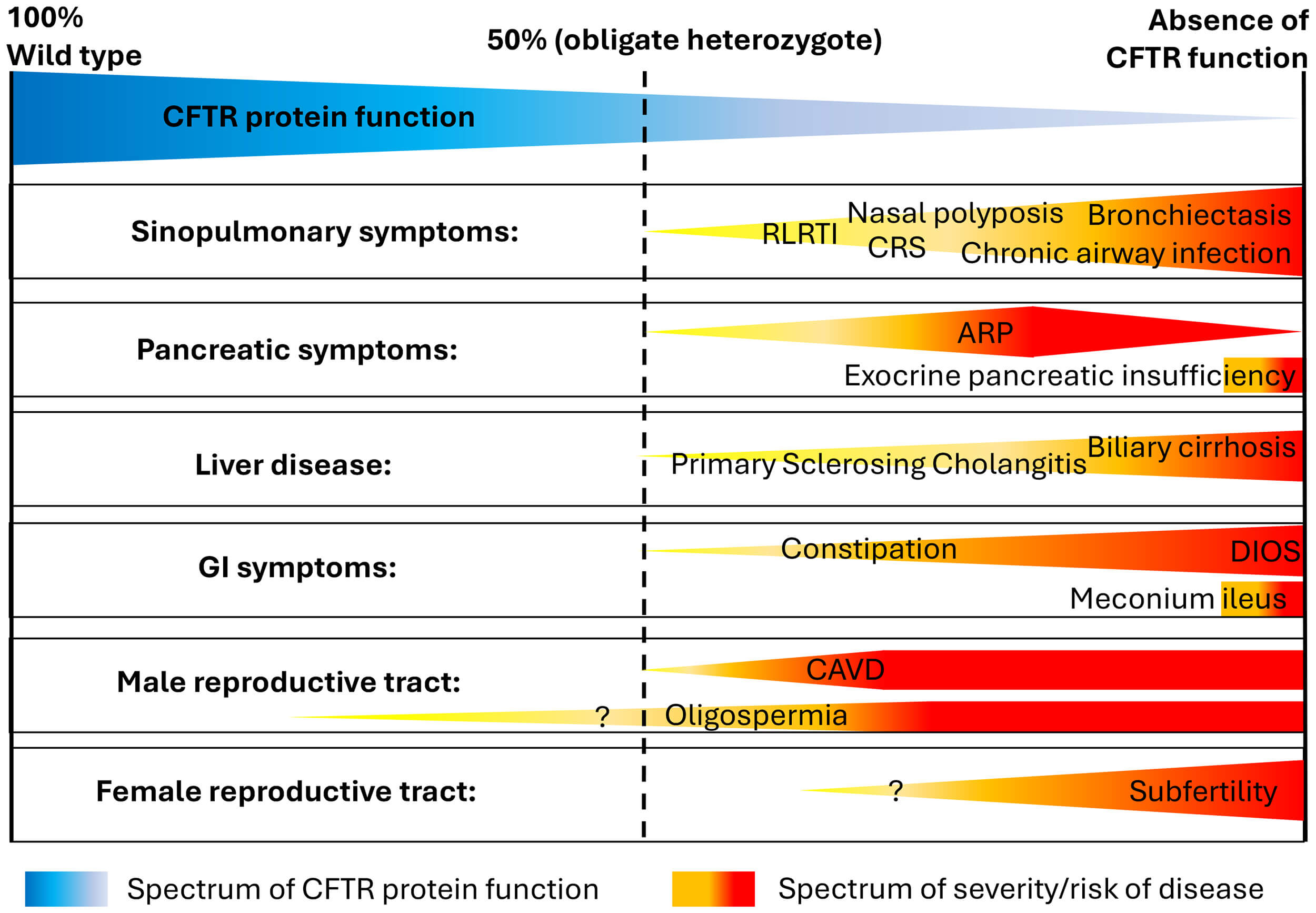 Fig. 1.
Fig. 1.
Schematic representation of the clinical spectrum of CFTR dysfunction and associated clinical conditions. In heterozygotes, CFTR protein function is approximately 50%. The question mark indicates that the association between CFTR dysfunction and these clinical symptoms has not been definitively established. Abbreviations: CFTR, cystic fibrosis transmembrane conductance regulator; CRS, chronic rhinosinusitis; RLRTI, recurrent lower respiratory tract infections; ARP, acute recurrent pancreatitis; GI, gastrointestinal; DIOS, distal intestinal obstruction syndrome; CAVD, congenital absence of vas deferens. Figure adapted from [18], available under the CC BY-NC license (https://creativecommons.org/licenses/by/3.0/).
