




 , Lijun Zhu 1, Yunmei Yang 1, Qin Zhang 1,*
, Lijun Zhu 1, Yunmei Yang 1, Qin Zhang 1,*
1 Department of Geriatrics, The First Affiliated Hospital, Zhejiang University School of Medicine, 310003 Hangzhou, Zhejiang, China
Abstract
Congenital adrenal hyperplasia (CAH) results from 21-hydroxylase deficiency (21-OHD), which is the most frequent form of CAH and often presents with atypical symptoms. Patients with non-classic 21-OHD (NC-21-OHD) are particularly susceptible to diagnostic challenges, including misdiagnosis and underdiagnosis. This study reports a case of NC-21-OHD and underscores the associated challenges in its diagnosis, treatment, and clinical management.
This study reports a 56-year-old male patient who presented with fatigue lasting over a year. Initial ultrasound revealed a hypoechoic mass above the left kidney, and further evaluation, including renal tumor evaluation and computed tomography angiography, indicated thickening of the adrenal glands and multiple lesions. Endocrinological assessment revealed reduced luteinizing and follicle-stimulating hormone levels, along with elevated dehydroepiandrosterone sulfate and abnormal corticotropin, ultimately diagnosing the patient with primary adrenal insufficiency. Furthermore, genetic screening identified heterozygous mutations in the CYP21A2 (cytochrome P450, family 21, sub-family A, polypeptide 2) gene, confirming CAH due to NC-21-OHD. The patient was treated with hydrocortisone at a dosage of 20 mg twice a day.
Hydrocortisone therapy resulted in a significant alleviation of fatigue symptoms and a substantial reduction in 17α-hydroxyprogesterone (17-OHP) levels, thereby enhancing the patient’s confidence in disease management.
This case emphasizes the significance of early recognition of nonspecific symptoms, prompt diagnosis, and timely treatment in managing CAH to enhance the overall quality of life and reproductive health in affected individuals.
Keywords
- congenital adrenal hyperplasia
- 21-hydroxylase deficiency
- 17α-hydroxyprogesterone
- fatigue
- case report
Congenital adrenal hyperplasia (CAH) is a group of autosomal recessive disorders
resulting from enzyme deficiencies in the adrenal steroidogenesis pathway. Among
these, 21-hydroxylase deficiency (21-OHD) is the most common type, accounting for
approximately 95% of cases [1]. The clinical spectrum of CAH varies widely; in
particular, non-classic CAH (NCCAH) poses significant diagnostic challenges
attributed to its subtle endocrine manifestations and subclinical impairments in
spermatogenesis; therefore, early diagnosis and management are crucial to prevent
long-term complications, such as infertility and metabolic disorders [1, 2].
Diagnosis of NCCAH depends on a combination of biochemical and genetic
evaluations, including elevated 17
In this case report, we present a 56-year-old male patient diagnosed with CAH secondary to non-classic 21-OHD (NC-21-OHD), prepared following the case report (CARE) framework (Supplementary Material). This case is particularly noteworthy due to the patient’s atypical presentation, including infertility and fatigue, accompanied by significant endocrine abnormalities. Identifying such cases is essential, as they highlight the need for clinicians to consider CAH in adults presenting with unexplained symptoms such as fatigue, skin pigmentation changes, and reproductive issues, or adrenal masses [5, 6]. The significance of this case lies in its potential to enhance awareness of CAH as a differential diagnosis in adult patients, particularly those with a history of infertility and endocrine dysfunction. Furthermore, it underscores the significance of genetic screening and detailed family history in understanding the hereditary nature of this disorder, which can inform tailored treatment strategies and ultimately improve patient outcomes [7].
One year ago, a 56-year-old Chinese male from Zhejiang province first experienced fatigue, which he did not consider crucial and did not seek any medical attention. Over the past month, his fatigue symptoms progressively aggravated and he visited Xiaoshan Mercy Hospital, where an ultrasound examination revealed a hypoechoic mass above the left kidney. He was advised to consult the department of urology and undergo an adrenalectomy if necessary.
Subsequently, the patient independently sought medical attention at the First Affiliated Hospital, Zhejiang University School of Medicine. Renal tumor evaluation and computed tomography angiography (CTA) showed thickening of the right adrenal gland and multiple lesions in the left adrenal gland, meeting the criteria for adrenalectomy (Fig. 1). During consultation, the patient incidentally mentioned the recent development of cutaneous hyperpigmentation, prompting targeted endocrine evaluation. Therefore, the patient was admitted to the geriatric department for further assessment and management.
