






 , Ian Beeton 2
, Ian Beeton 21 Department of Medicine, Basildon University Hospital, Mid and South Essex NHS Foundation Trust, SS16 5NL Basildon, UK
2 Department of Cardiology, St. Peter’s Hospital, Ashford and St. Peter’s Hospitals NHS Foundation Trust, KT16 0PZ Chertsey, UK
Abstract
Phaeochromocytoma is a rare catecholamine-secreting neuroendocrine tumour arising from chromaffin cells of the adrenal medulla or from extra-adrenal paraganglionic tissue, in which case it is termed paraganglioma. Its clinical presentations are highly variable with up to 10–20% of patients being asymptomatic or having non-specific symptoms, therefore leading to frequent misdiagnosis. The aim of this case report is to present a rare and diagnostically challenging presentation as well as highlight the need to maintain a high index of suspicion in patients with unexplained cardiovascular symptoms.
A 54-year-old female presented with acute chest pain and atrial fibrillation, initially suggestive of acute coronary syndrome. However, coronary angiography demonstrated unobstructed coronary arteries, and electrocardiogram (ECG) changes resolved. Persistent symptoms and refractory hypertension prompted further investigation, which revealed a left adrenal mass and significantly elevated urinary metanephrines, consistent with a diagnosis of phaeochromocytoma.
After the diagnosis was confirmed by Endocrinology, treatment with phenoxybenzamine was initiated and titrated to 10 mg twice daily, resulting in stabilisation of her blood pressure and heart rate. Bisoprolol was added for further rate control. Following preoperative optimisation, the patient later underwent an uncomplicated laparoscopic adrenalectomy. At follow-up, she remained in sinus rhythm with a normalised blood pressure and resolution of symptoms.
This case highlights the diagnostic challenge posed by phaeochromocytoma, which can mimic myocardial infarction and myocarditis due to catecholamine-induced cardiac effects.
Keywords
- phaeochromocytoma
- myocarditis
- acute coronary syndrome
- catecholamines
- case report
Phaeochromocytoma is a rare catecholamine-secreting neuroendocrine tumour arising from chromaffin cells of the adrenal medulla or from extra-adrenal paraganglionic tissue, in which case it is termed paraganglioma [1]. These tumours produce and release varying amounts of catecholamines, particularly epinephrine, norepinephrine, and dopamine, which may be secreted in continuous, paroxysmal, or mixed patterns. The paroxysmal and sustained actions of norepinephrine release commonly result in hypertension and tachycardic states via alpha-1-adrenergic and beta-1-adrenergic receptor activation, respectively [2]. Sustained norepinephrine release commonly results in persistent hypertension, whereas paroxysmal epinephrine surges may precipitate tachyarrhythmias [3]. The classic clinical triad of phaeochromocytoma includes episodic headache, tachycardia, and diaphoresis; however, clinical presentations are highly variable. Up to 10–20% of patients may be asymptomatic, and many exhibit non-specific or intermittent symptoms, leading to frequent misdiagnosis. Phaeochromocytoma is thus often referred to as “the great mimic” due to its protean manifestations [4].
Myocarditis, defined as inflammation of the myocardium, presents with a similarly broad clinical spectrum, ranging from mild chest pain and palpitations with transient electrocardiographic changes to fulminant heart failure, cardiogenic shock, and life-threatening arrhythmias [5]. Given the overlap in symptom presentation, including chest pain, palpitations, dyspnoea, and electrocardiogram (ECG) abnormalities, patients with phaeochromocytoma may be misdiagnosed with acute coronary syndromes (ACS) or myocarditis, particularly in the acute setting when transient atrial fibrillation (AF) and dynamic ST-segment changes are present. This therefore leads to increased risk of misdiagnosis and delays in the appropriate treatment.
This case report describes a 54-year-old female who initially presented with features suggestive of myocarditis but was subsequently diagnosed with phaeochromocytoma. The combination of transient AF and dynamic ST changes, as seen in this patient, represents a rare and diagnostically challenging presentation that has significant implications for clinical practice. It highlights the need for careful ECG interpretation and a high index of suspicion in patients with unexplained cardiovascular symptoms.
A 54-year-old female presented to the emergency department after waking up with symptoms of a dull, achy left-sided chest pain, which she rated a 6 out of 10 on the Numerical Rating Scale (0 = no pain, 10 = extreme pain) [6]. Shortly after, she became short of breath, sweaty, and developed diarrhoea, nausea and vomiting. She had a similar presentation 2 years ago. Her past medical history includes hypertension and type 2 diabetes mellitus, however, she has no family history of ischaemic heart disease, and is a non-smoker.
