



 , Louise Bowman 1
, Louise Bowman 11 Clinical Trial Service Unit, Oxford Population Health, University of Oxford, OX3 7LF Oxford, UK
Abstract
Low-density lipoprotein-cholesterol (LDL-C) is a causal factor in atherosclerotic cardiovascular disease (ASCVD). Decades of genetic, epidemiological and randomised evidence support LDL-C reduction as a central strategy for cardiovascular risk reduction. Statins remain the first choice of LDL-C-lowering therapy due to their proven efficacy and favourable safety profile. However, therapeutic options have now expanded with widespread availability of other agents including ezetimibe, bempedoic acid and proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors. These can be prescribed alone or in combination with statins to help achieve optimal LDL-C targets. More recently, RNA-based therapies, oral small molecule inhibitors, and gene-editing strategies have emerged, which may offer even greater LDL-C reduction. As evidence increasingly supports a “lower is better” approach, combining established and novel therapies offers opportunities to optimise ASCVD prevention. This review summarises the evolving landscape of LDL-C-lowering therapies, highlighting their mechanisms, evidence base, and implications for clinical practice.
Keywords
- hypercholesterolemia
- LDL-cholesterol
- 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors
- ezetimibe
- PCSK9 inhibitors
- small interfering RNA
Atherosclerotic cardiovascular disease (ASCVD) is one of the leading causes of morbidity and mortality worldwide [1]. Decades of research have firmly established the connection between atherosclerosis and circulating cholesterol, specifically that transported by apolipoprotein-B containing lipoproteins. Among these, low-density lipoprotein (LDL) has been irrefutably identified as a key driver of atherosclerosis [2]. Consequently, lowering serum LDL-cholesterol (LDL-C) is an effective strategy for mitigating cardiovascular (CV) risk [3]. Whilst lifestyle changes such as dietary modifications can contribute to LDL-C reduction, their impact is typically modest. Pharmacological treatments have thus played a pivotal role in addressing this modifiable risk factor. Statins have been the cornerstone of LDL-C-lowering treatment since the late 20th century, and remain first-line agents due to their proven efficacy and well-defined safety profile. However, recent pharmaceutical advancements targeting lipid metabolism have expanded the therapeutic landscape. This article provides an overview of LDL-C-lowering therapies, focusing on established treatments and emerging strategies poised to reshape the future of CV risk management.
Cholesterol can be derived from dietary sources or synthesised intracellularly before being packaged and transported in the blood. However, due to its hydrophobic nature and biological reactivity, cholesterol cannot be transported in its free state. Instead, lipoproteins act as carriers to transport cholesterol in its esterified form.
LDL forms part of the endogenous cholesterol transport pathway, facilitating delivery of cholesterol from the liver to peripheral tissues [4]. Each LDL particle contains a single apolipoprotein-B (ApoB) molecule, which stabilises LDL and functions as a receptor ligand, enabling its cellular uptake and metabolism. Binding of ApoB to the LDL receptor (LDLR) triggers receptor-mediated endocytosis; free cholesterol is then released via lysosomal degradation. The cholesterol content within LDL particles, referred to as LDL-C, is routinely measured in clinical laboratories and serves as the principal biomarker used to guide therapy.
Excess LDL can be cleared through endocytosis mediated by the hepatic LDLR, effectively removing it from the circulation. Expression of the LDLR is regulated by a negative feedback mechanism in response to intracellular cholesterol levels, where reduced intracellular cholesterol stimulates LDLR gene transcription. The LDLR is also negatively regulated by proprotein convertase subtilisin/kexin type 9 (PCSK9), which targets the LDLR for degradation. LDL metabolism is also regulated at numerous points within the lipoprotein pathway, including its interaction with other lipoproteins.
Over the past century, the relationship between cholesterol and cardiovascular disease has gradually been elucidated. In the early 1900s, examination of human blood vessels identified cholesterol-rich plaques, whilst a similar phenotype was observed in rabbits fed high-cholesterol diets [5]. Advances in lipoprotein research followed; after LDL was first isolated using ultracentrifugation in the late 1940s, Gofman et al. [6] demonstrated that individuals with a history of myocardial infarction often exhibited a pattern of elevated LDL and reduced high-density lipoprotein (HDL) concentrations. By the mid-to-late 1900s, large-scale epidemiological studies began demonstrating clear correlations between dietary cholesterol intake and increased risk of heart disease [7].
Since then, a multitude of genetic, clinical, and interventional studies have strengthened the argument for a causal link between LDL and atherosclerosis. Compelling results, some of which are explored below, have led the European Atherosclerosis Society to conclude that this evidence “unequivocally establishes that LDL causes ASCVD” [8].
LDL contributes to atherosclerosis primarily through ApoB-mediated retention and accumulation of LDL in the subendothelial space of the arterial intima [9]. Interaction of ApoB with extracellular matrix and local cells promotes retention and inflammation, leading to endothelial dysfunction and consequently atherosclerotic plaque formation. Although LDL represents the majority of measured ApoB, other ApoB-carrying lipoproteins share atherogenic properties with LDL. These include very-low-density lipoprotein (VLDL), intermediate density lipoprotein and lipoprotein(a), with each lipoprotein particle containing one ApoB protein.
Published in 1961, key findings from the seminal Framingham Heart Study demonstrated a positive dose-response relationship between increasing LDL-C and ASCVD risk [10]. This association has been consistently replicated in prospective studies, with meta-analysis of such studies confirming a log-linear relationship between LDL-C and ASCVD risk [11].
Familial hypercholesterolemia, an inherited autosomal co-dominant disorder, is characterised by increased LDL-C and ASCVD risk. Studies of this condition and its varying genotypes support that ASCVD risk is proportional to lifetime LDL-C burden (i.e., absolute level and duration of exposure) [12, 13]. Mendelian randomisation studies examining common polymorphisms known to influence circulating LDL-C concentration have provided clear evidence that lifetime exposure is crucial; the lower the LDL-C, the lower the ASCVD risk [14].