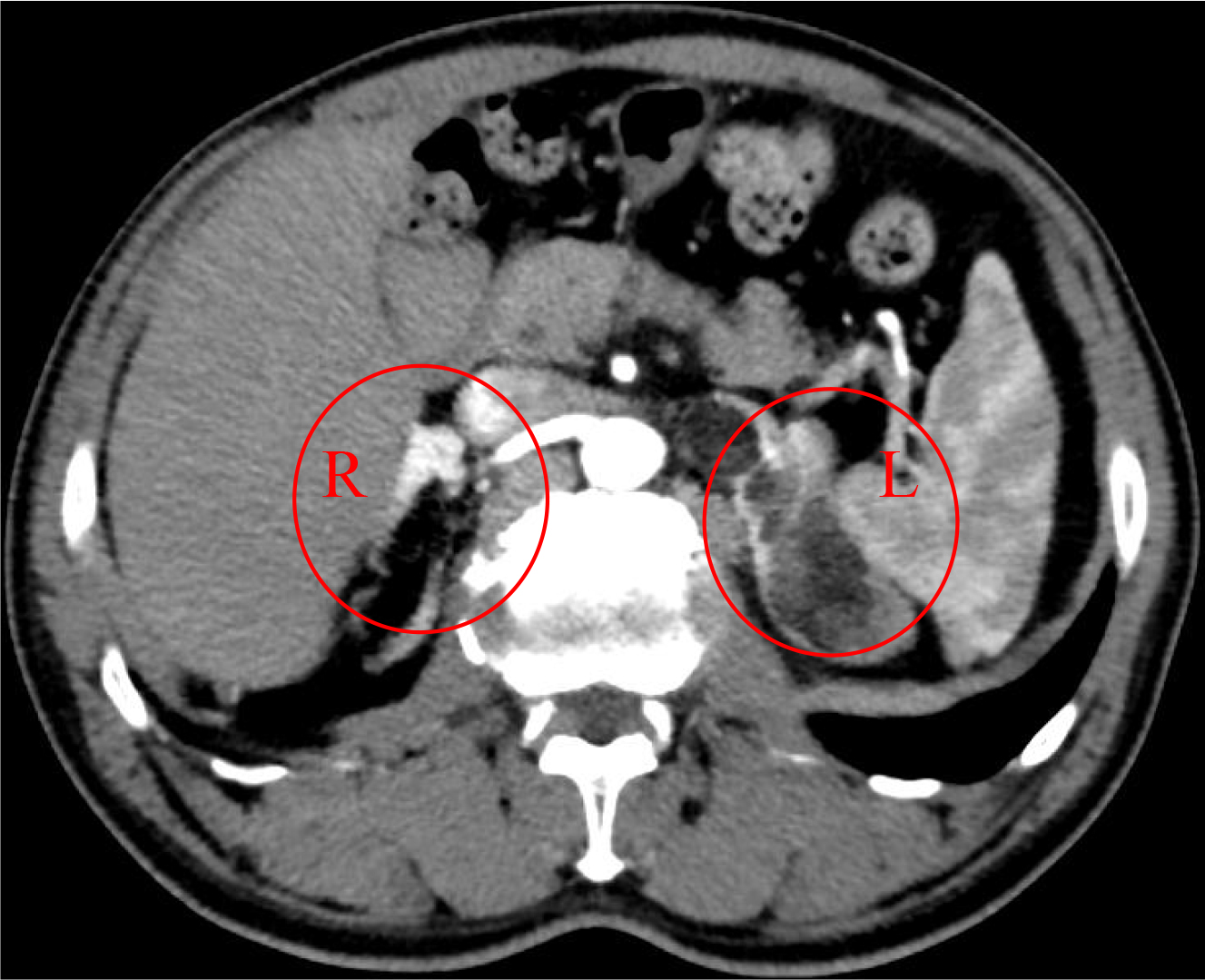 Fig. 1.
Fig. 1.
Renal tumor evaluation and computed tomography angiography (CTA). Thickening of the right adrenal gland and multiple lesions in the left adrenal gland (circles) were observed.
Both parents of the patient were deceased, preventing assessment of any family history of endocrine disorders. He has two married sisters with no living children.