On assessing the patient, her observations were stable with a heart rate of 77 bpm, blood pressure of 167/89 mmHg, oxygen saturation of 97% on air, respiratory rate of 17, and body temperature of 35.6 °C. Bedside examination was normal, no added heart sounds or murmurs, chest clear, abdomen soft and non-tender, and no peripheral oedema. Blood results showed a significant troponin rise post-symptom onset. Additional abnormalities included raised inflammatory markers, an elevated D-dimer (fibrin degradation product assay), electrolyte disturbances, and abnormal liver function tests. Among these, the troponin rise was considered the most clinically relevant finding, consistent with acute myocardial injury (Table 1).
| Parameter | Result | Reference range |
| Troponin (initial) | 245 ng/L | |
| Troponin (7 hrs later) | 6701 ng/L | |
| D-dimer | 1192 ng/mL | |
| Potassium | 3.3 mmol/L | 3.5–5.3 mmol/L |
| Magnesium | 0.61 mmol/L | 0.7–1.0 mmol/L |
| Alanine transaminase (ALT) | 55 U/L | 7–56 U/L |
| Alkaline phosphatase (ALP) | 180 U/L | 30–130 U/L |
| White cell count | 19.3 |
4.0–11.0 |
| C-reactive protein | 42 mg/L |
An immediate ECG showed AF and ST-segment elevation (STE) in V1 and V2 [7, 8]. A repeat ECG shortly after showed sinus rhythm with STE in V2 (Fig. 1). With this information, an initial diagnosis of ACS was made, therefore the patient was started on the initial protocol, which included loading doses of Aspirin and Clopidogrel.
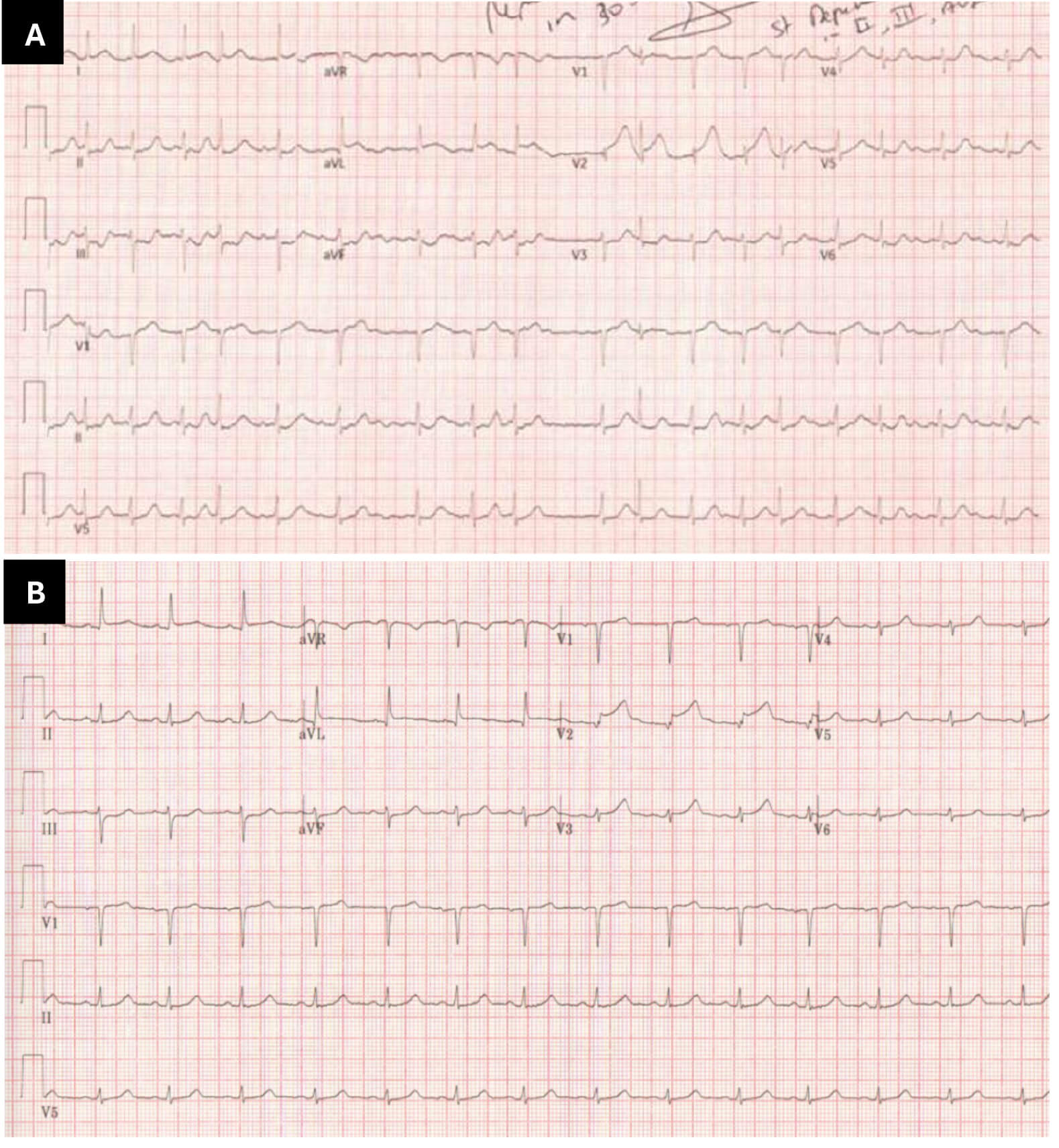 Fig. 1.