The classic CF phenotype—characterised by meconium ileus in the neonatal period, severe pancreatic insufficiency leading to failure to thrive in infancy, and suppurative lung disease from a very young age—is usually associated with CFTR gene variants that result in a loss of CFTR protein function below 5% of wild type [21]. When CFTR protein function is approximately 10% of wild type, a milder CF phenotype is usually present, with preserved pancreatic function and less severe (or delayed onset) lung disease [13, 22, 23]. Above the threshold of 10% wild type, ex vivo cell studies show much improved transepithelial chloride transport [19, 24], although still at a level where pathological features can develop, but usually with a milder phenotype or CFTR-RD [10].
In pwCF diagnosed in adulthood, clinical symptoms may therefore be less characteristic. However, if somebody presents with symptoms typically associated with CFTR dysfunction affecting several organ systems (Fig. 1) or in patients with severe mono-organ disease (e.g., severe bronchiectasis, idiopathic pancreatic exocrine insufficiency or acute recurrent pancreatitis), CF and CFTR-RD need to be ruled out.
Debate continues over whether CFTR protein function
It is therefore important to note that despite CF being a monogenetic disease, a direct correlation of genotype to phenotype is usually not possible, with various and sometimes unknown mechanisms at each level (genetic expression, protein expression, and phenotype; see Fig. 2).
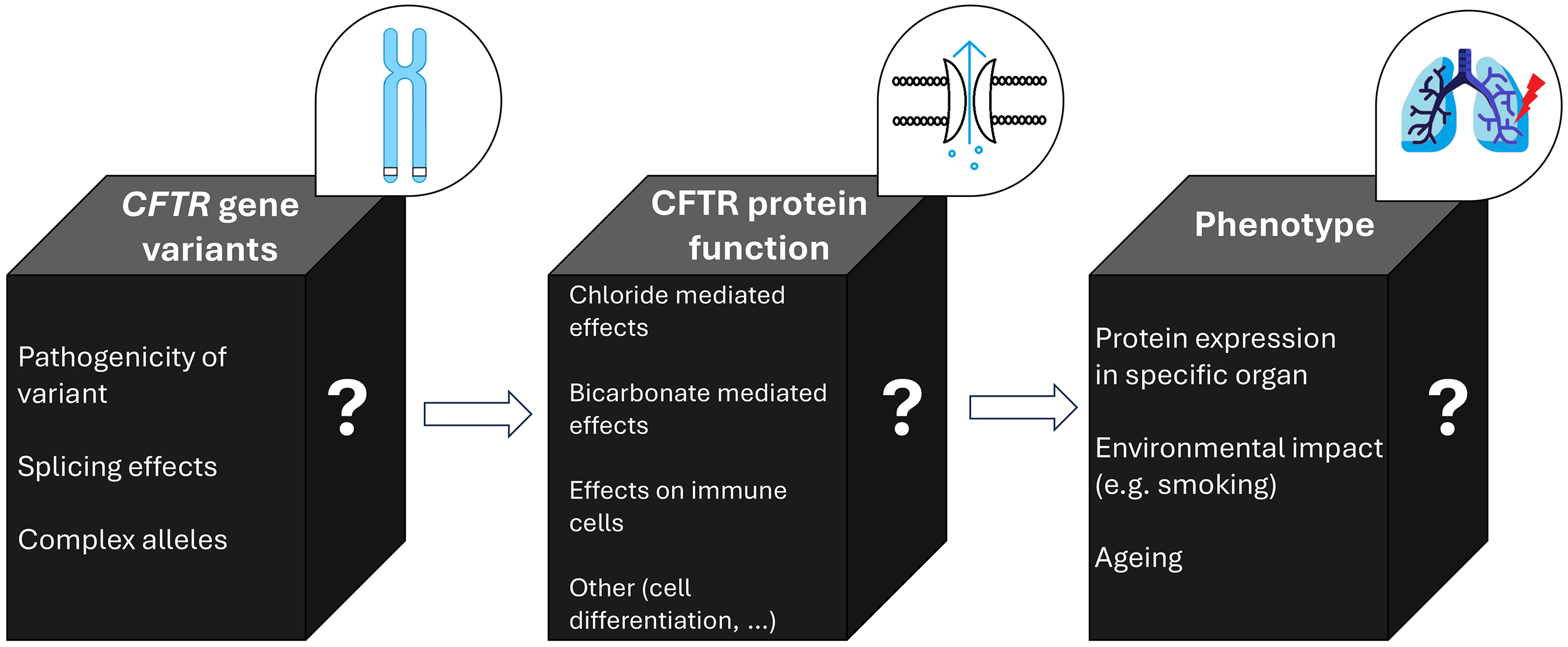 Fig. 2.