On admission, we recorded the patient’s height (154 cm), body weight (53.6 kg), and body mass index (BMI, 22.6 kg/m2), with his height remaining unchanged since age 12. He has experienced a weight loss of 5 kg over the past year. Vital signs showed a heart rate of 66 beats/minute, a body temperature of 36.3 °C, and a blood pressure of 100/46 mmHg. Physical examination revealed cutaneous hyperpigmentation and genital atrophy, although pubic hair was observed. Furthermore, the patient also reported reduced sexual desire. However, cardiovascular, respiratory, and abdominal assessment showed no observable abnormalities. Laboratory examinations and clinical assessments revealed impaired renal function, with a serum creatinine (Cr) of 112 µmol/L and estimated glomerular filtration rate (eGFR) of 62.5 mL/min (Table 1).
| Peripheral Blood | |||||
| Parameter | Value | Reference range | Parameter | Value | Reference range |
| Red blood cells | 4.55 |
4.09–5.74 | Lymphocytes | 38% | 20.0–40.0 |
| Hemoglobin | 147 g/L | 131–172 | Monocytes | 5.8% | 3.0–10.0 |
| White blood cells | 4.28 |
4.0–10.0 | Eosinophils | 1.9% | 0.5–5.0 |
| Neutrophils | 53.8% | 50.0–70.0 | Platelets | 175 |
83–303 |
| Blood Biochemistry | |||||
| Parameter | Value | Reference range | Parameter | Value | Reference range |
| Total protein | 66.5 g/L | 65.0–85.0 | HDL-C | 0.81 mmol/L | 0.78–1.81 |
| Albumin | 43.3 g/L | 40.0–55.0 | BUN | 4.26 mmol/L | 3.10–8.00 |
| Total bilirubin | 23.3 µmol/L | 20.0–40.0 | UA | 378 µmol/L | 208–428 |
| AST | 8 U/L | 9–50 | Cr | 112 µmol/L | 57–97 |
| ALT | 12 U/L | 15–40 | eGFR | 62.5 mL/min | |
| 12 U/L | 10–60 | Electrolytes | |||
| ALP | 74 U/L | 45–125 | Potassium | 3.65 mmol/L | 3.50–5.30 |
| LDH | 172 U/L | 120–250 | Sodium | 146 mmol/L | 137–147 |
| Plasma glucose | 4.06 mmol/L | 3.90–6.10 | Chloride | 107 mmol/L | 99–110 |
| Triglyceride | 2.09 mmol/L | 0.30–1.70 | Calcium | 2.29 mmol/L | 2.11–2.52 |
| Cholesterol | 3.31 mmol/L | 3.14–5.86 | Phosphorus | 1.23 mmol/L | 0.85–1.51 |
| LDL-C | 1.45 mmol/L | 1.31–3.29 | |||
| Endocrine Markers | |||||
| Parameter | Value | Reference range | Parameter | Value | Reference range |
| TSH | 1.131 mIU/L | 0.35–4.94 | Estradiol | 26.42 pg/mL | 11.0–44.0 |
| FT3 | 4.66 pmol/L | 2.43–6.01 | FSH | 0.60 mIU/mL | 0.95–11.95 |
| FT4 | 13.15 pmol/L | 9.01–19.05 | LH | 0.04 mIU/mL | 0.57–12.07 |
| GH | 0.07 ng/mL | 0.0–3.0 | Prolactin | 12.07 ng/mL | 3.46–19.40 |