Fig. 1.
Dynamic electrographic changes demonstrating transient ST-segment abnormalities and atrial fibrillation (AF) on admission with subsequent resolution. (A) Immediate electrocardiogram on admission, showing AF and ST elevation in leads V1 and V2. (B) Repeat electrocardiography 30 minutes later, showing subtle ST depression in leads III and augmented vector foot (aVF), as well as high take off in leads I, augmented vector left (aVL) and V2; the AF had resolved at the time of this electrocardiogram (ECG). These dynamic changes suggest possible early anterior wall ischaemia. aVR, augmented vector right.
A previous coronary angiogram showed unobstructed coronaries, and a cardiac magnetic resonance imaging (MRI) scan in 2020 reported concentric left ventricular remodelling and the absence of abnormal late gadolinium enhancement or myocardial oedema (Fig. 2). The angiogram subsequently carried out during this admission (Fig. 3) showed no changes from previous angiogram.
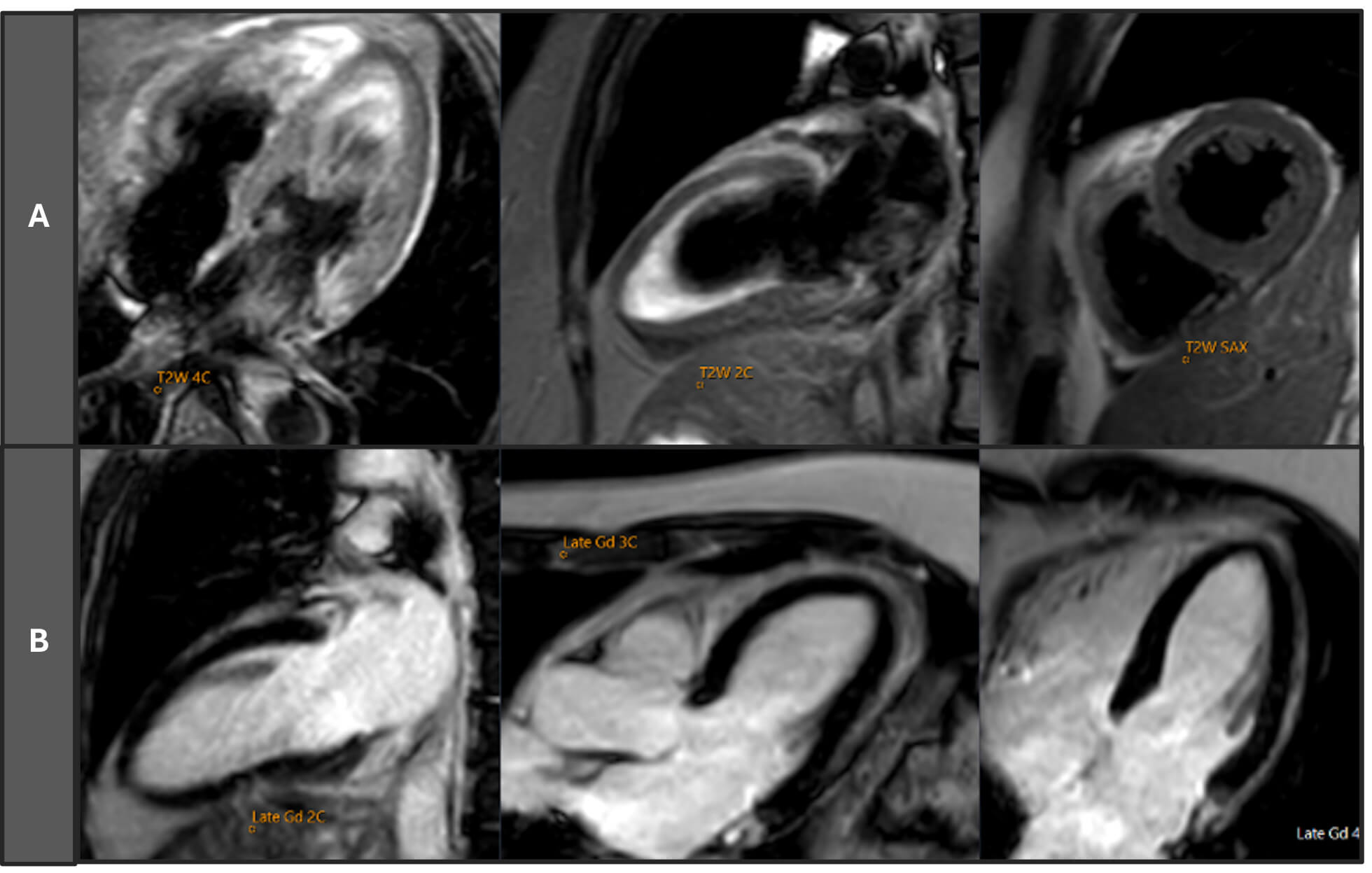 Fig. 2.