Fig. 2.
Schematic representation of the relationship of CFTR gene variants, CFTR protein function and the resulting phenotype. Despite CF being a monogenetic disease, the genotype-phenotype correlation is variable, as each of the steps (gene variant, protein function, clinical phenotype) is influenced by various and sometimes unknown mechanisms (illustrated by the question marks). In the black boxes, known influences on CFTR gene variant expression, CFTR protein function, and clinical phenotype, are listed. Abbreviations: CF, cystic fibrosis; CFTR, cystic fibrosis transmembrane conductance regulator. The figure was created using Microsoft PowerPoint 2019 and Paint (Windows 10), both Microsoft Corporation, Redmond, Washington, USA.
In several countries, national CF registries have been established, which have
been crucial for improving our understanding of the epidemiology and clinical
characteristics of the disease. For example, the UK CF Registry reported on
11,381 pwCF in 2024, highlighting that 65.2% are currently aged
In the following section, we discuss the stepwise approach to assessing a patient in whom a diagnosis of CF or CFTR-RD is suspected and how to reach the final diagnosis, presenting a pragmatic diagnostic algorithm (Fig. 3). Furthermore, we explain the different CFTR functional tests and their application, discuss the challenges of CFTR genetics, and provide a systematic approach to the clinical evaluation and eventual diagnosis. This is also illustrated using two clinical cases.
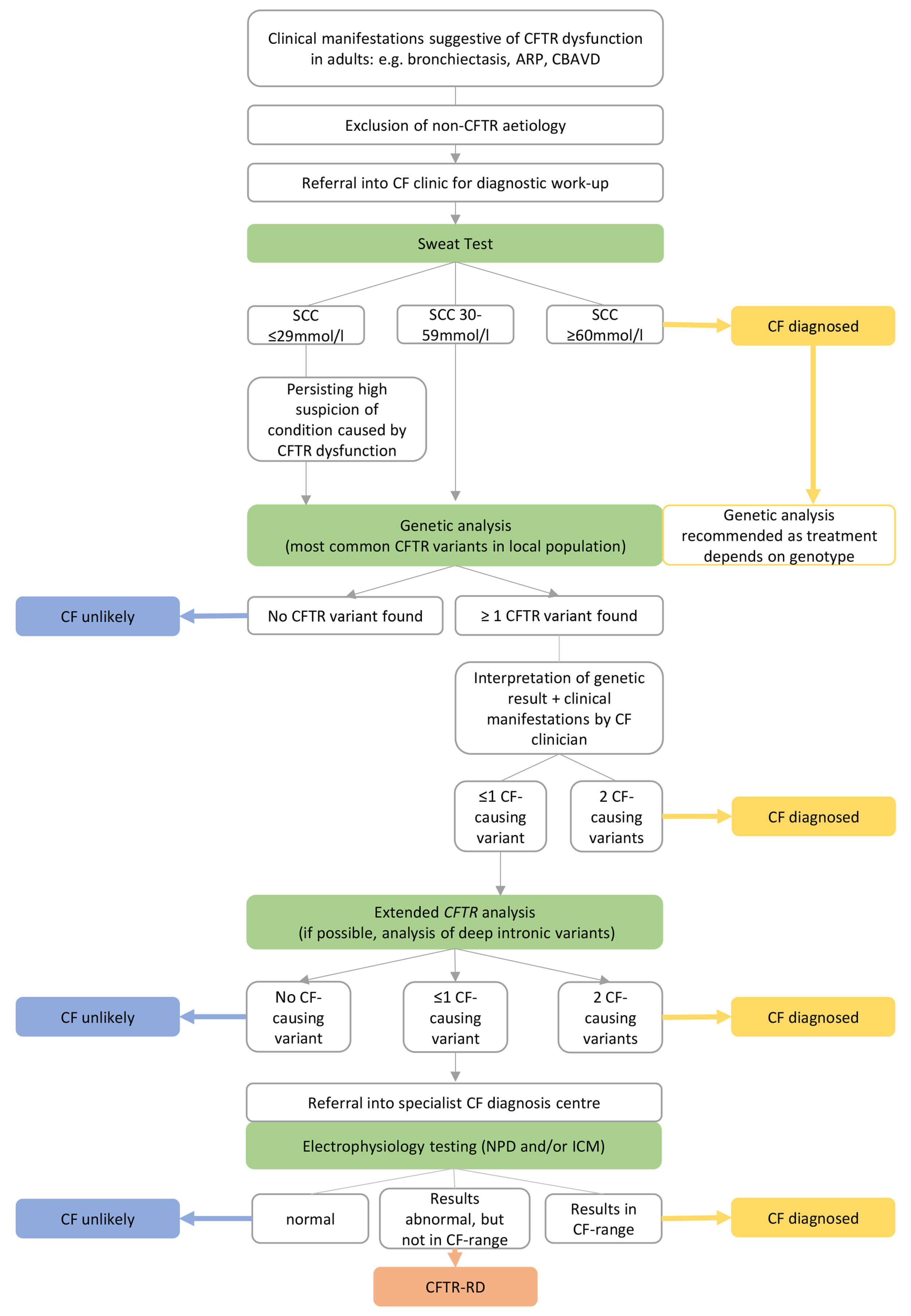 Fig. 3.
Fig. 3.
Schematic work-up for a patient presenting with clinical manifestations in whom a condition caused by CFTR dysfunction is suspected. Abbreviations: CFTR, cystic fibrosis transmembrane conductance regulator; ARP, acute recurrent pancreatitis; CBAVD, congenital bilateral absence of the vas deferens; CF, cystic fibrosis; SCC, sweat chloride concentration; NPD, nasal potential difference; ICM, intestinal current measurement; CFTR-RD, CFTR-related disorder. The figure was created using Microsoft PowerPoint 2019, Microsoft Corporation, Redmond, Washington, USA.
As outlined above, CFTR is expressed widely and functions as both a chloride and bicarbonate channel, so it is challenging to develop a clinical test that can reflect all these different characteristics. Here and in Table 1, we describe the available diagnostic tests.
| Main features | Indications | Advantages | Disadvantages/Limitations | Clinical application | |
| Sweat test | Assessment of sweat chloride concentration. Sweat is collected after stimulating sweat production by iontophoresis. | Most widely used biomarker for assessing CFTR function. Used for diagnosing CF, CFTR-RD. | -Non-invasive and well-tolerated. -Relatively inexpensive. -Extensively validated. -Single-visit test (although sometimes needs to be repeated). |
-Not able to definitely distinguish between CF/CFTR-RD/healthy controls. | First-line test. CF diagnosis if SCC |
| -Some CF-causing variants are associated with low SCC (e.g., D1152H) so a CF diagnosis might be missed. | |||||