| IGF-1 | 156 ng/mL | 45.0–210.0 | Pregnanediol | 6.45 ng/mL | 0.0–0.2 |
| Adrenaline | 50.67 pg/mL | 14–90 | Testosterone | 864.31 ng/dL | 142.39–923.14 |
| Noradrenaline | 110.81 pg/mL | 19–121 | DHEAS | 559.2 µg/dL | 48.6–361.8 |
| Androstenedione | 0.6–3.1 | SHBG | 55.50 nmol/L | 17.1–77.6 | |
| 17-OHP | 356.19 nmol/L | FTI | 54% | 2.40–81.20% | |
| Cortisol | ACTH | ||||
| 8 AM | 5.85 µg/dL | 5–25 | 8 AM | 61.00 pg/mL | 0–46 |
| 4 PM | 3.32 µg/dL | 4 PM | 30.90 pg/mL | ||
| 30 min after | 5.07 µg/dL | 30 min after | 79.80 pg/mL | ||
| Urine Collection Test | |||||
| Parameter | Value | Reference range | Parameter | Value | Reference range |
| Cortisol | 4.3–176.0 | Aldosterone | 39.4 µg/24 h | 1.2–28.0 | |
| Potassium | 23.19 mmol/d | 25.00–100.00 | Calcium | 2.16 mmol/d | 2.50–7.50 |
| Sodium | 114 mmol/d | 130.00–260.00 | Magnesium | 2.25 mmol/d | 2.50–8.50 |
| Chloride | 88.8 mmol/d | 170.00–250.00 | Phosphorus | 14.7 mmol/d | 12.9–42.0 |
AST, aspartate aminotransferase; ALT, alanine aminotransferase;
The patient’s electrolyte levels were within normal range. Gonadotropins were
substantially suppressed, with luteinizing hormone (LH) at 0.04 mIU/mL and
follicle-stimulating hormone (FSH) at 0.6 mIU/mL. In contrast, adrenal androgen
levels were significantly increased, including dehydroepiandrosterone sulfate
(DHEAS) at 559.2 µg/dL and androstenedione
Subsequently, the diurnal fluctuations in cortisol and ACTH levels were
examined. Cortisol levels were 5.85 µg/dL at 8 AM and 3.32 µg/dL at 4
PM, whereas ACTH was elevated in the morning (61.00 pg/mL at 8 AM) and normalized
in the afternoon (30.90 pg/mL at 4 PM) (Table 1), suggesting that both cortisol
and ACTH maintained their diurnal rhythm. Following this, an exercise experiment
trial was performed to assess the levels of cortisol and ACTH after 30 min of
physical activity, serving as an adjunctive method to simulate physiological
stress and evaluate adrenal stress response capacity [8, 9]. Post-exercise
results revealed a modest increase in ACTH to 79.80 pg/mL at 8 AM, whereas the
cortisol levels did not increase, remaining lower than the baseline level at 8 AM
without exercise (5.07 µg/dL). Furthermore, 24-h urinary free cortisol
levels were reduced (
In 56-year-old males, normal testicular dimensions are generally in the range of
32–50
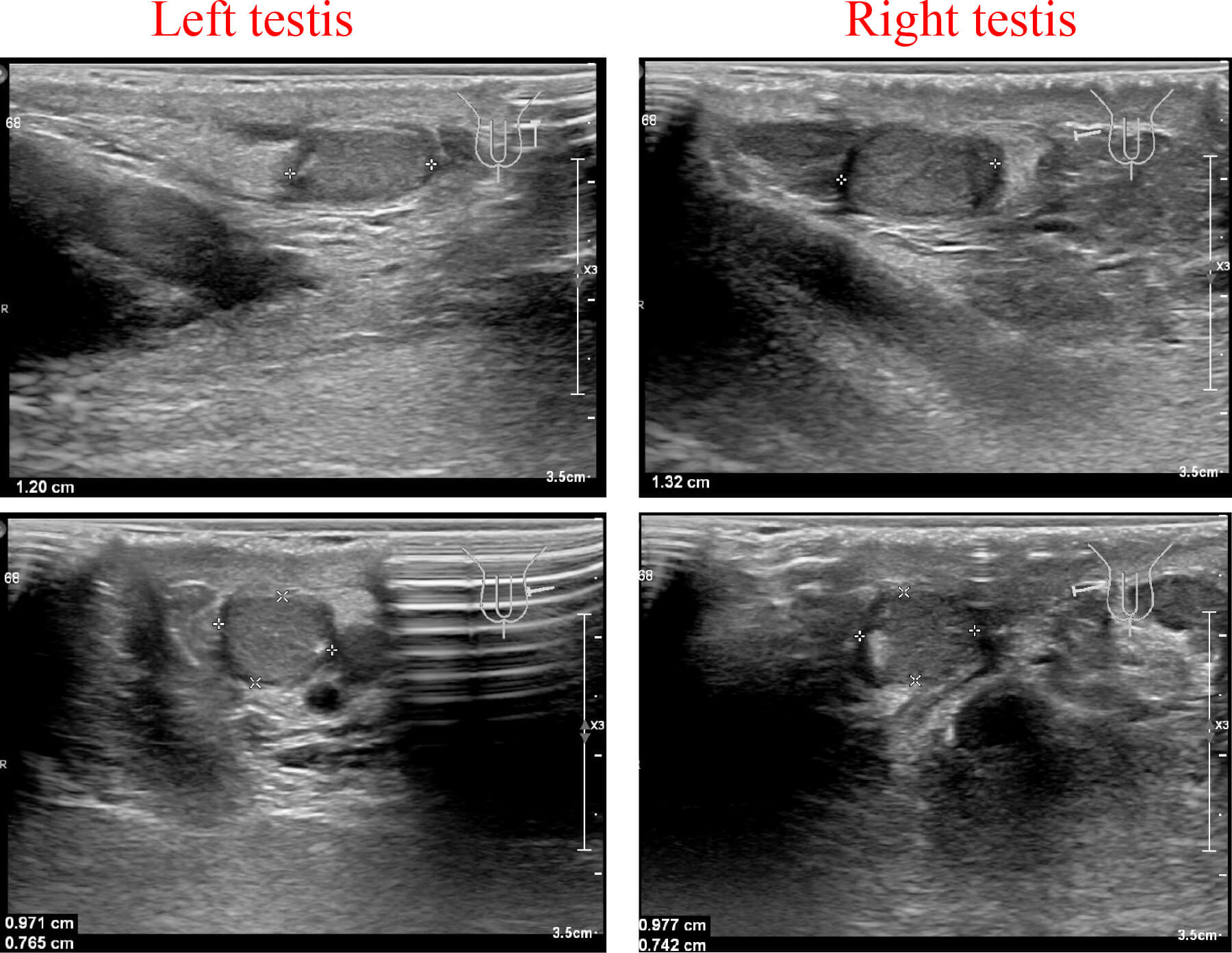 Fig. 2.