Fig. 2.
Cardiac magnetic resonance imaging (MRI) scan of the patient (2020). Row (A) T2-weighted (T2W) short axis (SAX), two-chamber (2C), and four-chamber (4C) views, showing no evidence of myocardial oedema or increased signal intensity. Row (B) Late gadolinium enhancement (LGE) images in 2C, three-chamber (3C), and 4C views, revealing no areas of abnormal delayed enhancement, consistent with the absence of myocardial fibrosis or infarction. Cardiac MRI findings confirm a maximal septal thickness of 10 mm, and a functionally normal heart with preserved biventricular function (left ventricular ejection fraction [LVEF] 68%, right ventricular ejection fraction [RVEF] 53%) and no features of myocarditis, myocardial infarction, or cardiomyopathy. Gd, gadolinium.
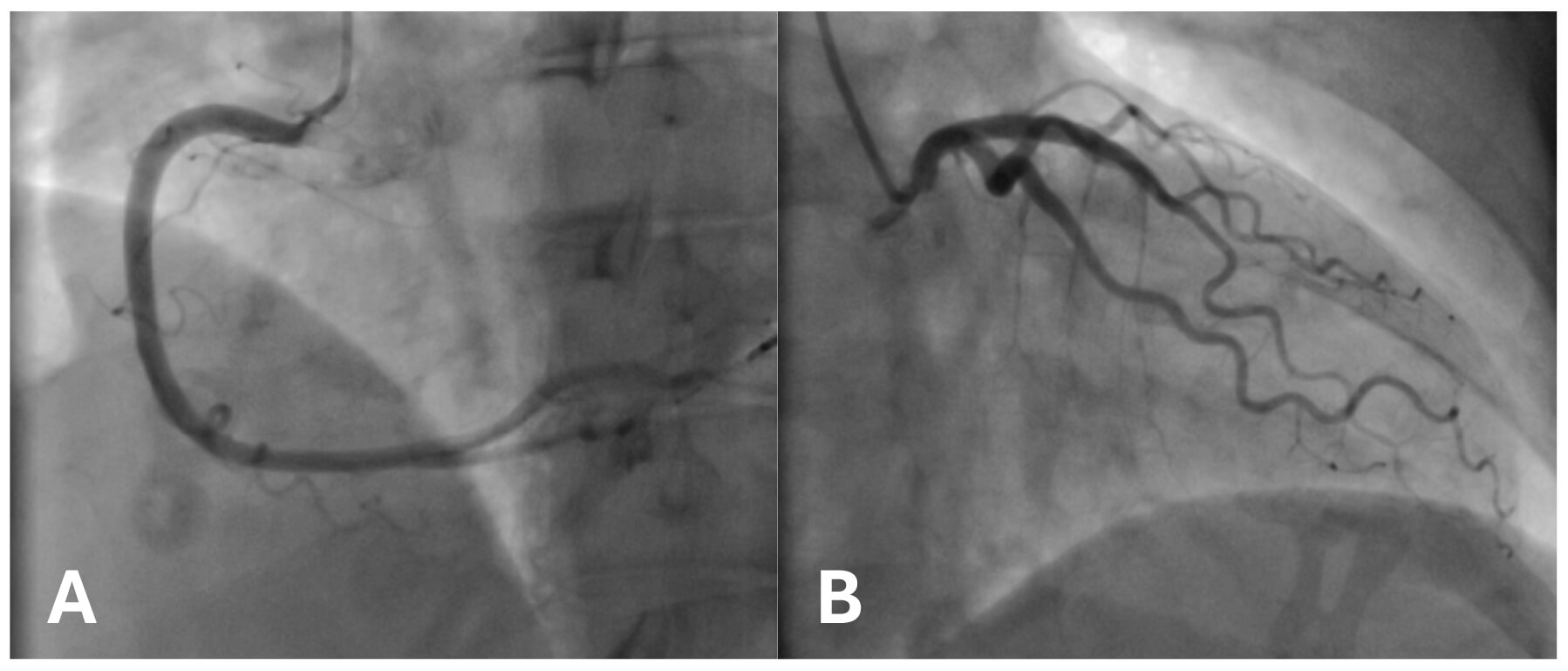 Fig. 3.