Randomised controlled trials (RCTs) assessing the effects of a wide range of LDL-C-lowering therapies consistently demonstrate a reduction in ASCVD risk, with risk reduction directly proportional to magnitude of LDL-C-lowering [3]. In light of this evidence, international guidelines are increasingly adopting a ‘lower is better’ approach for LDL-C targets, particularly in patients at high risk of CV events [15, 16, 17].
Over the past several decades, a number of LDL-C-lowering therapies have become available. These range from well-established agents, such as statins, to newer drug classes targeting alternative molecular pathways in lipid metabolism. The key pharmacologically relevant pathways are illustrated in Fig. 1, alongside the molecular targets of major current and emerging therapies discussed in this review. The following section outlines their mechanisms of action, clinical efficacy, and positioning within lipid management guidelines, with key characteristics summarised in Table 1 (Ref. [18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44]).
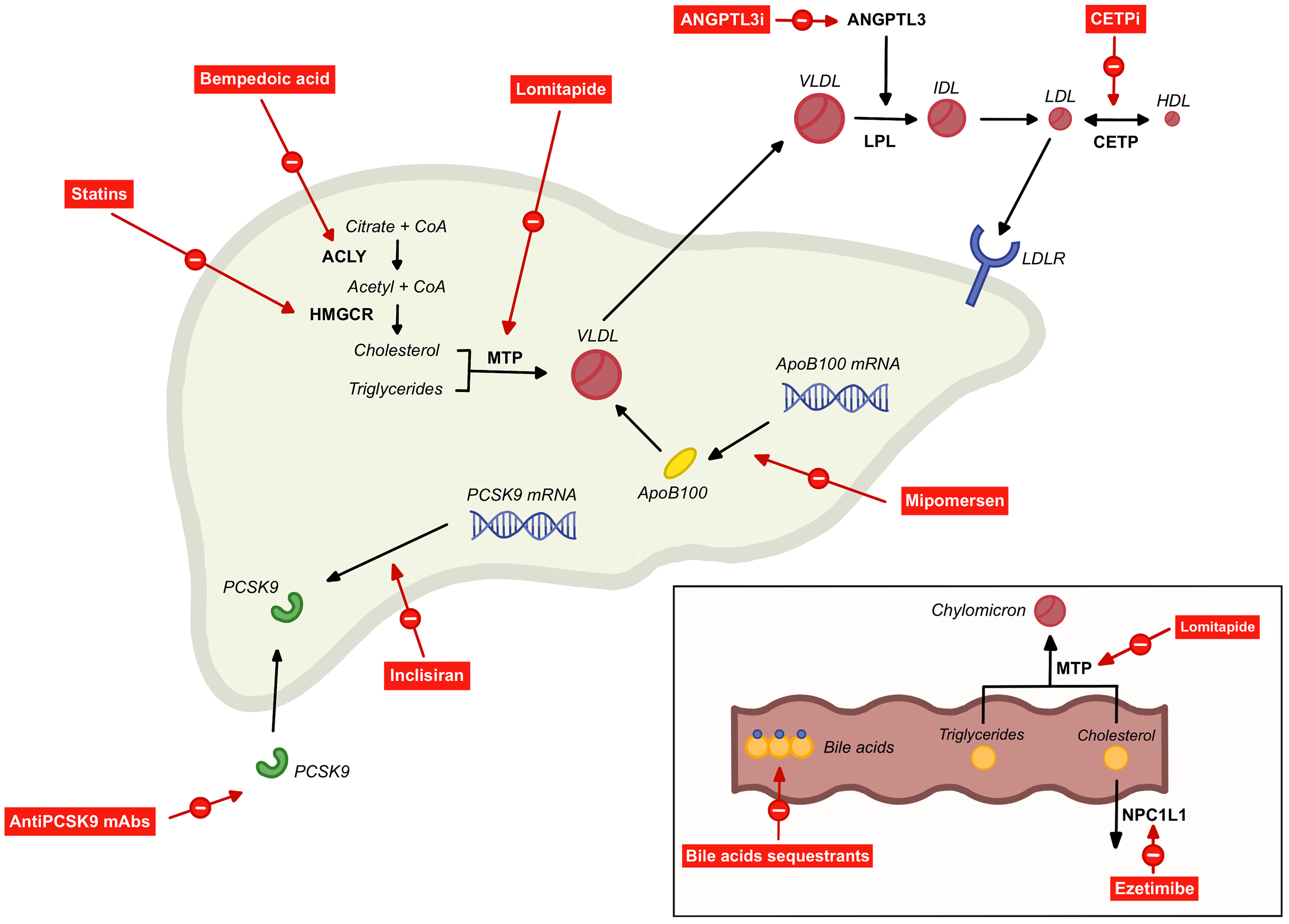 Fig. 1.
Fig. 1.
Mechanisms of low-density lipoprotein (LDL)-cholesterol-lowering therapies. Statins - inhibit the rate-limiting enzyme in hepatic cholesterol synthesis, 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR). Bempedoic acid - inhibits an upstream enzyme in the same pathway, adenosine triphosphate-citrate lyase (ACLY). Ezetimibe - blocks the intestinal cholesterol transporter Niemann-Pick C1-Like 1 (NPC1L1). Bile acid sequestrants - bind bile acids in the intestinal lumen and prevent their reabsorption. Anti-PCSK9 mAbs - bind circulating proprotein convertase subtilisin/kexin type 9 (PCSK9). Inclisiran - small interfering RNA targeting PCSK9 messenger RNA (mRNA). Lomitapide - inhibits microsomal triglyceride transfer protein (MTP) in the small intestine and liver. Angiopoietin-like 3 protein inhibitors (ANGPTL3i) - enhance lipoprotein lipase (LPL) activity, increasing catabolism of very-low-density lipoprotein (VLDL). Mipomersen - antisense oligonucleotide targeting ApoB-100 mRNA. Cholesteryl ester transfer protein inhibitors (CETPi) - inhibit cholesteryl ester transfer protein (CETP) action, reducing transfer of cholesteryl esters from high-density lipoprotein (HDL) to LDL. Figure was created using Procreate (version 5.4.4; Savage Interactive Pty Ltd., Hobart, Tasmania, Australia). The authors have no financial or personal relationship with Procreate, and the use of this tool does not imply any endorsement. CoA, coenzyme A; ANGPTL3, angiopoietin-like 3 protein; IDL, intermediate-density lipoprotein; mAb, monoclonal antibody; LDLR, LDL receptor.