| Nasal potential difference | Measures the electrical potential difference across nasal epithelium in vivo by applying pharmacological solutions to the nasal surface. | In situations where sweat tests are non-diagnostic ( |
Able to distinguish between healthy controls, obligate heterozygotes, CF and CFTR-RD (although overlap can still occur). | -Operator-dependent, technically demanding. -Uncomfortable and time-consuming for the patient. -Nasal inflammation can lead to inconclusive findings. -Only available in specialised centres. |
Diagnostic second line test if CF/CFTR-RD cannot be determined with sweat test and genetic analysis. |
| Intestinal current measurement | Ussing chamber assay using rectal epithelium (from a rectal biopsy). CFTR function is assessed ex vivo after pharmacological stimulation of CFTR. | In situations where sweat tests are non-diagnostic ( |
Able to distinguish between healthy controls, obligate heterozygotes, CF and CFTR-RD (although overlap can still occur). | -Invasive intestinal biopsy. -Requires highly specialised set-up. |
Diagnostic second line test if CF/CFTR-RD cannot be determined with sweat test and genetic analysis. |
| CFTR-dependent pathways for sweat secretion are stimulated by |
Further complementary diagnostic test—differentiates heterozygotes from CF/CFTR-RD, from healthy controls. | Delivers immediate results as point of care testing. | -No standardised commercial kits available. -Requires subcutaneous injections. |
-So far mostly used in research settings. -Emerging evidence that test result may be independent of CFTRm exposure. | |
| Patient-derived organoids (e.g., intestinal or airway organoids) | 3D-cultures grown from patient stem cells that serve as a model for the organ cell architecture. When stimulating CFTR (e.g., with forskolin), organoids swell. The amount of swelling is a measure of CFTR function. The morphology (unstimulated) of the organoid is under development as a diagnostic test. | -Functional testing of rare CFTR variants, mostly to predict response to CFTRm. -Emerging data to support its role in diagnostics. |
-In vitro model of a patient’s own tissue. -Testing of several compounds simultaneously. |
-Invasive technique as requires biopsy of target organ. -Cost-intensive and only in specialised research facilities. |
So far mostly used in research settings to predict responsiveness to CFTRm and early work on morphological assessment and diagnosis. |
| Pilocarpine microneedle patches | Pilocarpine (parasympathomimetic agent which induces sweating) is directly applied in the epidermis with microneedles. | Rapid, point-of-care, sweat induction for CF testing. | -Painless, portable equipment/procedure. -Suitable for low-resource settings. |
-No standardised commercial kits available. -Limited validation so far. -Regulatory approval in many regions pending. -Accuracy for CFTR-RD diagnosis unknown. |
Relatively new alternative to sweat testing, especially suitable for low-resource settings. |
Abbreviations: CFTR, cystic fibrosis transmembrane conductance regulator; CF, cystic fibrosis; CFTR-RD, CFTR-related disorder; SCC, sweat chloride concentration; CFTRm, CFTR modulator therapy.