Fig. 2.
Testicular ultrasound report of the patient. Ultrasound examination showed bilateral testicular atrophy.
Additionally, enhanced magnetic resonance imaging (MRI) of the adrenal gland revealed multiple medullary lipomas in both the inner and outer limbs of the left adrenal gland, an adenoma in the outer limb of the left adrenal gland, and thickening of the right adrenal gland, consistent with adrenal hyperplasia (Fig. 3). No significant retroperitoneal lymphadenopathy was observed, and pituitary MRI did not reveal any apparent abnormalities. Based on these comprehensive clinical and imaging findings, a provisional diagnosis of CAH was established.
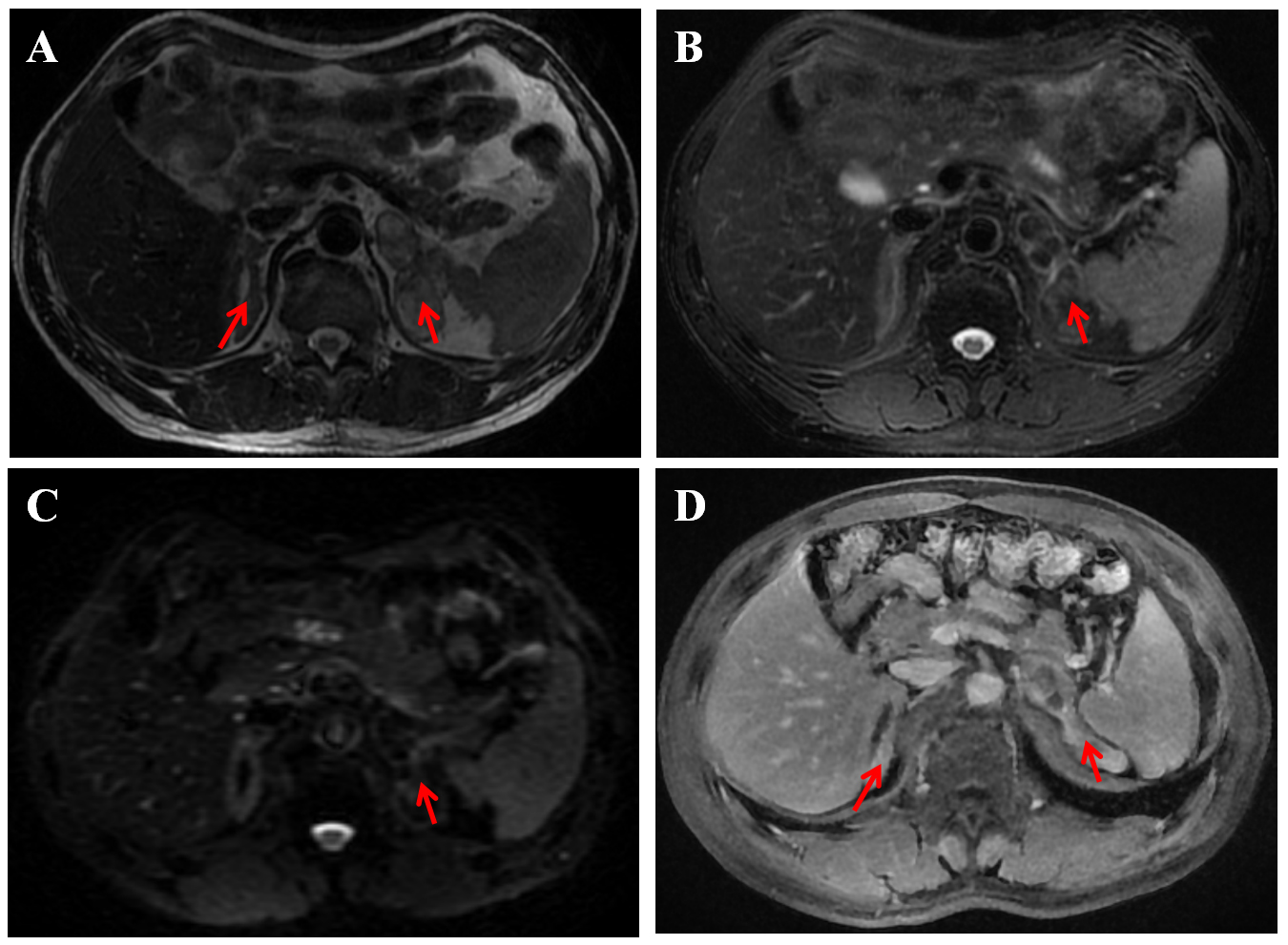 Fig. 3.