| Target | Drug | Additional therapy (statin/ezetimibe/maximally tolerated lipid-lowering therapy [MTLLT]) | Approx. expected LDL-C reduction (additional reduction denoted by ‘+’ where in combination) | Regulatory status (where ‘licensed’ indicates regulatory approval for use in US/EU/UK) | Dosing route and typical frequency |
| HMG-CoA reductase inhibition | Statin | - | 20–50% (see Table 2) | Licensed | Oral tablet, once daily |
| ACLY inhibition | Bempedoic acid | - | 20% [18] | Licensed | Oral tablet, once daily |
| + statin | + 15% [19] | ||||
| + ezetimibe | + 10–20% [20] | ||||
| NPC1L1 inhibition | Ezetimibe | - | 20% [21] | Licensed | Oral tablet, once daily |
| + statin | + 15% [22] | ||||
| Bile acid sequestrants | Cholestyramine | - | 11% [23] | Licensed | Oral tablet or powder, multiple doses daily |
| Colestipol | - | 16–27% [24] | |||
| Colesevelam | - | 9–18% [25] | |||
| Collectively | + statin | + 16% [26] | |||
| PCSK9 protein mAbs | Evolocumab | - | 50–60% [27] | Licensed | Subcutaneous injection, once every 2–4 weeks |
| + statin | + 50–70% [28] | ||||
| Alirocumab | + statin | + 50–70% [29] | Licensed | ||
| PCSK9 siRNA | Inclisiran | + MTLLT | + 50% [30] | Licensed | Subcutaneous injection, once every 6 months |
| CETP inhibition | Dalcetrapib | + MTLLT | No significant effect [31] | Never marketed (lack of efficacy) | Oral tablet, once daily |
| Evacetrapib | + MTLLT | + 31% [32] | Never marketed (lack of efficacy) | ||
| Anacetrapib | + statin | + 17% [33] | Never marketed (manufacturer decision) | ||
| Obicetrapib | + MTLLT | + 30% [34] | Not yet licensed. Phase 3 CVOT ongoing (PREVAIL) | ||
| MTP inhibition | Lomitapide | - | 20–30% [35] | Licensed for homozygous FH | Oral capsule, once daily |
| + MTLLT | + 40–50% [36, 37] | ||||
| ApoB inhibition | Mipomersen | + MTLLT | + 36% [38] | US license granted in 2013. Since discontinued by manufacturer | Subcutaneous injection, once weekly |
| ANGPTL3 mAb | Evinacumab | + MTLLT | + Up to 50% [39] | Licensed for homozygous FH | Intravenous infusion, once every 4 weeks |
| ANGPTL3 siRNA | Vupanorsen | + statin | + 8–16% [40] | Never marketed (efficacy and safety concerns) | Subcutaneous injection |
| Zodasiran | + statin | + 15–20% [41] | Not yet licensed. Phase 2 trials completed | ||
| Anti-PCSK9 oral peptide | Enclitide | + MTLLT | + 40–60% [42] | Not yet licensed. Phase 3 CVOT ongoing | Oral tablet, once daily |
| Anti-PCSK9 vaccine | AT04A | - | + 11–13% [43] | Not yet licensed. Early phase 2 trial underway | Subcutaneous injection |
| Anti-PCSK9 small binding protein | Lerodalcibep | + MTLLT | + 60% [44] | Licensing applications in progress | Subcutaneous injection, once monthly |
HMG-CoA, 3-hydroxy-3-methylglutaryl-coenzyme A; ApoB, apolipoprotein B; PCSK9, proprotein convertase subtilisin/kexin type 9; siRNA, small interfering RNA; NPC1L1, Niemann-Pick C1-Like 1; mAb, monoclonal antibody; CETP, cholesteryl ester transfer protein; MTP, microsomal triglyceride transfer protein; ACLY, adenosine triphosphate-citrate lyase; LDL-C, low-density lipoprotein-cholesterol; CVOT, cardiovascular outcome trial; FH, familial hypercholesterolaemia.
Since lovastatin was licensed in the 1980s, statins have become one of the most widely prescribed medications worldwide [45]. As shown in Fig. 1, these drugs competitively inhibit the rate-limiting enzyme in the cholesterol synthesis pathway, 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase [46]. Primarily exerting their effects in the hepatocyte, subsequent reduction in intracellular cholesterol metabolites leads to upregulation of the LDLR. This enhances the uptake of ApoB-containing lipoproteins from the circulation, resulting in decreased circulating LDL. The LDL-C-lowering effect of statins varies dependent on dose and type used. Broadly, statins are categorised according to their ‘intensity’, as determined by their LDL-C-lowering effect (Table 2, Ref. [15]).
| Intensity (LDL-C-lowering) | Drug (dose) |
| High ( |
Atorvastatin (40–80 mg) |
| Rosuvastatin (20–40 mg) | |
| Moderate (30–49%) | Atorvastatin (10–20 mg) |
| Rosuvastatin (5–10 mg) | |
| Simvastatin (20–40 mg) | |
| Pravastatin (40–80 mg) | |
| Lovastatin (40–80 mg) | |
| Fluvastatin (80 mg) | |
| Low ( |
Simvastatin (10 mg) |
| Pravastatin (10–20 mg) | |
| Lovastatin (20 mg) | |
| Fluvastatin (20–40 mg) |
ACC, American College of Cardiology; AHA, American Heart Association.