The sweat test was the first diagnostic test developed for CF in the 1950s, following the observation that affected children had abnormally salty skin [40]. CFTR protein, expressed in sweat glands, plays a crucial role in reabsorbing chloride ions, thereby minimising salt loss through sweat. Consequently, CFTR dysfunction in pwCF, leads to an elevated sweat chloride concentration [41]. The sweat test remains the most widely used biomarker for CFTR function. The procedure involves placing electrodes on the forearm to generate sweat production via a mild electrical current, stimulated by pilocarpine (‘iontophoresis’). The collected sweat is then analysed for chloride concentration in the laboratory [42].
A sweat chloride concentration in the pathological range (
When the sweat test is not diagnostic (
NPD evaluates CFTR function by measuring the electrical potential difference across the nasal epithelium. This is done by sequentially applying solutions with two main purposes—first to inhibit sodium absorption via ENaC and then to induce chloride secretion. The chloride response in a healthy (non-CF) individual should be at least 5 mV. In pwCF, chloride secretion is absent or reduced [45]. The principle of ICM is similar to NPD, but conducted on rectal biopsy samples ex vivo, measuring transepithelial membrane potential difference in response to a series of inhibitors and secretagogues. Due to their complexity and need for highly trained personnel, both NPD and ICM are only performed in select specialist CF centres worldwide with sufficient expertise and experience [6, 13].
Both ICM and NPD help to distinguish between healthy controls, obligate heterozygotes, CF and CFTR-RD, although even with these tests, overlap exists [13].
To address the limitations of the currently used CFTR functional tests, several
new diagnostic approaches are under development, although they are currently
limited to research settings. The
Other approaches include ex vivo studies using patient-derived organoids—three-dimensional structures formed from stem cells. When wild type CFTR channels are stimulated (e.g., by forskolin), the organoids swell, but in the presence of CFTR dysfunction, cells fail to reabsorb chloride, thus swelling is reduced or absent completely. This technique is particularly promising for personalised medicine, as it also enables the assessment of individual responses to novel CFTR-targeted therapies. Recent progress in the diagnostic field has shown that the shape (‘circularity’) and luminal properties of the organoid correlate well with diagnostic outcomes, including people with an unclear diagnosis by conventional methods [49, 50, 51].
Of these emerging tests, the
Finally, one of the biggest challenges with each of the functional tests is that they are designed to differentiate between healthy controls and CF, but an overlap between CFTR-RD and carrier status or CF still exists. Therefore, in this situation, added benefit could be derived by sequentially testing with different methodologies (sweat test, NPD, ICM) across different tissue types (skin, nasal mucosa, rectal mucosa), to provide complementary data to maximise the diagnostic potential [55]. Combining multiple tests may yield the most accurate assessment of CFTR function, though further research is needed [13].