Fig. 3.
Enhanced magnetic resonance imaging of the adrenal gland. The report of the patient indicated multiple medullary lipomas within the inner and outer limbs of the left adrenal gland, an adenoma in the outer limb of the same gland, and thickening of the right adrenal gland, which is suggestive of adrenal hyperplasia. (A) On T2-weighted imaging (T2WI), the right adrenal gland is thickened and multiple abnormal masses in the left adrenal gland showed high signal intensity (the red arrows). (B) On the T2WI fat-saturated sequence, the left adrenal masses presented low signal intensity (the red arrow). (C) The diffusion-weighted imaging (DWI) signal was not high (the red arrow). (D) After contrast enhancement, the right adrenal gland showed obvious enhancement, and the left adrenal gland showed marginal enhancement (the red arrows).
The intermediate metabolites involved in steroidogenesis were also examined. As
shown in Table 1, the level of 17-OHP, a substrate of 21-hydroxylase, was
significantly elevated at 356.19 nmol/L. Chromosomal karyotyping confirmed a 46XY
karyotype. Furthermore, comprehensive whole-exome sequencing (WES) unveiled
compound heterozygous mutations in the CYP21A2 (cytochrome P450, family
21, sub-family A, polypeptide 2) gene, including a severe loss-of-function
allele (c.293-13C
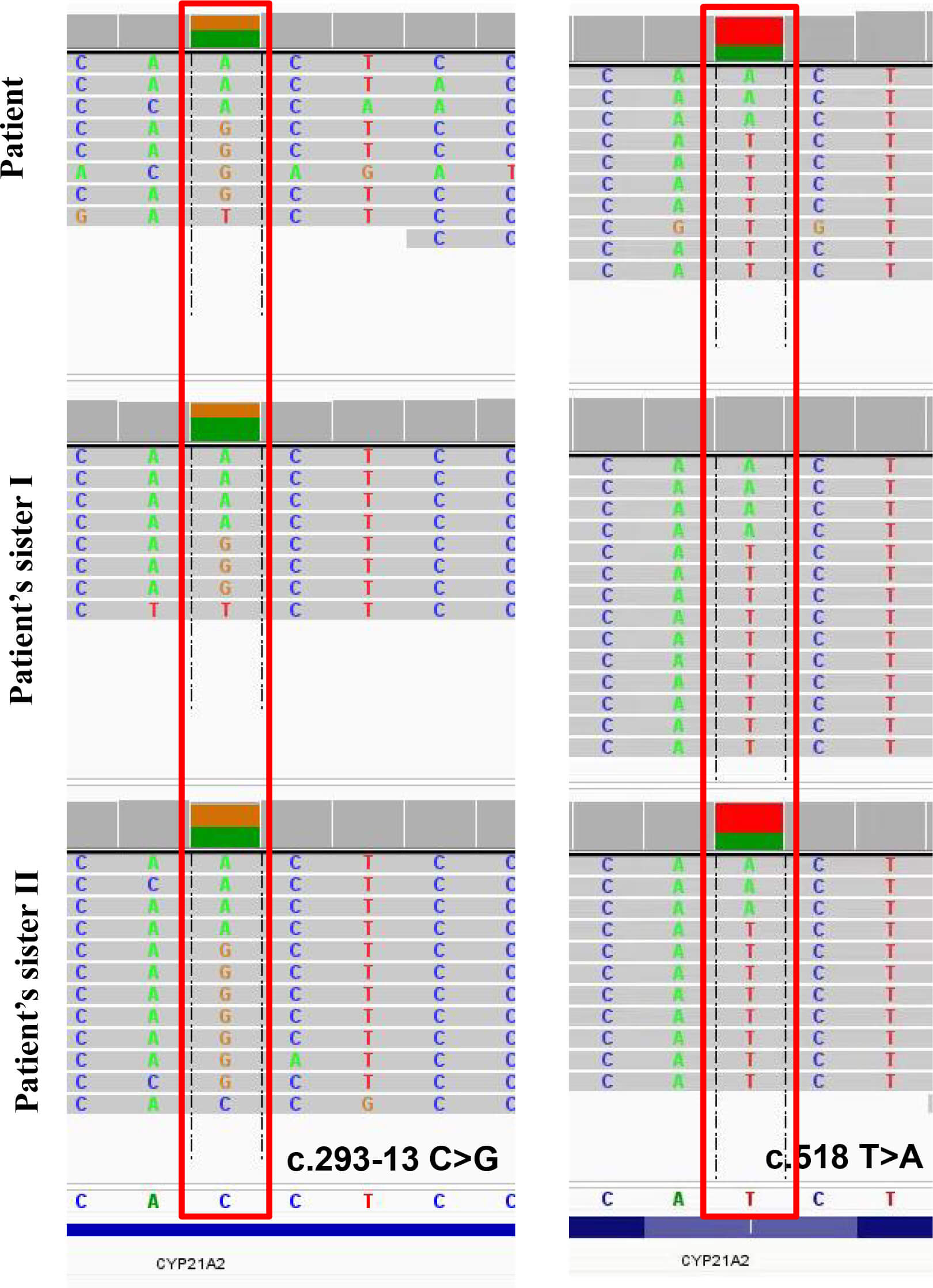 Fig. 4.
Fig. 4.
A comprehensive whole-exome sequencing of CYP21A2 (cytochrome P450, family 21, sub-family A, polypeptide 2) gene. The
following heterozygous mutations were observed: c.518T
After the diagnosis, the patient started receiving hydrocortisone 20 mg twice daily [10]. At a one-month follow-up, a significant improvement in fatigue was observed compared to baseline. Biochemically, the level of 17-OHP significantly reduced to 136.22 nmol/L, whereas cortisol and ACTH levels at 8 AM were 3.37 µg/dL and 33.10 pg/mL, respectively. The patient was advised to continue oral hydrocortisone therapy and to adhere to regular follow-up examinations.
Atypical 21-hydroxylase deficiency (21-OHD) is a milder form of CAH characterized by hyperandrogenism. In this case report, we present a 56-year-old Chinese male patient with non-classic 21-OHD (NC-21-OHD). The patient reported atypical manifestations, including growth arrest from age 12 (final height 154 cm, –3.2 Standard Deviation Score (SDS)) and persistent