3.1.1.1 Major CV Outcome Trials
Spanning decades, multiple cardiovascular outcome trials (CVOTs) for different statin types and doses have been published. Meta-analysis of 26 randomised trials by the Cholesterol Treatment Trialists’ Collaboration, utilising individual participant data from over 170,000 participants, demonstrates that LDL-C-lowering with statin therapy reduces the relative risk of major CV events by around one-fifth for each 1 mmol/L reduction in LDL-C [3]. This is observed in a wide range of patient groups, and is largely independent of baseline LDL-C concentration with no evidence within the trials studied of a lower threshold at which benefits diminish [3, 47, 48].
3.1.1.2 Guidelines
Due to the wealth of evidence supporting the safety and efficacy of statin therapies, European Society of Cardiology (ESC) [16], American College of Cardiology/American Heart Association (ACC/AHA) [15] and UK National Institute for Health and Care Excellence (NICE) guidelines [17] universally recommend statins as first-line lipid-lowering therapy for primary hypercholesterolaemia and prevention of major cardiovascular events in at-risk groups.
3.1.1.3 Use in Practice
Statin therapy is widely acknowledged to be a cost-effective treatment for preventing ASCVD [49]. Concerns have been raised regarding side effects of statin therapies; however randomised evidence shows they are generally well tolerated [50]. The so-called “nocebo” effect appears to contribute to perceived adverse effects, whereby symptoms are attributable to patient expectations rather than the drug itself [51]. Although statins can, in rare cases, cause a clinically significant myopathy, including rhabdomyolysis, which occurs at an estimated rate of 1–3 cases per 100,000 patient-years, the widespread belief that statins commonly cause muscle symptoms is largely unsupported by evidence [52]. Meta-analysis of blinded, placebo-controlled trials indicates that over 90% of reported muscle symptoms are not attributable to statin use [52]. Statins are, however, associated with a small rise in blood glucose and a modest excess risk of new-onset diabetes, which predominantly occurs in individuals already close to the diagnostic threshold [53]. Additionally, statin therapy is known to be associated with mild, typically transient elevations in liver enzymes [54]. Whilst the clinical relevance of this remains unclear, severe liver injury caused by statin therapy is exceedingly rare [55]. Given their well-established benefits in reducing ASCVD risk, the overall benefit-risk profile of statins strongly favours their use in the majority of patients.
Bempedoic acid is a novel LDL-C-lowering therapy, acting via a similar mechanism to statins by modulating the intracellular cholesterol synthesis pathway [56]. This medication inhibits the enzyme adenosine triphosphate-citrate lyase (ACLY), an enzyme upstream of HMG-CoA reductase, with the effect of lowering intracellular cholesterol, upregulating the LDLR and increasing hepatic uptake of circulating LDL (Fig. 1). As monotherapy, bempedoic acid achieves a modest LDL-C reduction of around 20% [18, 57]. When added to statin therapy, an additional 15% LDL-C reduction compared with statin alone can be achieved [19, 58]. Pairing bempedoic acid with ezetimibe (in the absence of a statin) can result in a combined LDL-C reduction of around 40–50% [20].
3.1.2.1 Major CV Outcome Trials
To date, one major CVOT of bempedoic has been published. The CLEAR-outcomes trial randomised 13,970 patients with either established ASCVD or with high CV risk, and a history of statin intolerance, to bempedoic acid versus placebo for around 3.4 years [18]. Treatment with bempedoic acid resulted in a 13% relative risk reduction in major adverse cardiovascular events (MACE), a 4-point composite defined as CV death, nonfatal myocardial infarction (MI), nonfatal stroke and coronary revascularisation (hazard ratio [HR] 0.87; 95% confidence interval [CI] 0.79–0.96; p = 0.004).
3.1.2.2 Guidelines
ESC [16] and ACC/AHA [15] guidelines were both published prior to the 2020 approval of bempedoic acid by the European Medicines Agency (EMA) and Food and Drug Administration (FDA). However, the medication is now licensed for primary hypercholesterolaemia, with or without a history of CV disease, if LDL-C is not sufficiently controlled with statin therapy or if statin therapy cannot be used. In NICE guidelines, bempedoic acid is recommended with ezetimibe for hypercholesterolaemia, alongside lifestyle changes, if statins cannot be used and/or ezetimibe does not control LDL-C sufficiently [59].
3.1.2.3 Use in Practice
As a prodrug, bempedoic acid is converted to its active metabolite, bempedoyl-coenzyme A, by an enzyme which is not present in skeletal muscle [60]. Advantages over statin therapy are thus argued to include a lower likelihood of muscle symptoms, which has been demonstrated in clinical trials [60]. Metabolites of bempedoic acid are believed to competitively inhibit a common renal tubular transporter for uric acid excretion, leading to increased serum uric acid levels and an increased risk of gout in patients receiving this treatment [61].
Ezetimibe, first licensed in 2002, acts to inhibit the Niemann-Pick C1-Like 1 (NPC1L1) transporter in the small intestine [62]. NPC1L1 is the principal intestinal transporter mediating both dietary cholesterol absorption and re-absorption of cholesterol excreted in bile. Inhibition of this transporter leads to reduced cholesterol delivery to the liver, increasing hepatic uptake of LDL from the circulation via upregulated expression of the LDLR. As monotherapy, ezetimibe lowers LDL-C by around 20% [21]. When prescribed with concomitant statin therapy an additional 15% reduction may be achieved compared to statin alone [22]. Notably, individuals with relatively high absorption of dietary cholesterol, which could be explained by genetic variations, may experience even greater LDL-C reduction with ezetimibe [63].
3.2.1.1 Major CV Outcome Trials
There has been one major CVOT evaluating ezetimibe. The IMPROVE-IT trial randomised 18,144 participants with recent acute coronary syndrome to simvastatin + placebo versus simvastatin + ezetimibe [64]. After 6 years median follow-up, the addition of ezetimibe resulted in a modest 6.4% risk reduction in the primary endpoint, a 5-point MACE composite comprising CV death, non-fatal MI, unstable angina requiring hospitalization, coronary revascularization, and non-fatal stroke (HR 0.936; 95% CI 0.89–0.99; p = 0.016).