The CFTR gene is located on chromosome 7 and contains 27 exons, the coding regions of the gene. The exons are interspersed by introns which undergo splicing (i.e., removal/editing) to produce mature messenger RNA (mRNA) for protein translation. An individual’s phenotype is determined by the presence of at least two CFTR gene variants, one inherited from each parent. In rare cases, more than two variants are present, such as when a ‘complex allele’ is identified [56].
To date, over 2000 gene variants in CFTR have been described—they lead to varied clinical manifestations by affecting CFTR protein at different levels: mRNA and protein synthesis, protein maturation, protein trafficking (whether a protein reaches its target destination within the cell), and ion channel activity. ‘Nonsense’ variants introduce premature stop codons into the mRNA sequence, leading to mRNA degradation and an absence of protein production [9]. The molecular impact of many variants is difficult to predict—especially with rare variants where clinical correlations are limited. Consequently, interpretation is based on in vitro (cell lines) or in silico (computer-generated protein folding) data.
Following the discovery of CFTR in 1989, Welsh and Smith [57] proposed a functional classification for CFTR variants (this has since been extended from five to seven classes). The first three classes (I–III) include variants that result in defective protein synthesis, failed protein trafficking, or proteins with complete loss of function, and these are generally associated with severe disease phenotypes. In contrast, classes IV–VII generally result in milder disease manifestations due to reduced protein quantity, function, or stability [58]. However, this classification does not fully capture the broad spectrum of phenotypic variability associated with some variants. An alternative approach, which is particularly relevant to the CF diagnosis, is based on the potential of the variant to cause CF:
• Variants known to cause CF—when combined with another CF-causing variant, the individual will have CF.
• Variants with varying clinical consequences—when combined with another CF-causing variant, some individuals will have CF, some may have CFTR-RD and some will be healthy with neither condition.
• Variants that do not cause CF (when combined with a CF-causing variant) but could cause CFTR-RD in certain situations.
• Variants of unknown significance, for which to date insufficient data is available to determine their impact.
Large reference datasets to classify CFTR gene variants and assess their clinical significance have been developed [59, 60, 61]. One widely used database in clinical practice is ‘CFTR2’ (https://cftr2.org/) [62].
CFTR gene variants vary widely between different ethnic and geographical origins. The most common CF-causing variant in the Northern European population is F508del, affecting 81% of all CF-causing alleles in the UK, but its prevalence is very variable across the globe with only 20% of pwCF carrying this variant in Qatar, but 94.2% in Serbia [63]. Most commercial genetic screening kits test for only the most common CFTR variants, usually tailored to the population in which they are used. To detect rarer variants, extended analysis is implemented which includes exon sequencing and multiple ligation probe amplification (MLPA) for large deletions/duplications, although some laboratories analyse further with next generation sequencing of CFTR covering nearly the whole gene, including deep intronic regions [64].
A systematic approach to assess potential organ involvement in both CF and CFTR-RD is important [11]. Evaluation should include looking for evidence of pulmonary, sinus, gastrointestinal, pancreatic, hepatobiliary, dermatological, and reproductive pathology (Table 2). In the presence of chronic respiratory symptoms, it is advised to have a low threshold for a chest computed tomography (CT) and obtain a lower respiratory tract sample. In men, the guidelines suggest performing semen analysis [11]. Other investigations will be guided by clinical suspicion.
| Clinical manifestations | Further investigations | |
| Upper respiratory airways | -Chronic rhinosinusitis | -Sinus CT scan/MRI |
| -Polyps | -Nasoendoscopy | |
| Lower respiratory airways | -Chronic cough +/- sputum production | -High-resolution CT scan of thorax |
| -Recurrent lower respiratory tract infections | -Lower respiratory tract sample (sputum culture or cough swab [induced sputum culture or bronchoscopy if not available and/or more definitive culture required]) | |
| -Bronchiectasis | ||
| -ABPA | ||
| Gastrointestinal tract | -Pancreatic insufficiency | -Faecal elastase measurement |
| -Acute recurrent pancreatitis, sometimes resulting in exocrine pancreatic insufficiency | -Liver ultrasound | |
| -Liver fibrosis/cirrhosis | -MRCP and/or ERCP | |
| -Primary Sclerosing Cholangitis | -Blood tests assessing liver enzymes and function, coagulation, fat-soluble vitamin levels | |
| Uro-genital tract | -Uni- or bilateral congenital absence of the vas deferens | In men: |
| -Subfertility in women (e.g., history of fertility treatments) | -Semen analysis | |
| -Scrotal (or rectal) ultrasound imaging | ||
| -Pelvic MRI | ||
| Skin | -Aquagenic wrinkling of the palms | Based on clinical examination (biopsy not required) |
Abbreviations: CT, computed tomography; MRI, magnetic resonance imaging; MRCP, magnetic resonance cholangiopancreatography; ERCP, endoscopic retrograde cholangio-pancreatography; ABPA, allergic bronchopulmonary aspergillosis.