fatigue, accompanied by a left adrenal mass identified on imaging. Despite these outcomes, assessment for 21-OHD was not initially conducted until biochemical testing confirmed elevated 17-OHP and ACTH [11]. Integrating the hormonal profile (cortisol and ACTH 17-OHP levels), imaging findings, and genetic analysis, a diagnosis of NC-21-OHD was confirmed.
Clinical manifestations, endocrine profiles, and hormonal replacement therapy regimens vary significantly among male patients with NC-21-OHD. Gonadal function can be substantially compromised in men with CAH. A cross-sectional clinical study revealed that 14 out of 69 male patients demonstrated serum testosterone levels below the reference range [3]. For example, a 31-year-old male diagnosed with 21-OHD presented with infertility [12]. Unlike the case presented in this study, he did not show skin hyperpigmentation or genital atrophy. His ACTH level was high, cortisol was 6.9 µg/dL, and LH and FSH levels were both low, while free testosterone, estradiol, and 17-OHP levels were significantly elevated, which was consistent with the hormonal profile observed in our study. Despite administering 0.5 mg of long-acting dexamethasone, his infertility persisted, although sexual desire was moderately enhanced. Conversely, our patient received hydrocortisone 20 mg twice daily [10], leading to reduced fatigue and lowered 17-OHP levels. These findings highlight the significance of a comprehensive assessment and the development of tailored treatment plans for each male patient with NC-21-OHD.
A comprehensive preoperative endocrine evaluation should be considered standard practice for all adrenal lesions before surgical intervention, regardless of whether they are incidentally identified. This is essential to assess functional hyperplasia or tumors and to guide perioperative management. In our study, the patient was fortunate to receive a diagnosis of NC-21-OHD after a comprehensive evaluation, and hormonal therapy proved effective. This case underscores the critical importance of systematic endocrine evaluation for adrenal masses, including assessment of the cortisol axis, catecholamine secretion, and mineralocorticoid function, as these parameters directly impact surgical planning and postoperative care [6, 13]. Atypical symptoms are often misdiagnosed as metabolic diseases, such as hypothyroidism.