Complementing findings from the IMPROVE-IT trial, observational data from the SWEDEHEART registry has also suggested potential benefit from early ezetimibe initiation following MI [65]. Among 35,826 statin-treated patients after MI, starting ezetimibe within 12 weeks was associated with lower rates of MACE (composite of all-cause mortality, non-fatal MI, or non-fatal stroke), compared with late or no initiation. Using target-trial emulation methods, delayed (13 weeks–16 months) and no initiation was associated with 14% (HR 1.14; 95% CI 0.95–1.41) and 29% (HR 1.29; 95% CI 1.12–1.55) higher 3-year MACE risks respectively. While residual confounding inherent to observational analyses cannot be fully eliminated, and may lead to overestimation of benefit, these registry data support the principle that lower and earlier is better for LDL-C treatment, particularly after MI.
3.2.1.2 Guidelines
ESC [16], ACC/AHA [15] and NICE guidelines [17] universally recommend ezetimibe as an addition to statin therapy for treating primary hypercholesterolaemia where maximally tolerated statin monotherapy has not adequately controlled LDL-C. Where statin therapy is contraindicated or not tolerated, ezetimibe monotherapy is advocated for lipid-lowering.
3.2.1.3 Use in Practice
Ezetimibe is considered to be cost-effective for ASCVD prevention [66]. It is well tolerated with few reported side effects and is therefore a relatively safe, if modestly effective, option for LDL-C-lowering [67].
Bile acid sequestrants, comprising cholestyramine, colestipol and colesevelam, were among the first drugs approved for hypercholesterolemia management. They bind negatively charged bile acids in the gut, forming insoluble complexes that are excreted, preventing their recycling via enterohepatic circulation (Fig. 1) [68]. This depletion triggers hepatic bile acid synthesis, which requires cholesterol, thereby reducing intracellular cholesterol levels. Consequently, LDLR expression increases to restore cholesterol, leading to a reduction in circulating LDL. As monotherapy, bile acid sequestrants lower LDL-C levels by approximately 10% to 20%, depending on the specific agent and dose used [23, 69]. When added to statin therapy, they may confer an additional LDL-C reduction of around 16% [26].
3.2.2.1 Major CV Outcome Trials
Only one CVOT assessing bile acid sequestrants has been conducted; the Lipid
Research Clinics Coronary Primary Prevention Trial [70]. This primary prevention
trial recruited a population of 3800 men, who were randomised to cholestyramine
24 g/day versus placebo for around 7.4 years. Results demonstrated a 19%
relative risk reduction in the primary endpoint of coronary heart disease death,
or non-fatal MI (7.0% vs 8.6%; risk ratio [RR]
3.2.2.2 Guidelines
Likely reflecting the minimal RCT evidence and poor tolerability (see following section, ‘Use in Practice’), recommendations for bile acid sequestrants are limited. ESC [16] and AHA/ACC [15] suggest consideration when LDL-C remains suboptimal despite all other therapeutic options. In contrast, NICE advises prescription of bile acid sequestrants for patients with familial hypercholesterolaemia, if they are intolerant to other lipid-lowering therapies [71].
3.2.2.3 Use in Practice
Benefits of bile acid sequestrants include their low cost and established safety profile. Unlike many lipid-lowering therapies, they may be considered for cholesterol management in pregnant and paediatric populations [72]. However, a major limitation is the high incidence of gastrointestinal side effects, which contribute to poor tolerance, with up to 70% of patients discontinuing treatment within a year [73]. Also restricting their use, bile acid sequestrants may bind other medications in the gut which may impair their absorption and complicate dosing regimens [72]. Additionally, certain susceptible patients can experience severe hypertriglyceridaemia when taking these medications, necessitating caution in patients with elevated baseline triglycerides [72]. As a result, bile acid sequestrants have largely been superseded by more potent LDL-C-lowering agents with more robust CV outcome data.
First described in 2003, the PCSK9 protein binds the LDLR and targets it for lysosomal degradation, thus preventing its recycling to the cell membrane (Fig. 2) [74]. Gain-of-function mutations in PCSK9 cause familial hypercholesterolemia, with decreased LDLR expression, raised circulating LDL-C and increased ASCVD risk [75]. Conversely, loss-of-function mutations, as shown in Mendelian randomization studies, lead to lower LDL-C and reduced ASCVD risk [75]. These insights established PCSK9 as an important therapeutic target for LDL-C-lowering.