When patients present with a phenotype suspicious for CF or
CFTR-RD—particularly if presenting as an adult—the non-CF specialist should
first exclude common non-CFTR-related causes (e.g., of bronchiectasis) and then,
if negative, refer to the CF centre for a further diagnostic work-up. At the CF
centre, the patient will undergo sweat testing and appropriate genetic analysis
[11]. A CF diagnosis is unlikely when the sweat test is normal (sweat chloride
A 57-year-old woman of White British ethnicity was referred for a CF diagnostic work-up by the local respiratory clinic as she had bilateral bronchiectasis of unknown aetiology. In addition, she had a long-standing history of ABPA, chronic airway infection with Pseudomonas aeruginosa, and episodes of significant haemoptysis, prompting a further search for an underlying diagnosis, despite having had CF ‘excluded’ 30 years earlier. She did not report a history of sinusitis or nasal polyposis and had no history of pancreatitis or malabsorption. Of note, she underwent intrauterine insemination to conceive, and described aquagenic wrinkling of the palms (a condition associated with CFTR variants [14]).
Targeted first-line genotyping (50 variant panel) detected one variant only
(heterozygous F508del), but as her sweat chloride concentration came
back high (60 and 64 mmol/L), extended genetic analysis was initiated. This
confirmed a rare second pathogenic splice variant (c.579+3A
This case highlights the need to continually consider the underlying diagnosis, particularly as historical diagnostic criteria have changed (e.g., sweat test electrolytes and thresholds) and our knowledge of CFTR (and the technology to analyse it) has advanced significantly over the last few decades, i.e., the period since the patient’s original diagnostic work-up for CF (which would have been around the time of the discovery of the CF gene in 1989).
A 17-year-old adolescent male of White British ethnicity underwent diagnostic review due to prolonged excellent health despite a longstanding CF diagnosis (i.e., he failed to develop a CF phenotype). He was originally diagnosed with CF antenatally when his mother underwent chorionic villus sampling after the 20-week scan showed increased nuchal thickness. He was found to have two CFTR variants (F508del and R117H+7T) and was therefore followed up in the paediatric service where he remained well with stable (normal) lung function (although he did have a one-off growth of Pseudomonas aeruginosa aged 13 years for which he received antibiotic eradication therapy). Further testing at 17 years revealed sweat chloride concentrations in the equivocal range (37 mmol/L and 37 mmol/L). As his genotype was not conclusive for CF (R117H+7T is a variant of varying clinical consequences), he underwent NPD measurement. This confirmed CFTR function with evidence of chloride secretion. For social/psychological reasons he did not wish to undergo semen analysis at that stage in his life but did agree to a pelvic magnetic resonance scan which confirmed congenital bilateral absence of the vas deferens (CBAVD) [69]. Cross-sectional chest imaging of his chest was normal, and he was pancreatic sufficient.
In light of the findings, a diagnosis of CFTR-related disorder was considered appropriate due to the absence of the full clinical phenotype, borderline chloride transport on functional tests and the absence of a definitive CF-causing genotype. He has since had dornase alpha stopped (muco-active therapy which had been started earlier in life) and is followed up in a dedicated CFTR-RD clinic being seen on a less frequent (but appropriate) basis. This has reduced his treatment and visit burden, thus reducing risks of ‘over medicalisation’ and anxiety from a CF diagnosis, while still ensuring a balanced level of follow-up.