Fig. 3.
Coronary angiogram demonstrating unobstructed vessels with a smooth course. (A) Right coronary artery in the right anterior oblique (RAO) projection. (B) Left coronary artery in the left anterior oblique (LAO) projection.
Following the discovery of unobstructed coronary arteries, the working diagnosis was revised to paroxysmal AF secondary to myocarditis, an explanation consistent with the elevated troponin and inflammatory markers. Subsequent findings, including a repeat ECG demonstrating sinus rhythm (Fig. 4), a repeat angiogram confirming patent coronary arteries, and declining troponin levels, supported a recovering myocarditis.
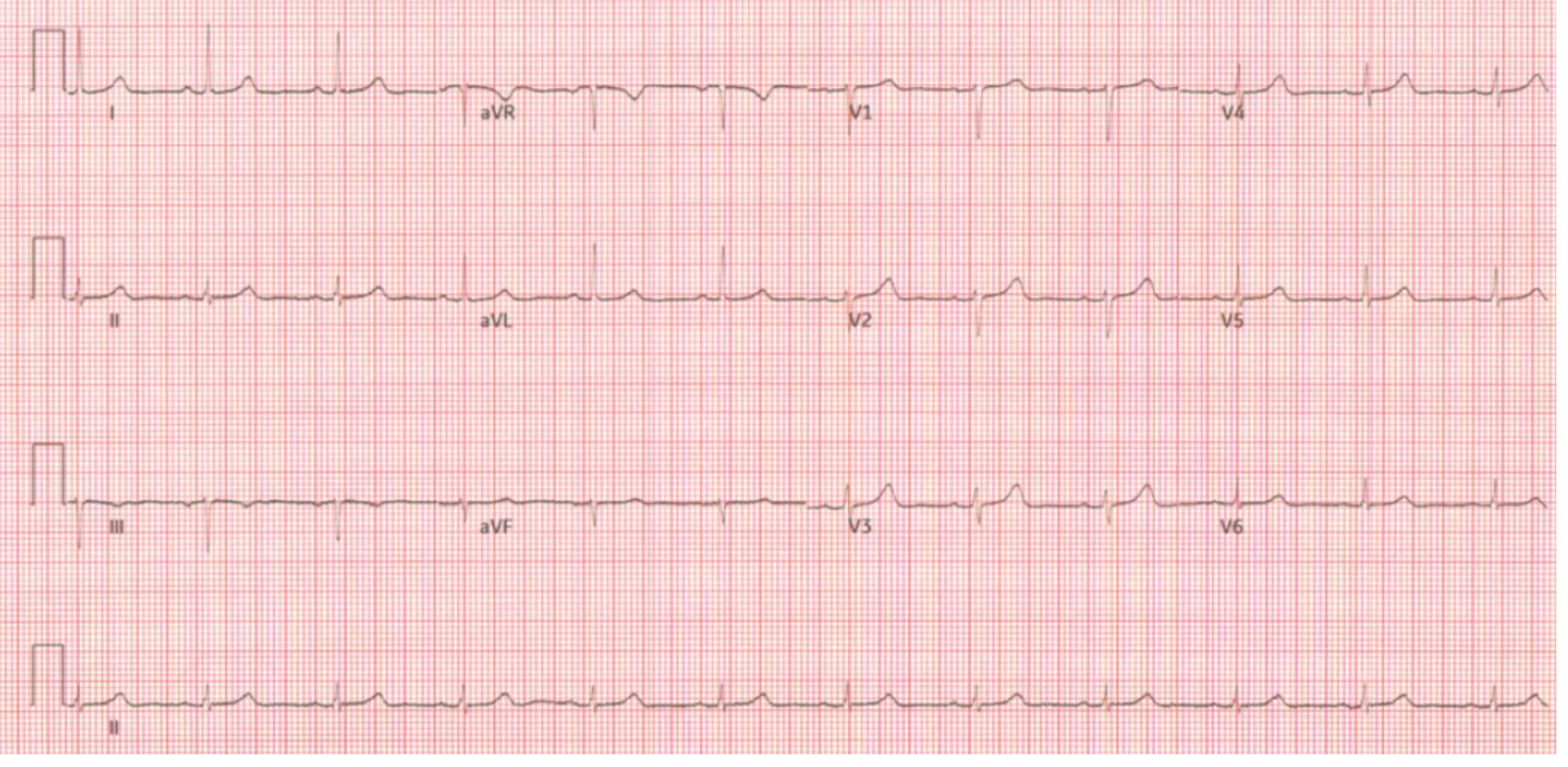 Fig. 4.