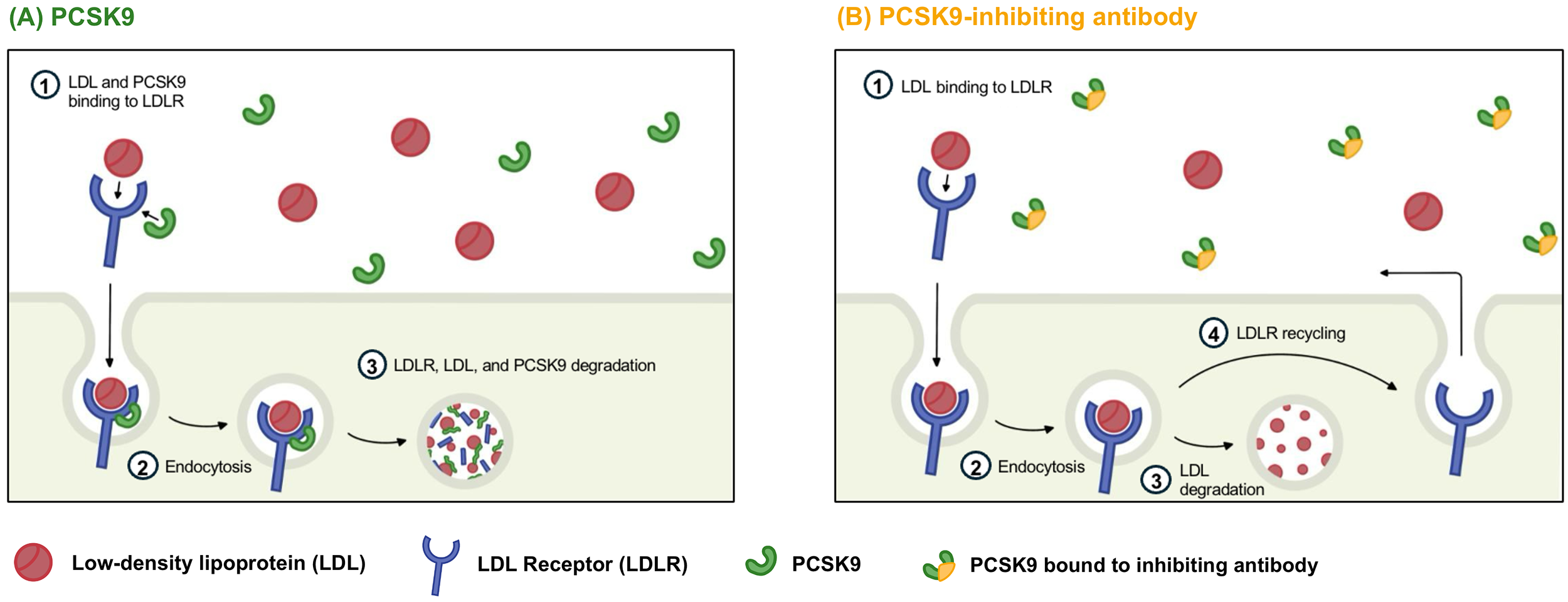 Fig. 2.
Fig. 2.
Proprotein convertase subtilisin/kexin type 9 (PCSK9) action and therapeutic inhibition. (A) PCSK9 inactivates the low-density lipoprotein receptor (LDLR) by binding to the receptor (1), targeting it for lysosomal degradation. The LDLR is then endocytosed (2) and degraded (3), preventing its normal recycling to the cell surface membrane. (B) PCKS9-inhibiting (monoclonal) antibody therapies bind PCSK9 in the circulation, preventing its binding to the LDLR. Circulating LDL is free to bind the LDLR (1), which is then endocytosed (2). LDL is subsequently degraded intracellularly (3) and, in the absence of PCSK9, the LDLR is recycled to the cell surface membrane (4). Figure was created using Procreate (version 5.4.4; Savage Interactive Pty Ltd., Hobart, Tasmania, Australia). The authors have no financial or personal relationship with Procreate, and the use of this tool does not imply any endorsement.
Designed to bind and modulate disease-related targets, monoclonal antibody (mAb) therapies are engineered, cloned and administered therapeutically. The first mAb against PCSK9 was licensed in 2015, and currently, two options—evolocumab and alirocumab—are available. These medications bind and inactivate the PCSK9 protein, allowing LDLR recycling to the hepatocyte cell membrane and increasing LDL uptake from the circulation (Fig. 2). When used in combination with a statin, PCSK9 mAb therapy can provide an additional LDL-C reduction of 50–70% [28, 29].
3.3.1.1 Major CV Outcome Trials
Both available PCSK9 mAb therapies have been evaluated in CVOTs. The FOURIER
trial randomised 27,564 patients with pre-existing ASCVD to evolocumab versus
placebo [28]. Patients also took maximally tolerated statin therapy. After
follow-up of 2.2 years, treatment with evolocumab resulted in a 15% reduction in
the relative risk of MACE, defined as a 5-point composite of CV death, MI, stroke,
hospitalisation for unstable angina and coronary revascularisation (HR 0.85; 95%
CI 0.79–0.92; p
3.3.1.2 Guidelines
ESC guidance [16] recommends addition of a PCSK9 inhibitor in ‘very-high-risk’
primary or secondary CV prevention patients (based on ASCVD history and/or risk
factors), with or without familial hypercholesterolaemia, if specific LDL-C
targets are not met with maximally tolerated lipid-lowering therapy (at minimum
3.3.1.3 Use in Practice
Although generally well tolerated, PCSK9 mAb therapies are associated with an
increased risk of injection site reactions. A meta-analysis of 32 trials reported
a relative risk ratio of 1.54 for such reactions (95% CI 1.38–1.71; p
Novel small interfering RNA (siRNA) therapies act to suppress protein expression by inhibiting messenger RNA (mRNA) translation. Inclisiran is currently the only licensed siRNA treatment for LDL-C management, targeting PCSK9 mRNA in the liver and reducing its synthesis. When added to maximally tolerated lipid-lowering therapy, it provides an additional approximately 50% reduction in LDL-C levels [30]. This is modestly lower than the PCSK9 monoclonal antibodies (approximately 60% reduction), likely because around 20% of circulating PCSK9 originates from extra-hepatic tissues not targeted by inclisiran [79].
3.3.2.1 Major CV Outcome Trials
No major CVOTs have yet been published, however the ORION-4 phase 3 trial has now completed recruitment, with results expected in 2027 [80]. This trial is evaluating inclisiran versus placebo in around 15,000 participants with established ASCVD, with average follow-up scheduled for five years. Results will provide crucial data on CV outcomes and long-term safety. A similar secondary prevention trial, VICTORION-2-PREVENT, is also underway [81].
3.3.2.2 Guidelines
Inclisiran received EMA and FDA approval for treatment of hyperlipidaemia in
2020 and 2021 respectively, and is therefore not included in the most recent ESC
[16] or ACC/AHA [15] guidance at the time of writing. In contrast, inclisiran is
recommended by NICE for people with established ASCVD and elevated LDL-C
(
3.3.2.3 Use in Practice
Inclisiran is administered infrequently at 6-monthly intervals. It is also well tolerated; pooled analysis of phase 3 trials comprising 3655 individuals reported injection site reactions as the only notable side effect, at a rate of 5% (n = 91) amongst inclisiran-treated participants versus 0.7% (n = 12) in the placebo group (RR 7.54; 95% CI 4.14–13.71) [83]. Convenient dosing and a favourable safety profile may improve long-term adherence compared with oral therapies such as statins. However, as with many newer agents, cost may restrict its global accessibility.