Diagnosing patients with CF gives them access to CF-specific multi-disciplinary team (MDT) care and treatments (e.g., CFTR modulator therapy [70, 71], dornase alpha [72] and CF-specific inhaled antimicrobials [73, 74, 75]). Novel CF treatments are highly effective, including for patients with low lung function and a high burden of multisystem disease [76]. This is important as an older age at diagnosis has been recognised to be a risk factor for death or transplantation [77]. CF care is usually organised in specialised CF centres where patients are regularly followed by an MDT consisting of specialised physicians, nurses, physiotherapists, dietitians, pharmacists and psychologists. It has been shown that newly diagnosed adult CF patients with regular CF-MDT follow-up have significant improvements in lung function [78]. As CF is associated with other health complications, such as diabetes [79] and CF-bone disease [80]—and importantly has an association with an increased cancer risk (especially bowel cancer with a 5-to-10-fold risk compared to the general population) [37]—a timely CF diagnosis facilitates the introduction of important preventative monitoring and treatment strategies.
CFTR-RD is a very heterogeneous group of conditions where the long-term prognosis is currently still unclear, and the management has not yet been standardised. The ECFS Standards of Care Committee recommends regular follow-up in specialised CF centres allowing for regular health monitoring and early intervention when indicated. People with CFTR-RD are currently not eligible for CF-specific treatments (e.g., CFTR modulator therapy, but also inhaled therapies such as dornase alfa and some inhaled antibiotics) [12]. In the context of CFTR modulator therapy, benefits have been reported in bronchiectasis patients carrying one CFTR variant with variable evidence of CFTR dysfunction, but more robust studies on well characterised cohorts with accurate diagnostics are urgently required, as are data on long-term outcomes for people with CFTR-RD [12, 81, 82].
Being newly burdened with the diagnosis of a life-long condition (with its life expectancy implications) can be daunting, so psychological support by the CF psychology team, and advocacy by regional or national patient associations can be helpful. As CF and CFTR-RD are genetic disorders, a new diagnosis also has an impact on the wider family and future family planning. Access to genetic counselling is therefore important for the patients and their families.
CF is often diagnosed in childhood, but an important proportion of patients are diagnosed as adults. Despite having strong historical associations with White populations, CF exists across all ethnicities. PwCF can present with different clinical phenotypes, from bronchiectasis to acute recurrent pancreatitis to fertility issues. More than 2000 gene variants have been identified in CFTR with variable worldwide distribution and effects on protein function. This is further complicated as disease expression is also affected by numerous other factors—some genetic, such as splicing effects and complex alleles—and some non-genetic, so-called ‘environmental’ factors.
Confirming a CF diagnosis in adults is often not straightforward and needs a stepwise approach. In patients with symptoms affecting several organ systems and/or severe mono-organ dysfunction of unknown aetiology, CFTR dysfunction should be suspected regardless of ethnicity. After excluding alternative, non-CFTR-related diagnoses, individuals should be referred to a CF centre where CFTR investigations, including sweat and genetic testing, will be performed. If the sweat test and genetic analysis are inconclusive, further tests to assess CFTR protein function, such as NPD or ICM, will be considered to further differentiate between conditions lying in the spectrum of CFTR dysfunction (CFTR-RD and CF). Confirmation (or exclusion) of the CF/CFTR-RD diagnosis is vital to ensure patients have access to appropriate care. Ultimately, patients need to be fully informed about their condition, so they have the knowledge and understanding, supported by an expert team, to effectively optimise their health status and reduce morbidity for the long-term.
• CF is not only diagnosed in children, but also in adults, often presenting with an ‘atypical’ phenotype.
• When clinical features which could be caused by CFTR dysfunction are present (e.g., unexplained bronchiectasis, azoospermia, recurrent pancreatitis, and chronic rhinosinusitis), CF must be considered, particularly when other, more common, causes have been excluded.
• The diagnosis of CF may not be straightforward, especially in patients with rare mutations or equivocal functional tests, necessitating referral to a CF centre for further essential assessment.
• Confirming CF or CFTR-RD is important as this not only provides access to specialist care (and therapies), but also has important implications for genetic counselling and family planning.
Not applicable.
Conceptualisation: LW and NJS, writing: LW and NJS, visualisation: LW, supervision: NJS. Both authors contributed to revising the manuscript critically for important intellectual content. Both authors read and approved the final manuscript. Both authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The study followed the principles of the Declaration of Helsinki. Both patients have provided their written informed consent to be included in the manuscript.
We are very grateful to both patients who consented to share their clinical histories for publication.
This research received no external funding.
LW does not have any conflicts of interest related to this work. NJS has received honoraria for advisory boards from Vertex, Gilead, Chiesi, Pulmocide, Menarini, Roche and Boehringer Ingelheim. He has also received honoraria for educational activities from Vertex, Chiesi, Gilead, Medison, Teva and Zambon.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.