Fig. 4.
Repeat electrocardiogram later during admission showing normal sinus rhythm with resolution of ST changes.
4 days after admission, her chest pain and diarrhoea had settled. However, during the ward round, she brought up concerns of nocturnal palpitations associated with high blood pressure, despite being on two antihypertensive medications (Ramipril and Bisoprolol). Upon further questioning, it was found that she had been episodically waking up with a headache, hot, sweaty, and anxious, which lasts 30 minutes to 2 hours, for the last 2 years. A secondary hypertension screen was then carried out, involving a computerised tomography (CT) scan and renal artery doppler, which showed a cystic lesion at the tail of the pancreas close to the left renal vessels. Further investigations included CT of the thorax, abdomen and pelvis, and 24-hour urinary catecholamines. Abdominal MRI demonstrated a mass, arising from the left adrenal gland rather than the pancreas, and measuring 5 cm in maximum diameter (Fig. 5), and 24-hour urinary metanephrines were significantly raised (Table 2).
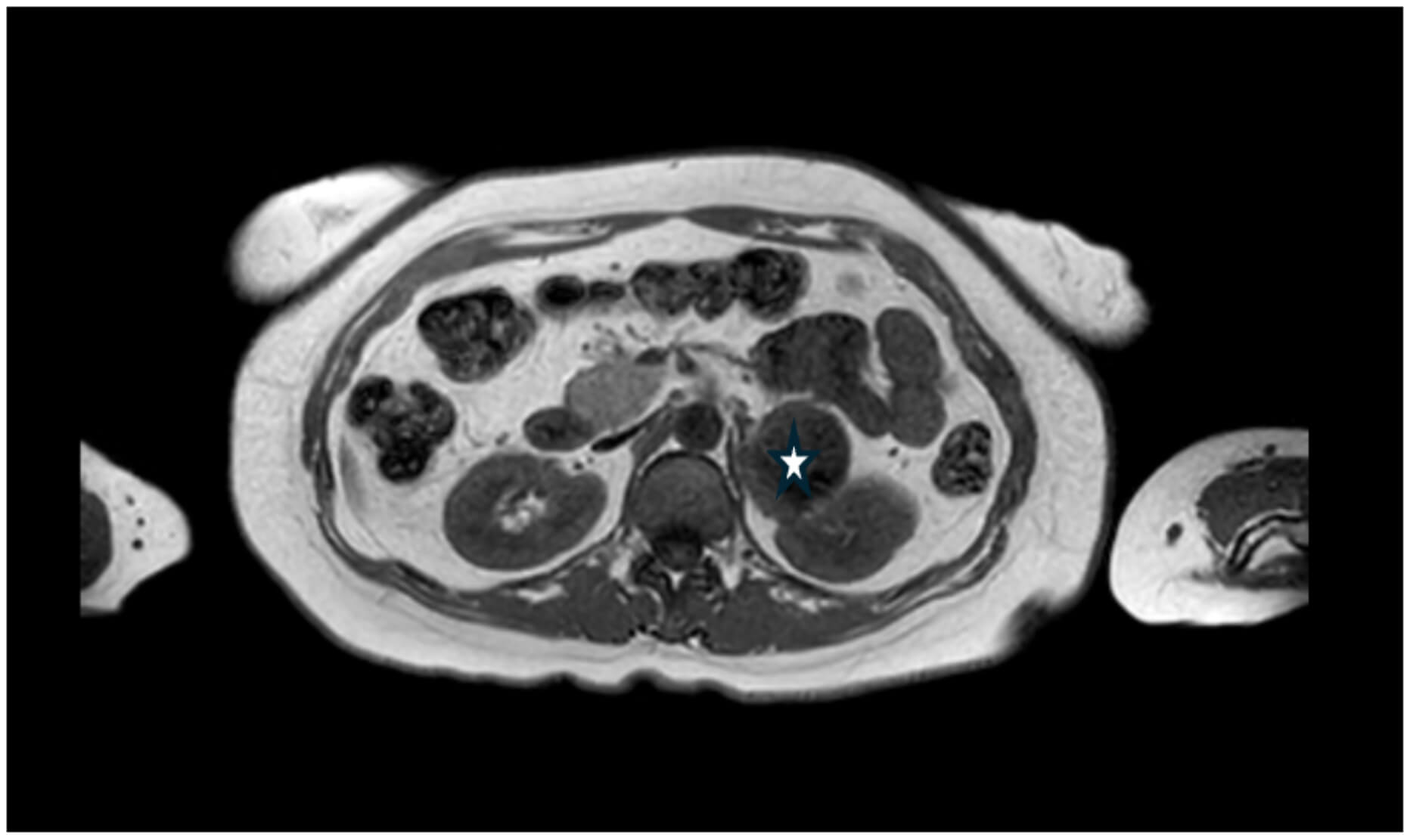 Fig. 5.