21-OHD results from mutations in the CYP21A2 gene, located on
chromosome 6p21.3, which encodes the 21-hydroxylase (P450c21) enzyme [14]. The
clinical manifestations and severity of 21-OHD correlate with the degree of
residual CYP21 (cytochrome P450, family 21) enzymatic activity determined
by specific allelic variant [15]. In this patient, the presence of compound
heterozygous mutations, which are a known hotspot in the Chinese population with
CAH due to 21-OHD, explains the mild biochemical phenotype, consistent with
heterozygote effects [16]. Notably, the c.518T
In this patient, the elevated 17-OHP levels (356.19 nmol/L), accompanied by low cortisol and high ACTH, are consistent with the pathophysiology of CAH, in which steroidogenesis is disrupted due to 21-hydroxylase deficiency. These biomarkers serve as critical indicators in diagnosing CAH and distinguishing it from other adrenal pathologies. The significant increase in 17-OHP supports its diagnostic potential in clinical practice [18].
Recent multi-center studies have highlighted persistent controversies regarding
the optimal basal 17-OHP cutoff for screening NC-21-OHD. Proposed thresholds
range from 1.7 to 3.19 ng/mL across different populations, each demonstrating
optimal sensitivity and specificity profiles (85–93%/92–97%) [19]. In this
context, the cutoff of 2.02 ng/mL has been implemented. A growing number of
research findings indicate that cutoff values can differ among distinct
populations and geographic regions, which highlights an urgent need for
region-specific validation attributable to ethnic differences in the CYP21A2
mutation spectrum and baseline hormonal levels [19, 20]. A threshold of 2.02
ng/mL reduces false positives compared with 1.7 ng/mL, while enhancing
sensitivity to 3.19 ng/mL. For neonatal mass screening of 21-OHD, the primary
concern remains the high false-positive rate (low positive predictive value)
[21]. However, using liquid chromatography- tandem mass spectrometry (LC-MS) has
effectively enhanced diagnostic performance [22]. Furthermore, a strong
correlation between 17-OHP and progesterone levels has been observed in NC-21-OHD
patients, particularly during the early follicular phase, when luteal phase
progesterone interference is minimal [23]. Preliminary studies reveal that an
early follicular progesterone level
Despite certain promising outcomes, our study has some limitations. Due to the lack of long-term follow-up, the sustained efficacy of hormonal therapy in this case could not be comprehensively assessed. In our future investigations, we will strive to extend the follow-up duration and systematically monitor the dynamic changes of various biochemical indicators and clinical symptoms, thereby offering a more comprehensive understanding of the long-term efficacy and prognosis of hormonal therapy in male patients with NC-21-OHD.
This case demonstrates that endocrine disorders can manifest with nonspecific symptoms such as fatigue. Clinically, vigilance for hormonal abnormalities and a comprehensive endocrine evaluation are imperative, providing a basis for routine endocrine assessment in cases with similar presentations to facilitate early diagnosis and optimal management of conditions like CAH. The diagnosis and management of NC-21-OHD remain challenging; however, multidisciplinary collaboration, standardized neonatal screening, and long-term monitoring of hormonal replacement therapy can help reduce complications, including adrenal crisis and infertility, thereby improving long-term patient prognosis.
• Routine endocrine assessment is pivotal for early diagnosis of CAH and related disorders.
• NC-21-OHD poses substantial diagnostic and management challenges, demanding heightened clinical vigilance.
•Multidisciplinary collaboration and long-term monitoring of hormonal replacement therapy can minimize long-term complications.
CAH, congenital adrenal hyperplasia; 21-OHD, 21-hydroxylase deficiency; NC-21-OHD, non-classic 21-OHD; 17-OHP, 17
The datasets used or analyzed in the current study are available from the corresponding author upon reasonable request.
SMH, WZW, YW, and JQW collected and analyzed data and wrote the manuscript. SMH, LJZ, YMY, and QZ made substantial contributions to the conception and design of the study, data acquisition, data analysis and interpretation, and contributed to the discussion. All authors contributed to revising the manuscript critically for important intellectual content. All authors have read and agreed to the published version of the manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
This study was conducted in accordance with the Declaration of Helsinki and was approved by the Ethics Committee of the First Affiliated Hospital, Zhejiang University School of Medicine (approval number: 2025B.No.0947). All methods were carried out in accordance with relevant guidelines and regulations. Written informed consent was obtained from the patient for the publication of this case report.
We would like to express our gratitude for the patient’s cooperation throughout the treatment process and for the collaborative efforts of the entire medical team.
This research received no external funding.
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/BJHM53026.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.