3.3.3.1 Oral Peptide Therapies
Easily administered tablet forms of PCSK9 inhibitors are currently in development. Among these, the peptide enclitide (until recently named MK-0616) is the most advanced candidate, demonstrating a promising 61% LDL-C reduction in a phase 2 trial [42]. A phase 3, randomised, double-blind, placebo-controlled CVOT trial—CORALreef outcomes—is currently underway to further evaluate its efficacy and safety [84].
3.3.3.2 Small Binding Protein Therapies
Lerodalcibep is a PCSK9-binding fusion protein (adnectin-albumin), mechanistically similar to anti-PCSK9 mAb therapy, and is administered once monthly by subcutaneous injection. License applications are underway after initial phase 3 efficacy and safety trials have demonstrated significant LDL-C reduction at approximately 55–60% beyond background lipid-lowering therapy, and a favourable safety profile [44].
3.3.3.3 Vaccines
Vaccines designed to modulate the immune system towards a self-antigen, targeted at PCSK9, may offer sustained reductions in LDL-C with a more convenient dosing schedule than existing injectable PCSK9 inhibitor therapies. A phase 1 trial of a proposed therapy, AT04A, showed modest LDL-C-lowering and is being evaluated in a phase 2 trial [43]. Further studies will establish if this approach will prove successful.
3.3.3.4 Gene Editing
A novel approach with wide-ranging therapeutic prospects, gene editing technologies targeting PCSK9 are currently undergoing early phase trials. Verve therapeutics have developed drugs which employ gene editing techniques with lipid nanoparticle delivery systems [85]. Preliminary results from a first-in-human trial of the candidate VERVE-101 reported significant LDL-C reduction, however the trial was halted after safety concerns arose [86]. A second therapy, VERVE-102, has recently been evaluated in a phase 1 trial. According to a company press release, pending peer-reviewed publication, the therapy achieved a mean LDL-C reduction of 53% and demonstrated a favourable short-term side effect profile [87].
Cholesteryl ester transfer protein (CETP) transfers cholesteryl esters from HDL to ApoB-expressing lipoproteins, in exchange for triglycerides [88]. Genetic studies have demonstrated that loss-of-function CETP mutations are associated with a favourable lipid profile, characterised by elevated high-density lipoprotein-cholesterol (HDL-C) and modestly reduced LDL-C, with lower ASCVD risk [89]. Multiple CETP inhibitors have been developed and evaluated in clinical trials, however none are currently licensed for use. Although initially hypothesised that their ASCVD protective effects were due to increased HDL, mounting evidence suggests any clinical benefit is more likely to be secondary to changes in LDL-C and ApoB [90].
Efficacy in LDL-C-lowering has shown variability across types of CETP inhibitors. Dalcetrapib, an early CETP inhibitor, has negligible LDL-C-lowering effect [31]. In contrast, the REVEAL trial demonstrated that anacetrapib reduced LDL-C by 17% when added to statin therapy [33]. Evacetrapib leads to around 30% LDL-C-lowering when administered with a statin [32]. Obicetrapib, the newest agent, achieved around 30% LDL-C reduction when added to maximally tolerated lipid-lowering therapy in the BROADWAY trial [34].
Major CV Outcome Trials
Four major CVOTs evaluating CETP inhibitors have been published. The earliest of these, ILLUMINATE, enrolled 15,067 participants who were randomised to torcetrapib versus placebo [91]. However, the trial was terminated early due to an unexpected increase in mortality and major CV events, which were attributed to off-target effects of the drug. Subsequent trials of the CETP inhibitors dalcetrapib and evacetrapib, in dal-Outcomes and ACCELERATE respectively, were stopped early for futility and as such reported no significant difference in CV outcomes [31, 32]. It is possible that these latter two trials had insufficient follow-up duration (at around 2–2.5 years) to observe a potentially delayed benefit of treatment that might be expected with lipid modifying therapy. Supporting this, the REVEAL trial of anacetrapib had a median follow-up of 4.1 years, and demonstrated a 9% relative risk reduction in 3-point MACE, defined as coronary death, MI and coronary revascularisation (HR 0.91; 95% CI 0.85–0.97; p = 0.004) [33]. This benefit only emerged after approximately two years of treatment with anacetrapib, but continued to be observed in long-term follow-up [92]. However, the manufacturer concluded that the clinical profile for anacetrapib did not support regulatory filings. Obicetrapib, the newest agent, was recently evaluated in the phase 3 BROADWAY trial which demonstrated a favourable safety profile, with no significant difference in adverse event rates between the obicetrapib and placebo groups [34]. The ongoing PREVAIL CVOT has recruited 9541 participants with established ASCVD, randomised to obicetrapib versus placebo [93]. Results from this study will show whether promising LDL-C reductions translate into clinical benefit, whilst also providing long-term safety data.
Microsomal triglyceride transfer protein (MTP) is crucial for the assembly and secretion of ApoB-containing lipoproteins, facilitating the production of chylomicrons in enterocytes and VLDL in the liver [94]. In the rare inherited condition abetalipoproteinaemia, loss of function mutations in the MTP gene result in a distinct lipid profile with markedly reduced levels of ApoB-containing lipoproteins, including LDL [95]. This provided the rationale for targeting MTP inhibition therapeutically, ultimately leading to the development of lomitapide, currently the only available medication in this class. Trials have focused on homozygous familial hypercholesterolaemia (hoFH) patients, where lomitapide has demonstrated an LDL-C reduction of approximately 50% in both adult and paediatric populations when added to background lipid-lowering therapy [36, 37]. Smaller trials in non-hoFH patients have shown that lomitapide, when used as monotherapy, can achieve an LDL-C reduction of around 30% [35].