Fig. 5.
Magnetic resonance imaging (MRI) of the abdomen showing a left adrenal mass. There is a large mass anterior (✩) to the left kidney, measuring 5 cm in maximum diameter, and arising from the left adrenal gland.
| Parameter | Result | Reference range |
| Aldosterone | 272 pmol/L | 100–800 pmol/L |
| Renin | 28.3 mU/L | 5.4–60 mU/L |
| Aldosterone: Renin ratio | 10 | |
| Normetanephrine | *392 pg/mL | |
| Metanephrine | *180 pg/mL | |
| Urine normetanephrine | *7.39 µmol/24 h | |
| Urine metanephrine | *10.37 µmol/24 h | |
| Urine 3-methoxytyramine excretion | 1.70 µmol/24 h |
Abnormal results highlighted with a ‘*’; h, hours.
She was then referred to Endocrinology, where a formal diagnosis of phaeochromocytoma was confirmed. Treatment was initiated with the alpha-blocker phenoxybenzamine 10 mg once daily and, following good tolerance, the dose was increased to 10 mg twice daily after one week [1, 9]. This regimen effectively stabilised her blood pressure and heart rate, with average systolic readings between 110–120 mmHg and diastolic readings between 70–80 mmHg. She experienced mild dizziness and intermittent symptoms of anxiety but did not develop orthostatic hypotension. Given her history of AF and the new-found potential for catecholamine-induced tachyarrhythmias, bisoprolol 7.5 mg once daily was added for rate control [9]. Preoperative volume expansion was performed to reduce the risk of intraoperative hypotension. She subsequently underwent a laparoscopic adrenalectomy, which was completed without perioperative complications or the need for special interventions [1]. At follow-up, her ECG demonstrated sinus rhythm, with stable blood pressure and resolution of previous symptoms. She reported only rare episodes of mild anxiety, and no further cardiovascular abnormalities were identified. The patient herself expressed a high level of satisfaction with her care, noting a smooth recovery process and that she felt the process from admission to diagnosis and treatment was well-supported. She described feeling “back to normal” with significant improvement in her symptoms and overall well-being. No further follow-up investigations were conducted, and she is expected to make a full recovery, and no relapse is anticipated.
The case report (CARE) checklist has been attached as Supplementary Material associated with this article.
This case underscores the diagnostic complexity in differentiating phaeochromocytoma from myocarditis, particularly in the acute setting where both have widely overlapping cardiovascular manifestations. Both conditions can manifest with chest pain, dyspnoea, palpitations, diaphoresis, electrocardiographic abnormalities, diarrhoea, and elevated inflammatory markers; features commonly driven by catecholamine excess. In phaeochromocytoma, this arises from tumour-driven catecholamine overproduction, whereas in myocarditis, it reflects a physiological stress response mediated by endogenous catecholamine release. Such biochemical and clinical overlap often leads to initial misdiagnosis as primary cardiac pathology, particularly when supported by non-specific ECG and biomarker findings. Similar presentations have been described in other case reports, where transient arrhythmias, dynamic ST changes, and labile blood pressures delayed the recognition of phaeochromocytoma [10, 11, 12].