3.4.2.1 Guidelines
Lomitapide is licensed in the USA, Europe and UK for patients with hoFH only. It is not recommended or licensed for LDL-C-lowering in other patient groups and is therefore not included in generic CV risk guidelines.
3.4.2.2 Use in Practice
Due to its mechanism of action distinct from the LDLR, lomitapide is considered for patients with hoFH and null LDLR mutations, a group of patients with severe hypercholesterolaemia often refractory to traditional lipid-lowering therapies [95]. However, studies have reported significant gastrointestinal side effects, liver enzyme abnormalities, fat-soluble vitamin deficiencies and hepatic steatosis with lomitapide treatment [96]. Patients taking this medication require close monitoring of liver function and are advised to follow a very low-fat diet, reducing enterocyte chylomicron generation, in an attempt to mitigate gastrointestinal side effects. Long-term liver effects have not yet been defined, and continue to be monitored in a dedicated post-marketing registry [97]. These issues may explain why lomitapide has not been pursued in a broader population for LDL-C reduction, with no large-scale CVOTs having been published.
Angiopoietin-like 3 protein (ANGPTL3) plays an important role in lipid metabolism by inhibiting both lipoprotein lipase and endothelial lipase [98]. Targeted therapies designed to block ANGPTL3 aim to remove this inhibition, thereby enhancing lipase activity (Fig. 1). This accelerates VLDL metabolism, which is hypothesised to enable more rapid clearance of LDL and related lipoproteins via an LDLR-independent mechanism.
Evinacumab is a mAb targeting ANGPTL3, administered via monthly intravenous infusion. Clinical trials have primarily focused on patients with hoFH, with phase 3 studies demonstrating sustained LDL-C-lowering of approximately 45% [99]. One phase 2 trial in 96 patients with refractory hypercholesterolaemia has been published, demonstrating similar LDL-C-lowering to hoFH, however evidence in broader populations is not yet available [39].
Vupanorsen, an antisense oligonucleotide therapy targeting ANGPTL3, was evaluated in a phase 2 trial of 286 participants, demonstrating an LDL-C reduction of 8–16% [40]. However, dose-dependent elevations in transaminases and hepatic steatosis were observed in this study, leading to discontinuation of the drug’s development prior to licensing due to safety concerns. Since then, an alternative siRNA molecule, zodasiran, has undergone phase 2 trials demonstrating an LDL-C reduction of around 20% at a 200 mg dose, with no significant difference in liver-related adverse events or laboratory values reported [41]. Phase 3 trials are anticipated.
3.4.3.1 Guidelines
Evinacumab is licensed in the USA, Europe and UK for patients with hoFH only but is not recommended or licensed for LDL-C-lowering in other patient groups and is therefore not included in generic CV risk guidelines. Vupanorsen and zodasiran are not currently licensed for clinical use.
3.4.3.2 Use in Practice
With a mechanism of action independent of the LDLR, evinacumab may be a useful adjunct to other LDL-C-lowering therapies, particularly in patients with hoFH due to LDLR loss-of-function mutations. Phase 3 data indicate that evinacumab is generally well tolerated, with adverse and serious adverse events occurring at similar rates to placebo [39, 99]. However, trial populations have been relatively small, which may limit detection of rarer harms. Its use is currently limited to patients with hoFH, with trials having focused on this patient group. This limited scope may reflect the logistical, financial and safety challenges associated with intravenous infusion administration, particularly when other equally effective LDL-C-lowering therapies are available.
As previously discussed, ApoB is a key driver of atherosclerosis. Mipomersen, an antisense oligonucleotide, inhibits ApoB mRNA translation in hepatocytes, thereby reducing the production of ApoB-containing lipoproteins. Early trials demonstrated promising results, with an estimated LDL-C reduction of around 25% [100]. However, safety concerns, specifically elevated liver enzymes, hepatic steatosis and flu-like symptoms, led to high treatment discontinuation rates. Despite FDA approval in 2013 for the treatment of hoFH, the drug was rejected by the European Medicines Agency on safety grounds and has since been withdrawn by the manufacturer [100].
Conclusive evidence has established LDL-C reduction as a cornerstone of atherosclerotic cardiovascular disease prevention. Large-scale randomised trials have demonstrated that lowering LDL-C through diverse therapeutic strategies translates into significant reductions in cardiovascular risk. While statins remain the foundation of lipid management due to their efficacy, cost-effectiveness, and well-established safety profile, newer agents provide additional options for patients with inadequately controlled LDL-C despite statin therapy, or those unable or unwilling to take statins. The emergence of novel long-acting approaches, such as RNA-based therapies and vaccines, holds promise for sustained and potent LDL-C reduction. Integrating these advances into clinical practice, while ensuring accessibility and long-term safety, is likely to have a significant impact on the global burden of cardiovascular disease.
• LDL-C is an important modifiable risk factor for atherosclerotic cardiovascular disease (ASCVD), a leading global cause of morbidity and mortality.
• Evidence supports the principle that “lower is better” with regards to LDL-C-lowering.
• Established therapies such as statins and ezetimibe are effective first-line treatments, with well-defined safety profiles and robust evidence for cardiovascular risk reduction.
• Emerging treatments, including established PCSK9 inhibitors and early-phase therapies targeting novel pathways and mechanisms, offer promising new directions for LDL-C-lowering.
• Future care will likely combine established and novel therapies to optimise ASCVD risk reduction.
Not applicable.
RH was responsible for the literature review and manuscript writing. LB contributed to the conception and design of the review and interpretation of the evidence. Both authors contributed to revising the manuscript critically for important intellectual content. Both authors read and approved the final manuscript. Both authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
The authors thank Zoe Bowman for preparing the figures.
This research received no external funding.
LB has received unrestricted grants to the University of Oxford for running clinical trials from Novartis, Novo Nordisk, British Heart Foundation and Medical Research Council. Oxford Population Health has an explicit policy of not accepting any personal honoraria payments directly or indirectly from the pharmaceutical and food industries. RH has no conflicts of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.