The cardiovascular effects of catecholamines are multifactorial and contribute to a spectrum of cardiac manifestations that mimic more common conditions like acute coronary syndrome and myocarditis. Direct myocardial toxicity results from calcium overload and oxidative stress, while indirect mechanisms include coronary vasospasm, increased myocardial oxygen demand, elevated afterload, and structural myocardial changes. Chronic adrenergic stimulation leads to downregulation of adrenergic receptors, loss of viable myocardial contractile units, and eventual development of heart failure [13, 14, 15]. In this context, the patient’s elevated troponin, ST-segment changes, and AF were initially attributed to ACS, though they were likely secondary to a catecholamine surge rather than obstructive coronary disease. Coronary artery spasm is a recognised consequence of catecholamine excess, contributing to its ACS-like presentation [16].
Although transient AF is not uncommon in the setting of a ST-segment elevation myocardial infarction (STEMI), the STE seen in this patient was initially confined to leads V1–V2 and subsequently localised to V2 alone. This dynamic and limited distribution is atypical for a classical STEMI, where ECG changes usually follow a more consistent territorial distribution and characteristic evolutionary pattern [17]. In addition, the presence of AF can obscure subtle ischaemic changes and complicate ECG interpretation, while dynamic ST changes may fluctuate over short intervals, therefore also complicating interpretation of serial ECGs. In the context of labile hypertension and episodic symptoms, these findings may reflect transient myocardial injury secondary to catecholamine excess or transient coronary vasospasm [13, 18]. Nevertheless, transient coronary occlusion cannot be excluded, underscoring the importance of cautious interpretation to avoid misdiagnosis and delays in appropriate treatment.
Transthoracic echocardiography revealed left ventricular hypertrophy (LVH), initially attributed to chronic hypertension or aortic stenosis. However, emerging evidence suggests that catecholamines independently contribute to hypertrophic remodelling by promoting protein synthesis in cardiomyocytes. Studies have demonstrated a comparable prevalence of LVH in phaeochromocytoma and primary hypertension, even in the absence of sustained mechanical pressure overload [13, 19].
Clinicians should consider phaeochromocytoma in patients presenting with unexplained or refractory hypertension and episodic cardiovascular symptoms, particularly when these symptoms are unresponsive to standard therapy or lack a clear underlying cardiac cause. Alongside this, phaeochromocytoma is known to be associated with the familial cancer syndrome, Multiple Endocrine Neoplasia Type 2 (MEN-2), therefore may warrant screening of genetic syndromes based on their clinical and family history [20]. Early recognition and targeted screening are critical to avoid delays in definitive treatment and to prevent potentially life-threatening complications.
This report describes a single-patient experience, and therefore its findings may not be generalisable to all cases of phaeochromocytoma. Biochemical and imaging data were limited to what was clinically available at the time. Follow-up was limited to 6 months postoperatively, so longer-term outcomes and potential late complications could not be assessed. Nonetheless, this case highlights important diagnostic and management considerations that may support timely recognition and a multidisciplinary approach to treatment in similar clinical settings.
This case illustrates the diagnostic challenge of phaeochromocytoma presenting with acute cardiovascular manifestations. Maintaining a broad differential diagnosis in such presentations, even when initial investigations are inconclusive, ensures early recognition and timely intervention to prevent misdiagnosis and associated complications.
• Phaeochromocytoma can mimic acute coronary syndrome and myocarditis due to catecholamine-induced myocardial injury, often presenting with chest pain, arrhythmias, and elevated troponin.
• Persistent or episodic cardiovascular symptoms with refractory hypertension should prompt consideration of secondary causes, including phaeochromocytoma.
• Biochemical and imaging studies (e.g., urinary catecholamines, abdominal MRI) are crucial in differentiating phaeochromocytoma from primary cardiac pathology.
• Misdiagnosis of phaeochromocytoma is common due to its protean manifestations; maintaining a high index of suspicion is essential for timely diagnosis.
All data generated or analysed during this study are available from the corresponding author upon reasonable request.
ACM and IB were involved in conceptualisation of this case report. ACM drafted the manuscript and conducted the research related to the Discussion. Both authors contributed to revising the manuscript critically for important intellectual content. Both authors read and approved the final manuscript. Both authors participated sufficiently in the work, and agreed to be accountable for all aspects of the work.
Written informed consent was obtained from the patient for publication of this case report and any accompanying images, with all identifying details removed to ensure confidentiality. This case report describes anonymised clinical information obtained during routine care and does not involve research on human subjects; therefore, formal ethics committee approval was not required. The report adheres to the ethical principles of the Declaration of Helsinki.
The authors thank Rosanna Cheverton for assistance in obtaining the scan images used to prepare the figures.
This research received no external funding.
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/BJHM53981.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.