






 , Ricardo Madrigal-Burgaleta 2
, Ricardo Madrigal-Burgaleta 21 Department of Immunology and Immunogenetics, North Bristol NHS Trust, BS10 5NB Bristol, UK
2 Allergy & Severe Asthma Service, St Bartholomew’s Hospital, Barts Health NHS Trust, EC1A 7BE London, UK
Abstract
The term “allergy” is often used ambiguously, frequently applied to a broad spectrum of immune and non-immune reactions, which can create confusion in clinical practice. Whilst there is some controversy, the term “allergy” is now primarily reserved for describing immunoglobulin E (IgE)-mediated (Type I) hypersensitivity. Accurate diagnosis requires distinguishing Type I reactions from their mimics, especially given the clinical overlap with non-IgE-mediated conditions. This review focuses on Type I hypersensitivity, detailing its immunopathogenesis, clinical features, and diagnostic strategies, with particular emphasis on the role of serum tryptase in confirming mast cell activation. A comprehensive clinical history is crucial to distinguishing IgE-mediated allergy from its mimics, focusing on timing, symptom reproducibility, and the identification of potential triggers. Cofactors like non-steroidal anti-inflammatory drugs, alcohol, and exercise may exacerbate reactions, complicating diagnosis. Prompt intramuscular adrenaline administration is essential in cases of anaphylaxis, while patient education and specialist referral are key to long-term management. The review also examines conditions that may mimic Type I reactions, such as chronic spontaneous urticaria or isolated angioedema. In addition to classical allergy, clinicians must consider conditions such as hereditary angioedema, neuroendocrine tumours, and drug-induced pseudoallergic reactions. Recognising these mimics is vital to prevent misdiagnosis, ensuring patient safety, and avoiding unnecessary allergy labels or suboptimal management. This article provides a structured framework for evaluating suspected allergy, enhancing diagnostic accuracy, and guiding appropriate, patient-centred care across allergy and related clinical disciplines.
Keywords
- hypersensitivity
- tryptase
- urticaria
- angioedema
- mimic
Hypersensitivity reactions are common and can present across virtually all medical specialities. The diagnostic label of “allergy” is often applied when patients present with rashes, respiratory symptoms, or systemic reactions following exposure to drugs, foods, or environmental agents. However, not all such reactions are truly immunoglobulin E (IgE)-mediated. Understanding the mechanisms, clinical presentations, and management of non-IgE hypersensitivity reactions and their mimics is essential for accurate diagnosis and safe, targeted care.
Originally coined to describe altered reactivity to a stimulus, it has historically encompassed both immune responses to external allergens—such as foods, venoms, or drugs, and immune dysregulation against self-antigens, as seen in autoimmune diseases.
In modern clinical practice, allergy is frequently equated with IgE-mediated (Type I) hypersensitivity reactions [1], which are rapid in onset and can be life-threatening. However, IgE-mediated responses represent just one of several immune pathways capable of causing symptoms that mimic allergy. These mechanisms were first categorised by Gell and Coombs [2], who proposed four types (I–IV) of hypersensitivity based on the underlying pathophysiology. Recently, the European Academy of Allergy and Clinical Immunology (EAACI) expanded on the Gell and Coombs classification to include types V–VII [3]. See Table 1 for a summary.
| Type | Key effectors | Hallmark clinical features/examples | Preferred diagnostic tools | Management |
| Type I (IgE-mediated) | IgE, mast cells, basophils | Anaphylaxis, urticaria, allergic asthma, rhinitis, food allergy | Skin testing, specific IgE measurement, serum mast cell tryptase | Adrenaline (for anaphylaxis), antihistamine, corticosteroid, allergen avoidance, allergen immunotherapy (AIT), biologics (omalizumab) |
| Type II (Cytotoxic) | IgG, IgM antibodies, complement activation, antibody-dependent cellular cytotoxicity | Autoimmune haemolytic anaemia, thrombocytopenia, Goodpasture syndrome, transfusion reactions, oxaliplatin-induced immune syndrome (OIIS) | Coombs test, autoantibody detection | Immunosuppressive drugs, plasmapheresis, transfusion support as appropriate |
| Type III (Immune complex-mediated) | IgG, immune complex deposition, complement and neutrophil activation | Serum sickness, lupus nephritis, hypersensitivity pneumonitis | Low complement levels, inflammatory markers, biopsy | Corticosteroid, immunosuppressive treatment, antigen avoidance |
| Type IV (Delayed, T cell mediated) | T lymphocytes (Th1, Th2, Th17, cytotoxic T cells) | Contact dermatitis, hypersensitivity pneumonitis, coeliac disease, chronic asthma, eosinophilic esophagitis, Stevens-Johnson syndrome, toxic epidermal necrolysis | Patch testing, biopsy, lymphocyte transformation test | Antigen avoidance, corticosteroid, immunosuppressive agents, biologics |
| Type V |
T helper cells (Th1, Th2, Th17) | Asthma, chronic allergic rhinitis, atopic dermatitis, eosinophilic esophagitis, food protein-induced enterocolitis syndrome | Clinical features, inflammatory markers | Allergen avoidance, corticosteroid, immunosuppressants, biologics |
| Type VI (Metabolic-induced immune dysregulation) | Metabolic mediators (cytokines, chemokines), leading to increased airway remodelling, systemic inflammation | Obesity-associated asthma | Clinical features, inflammatory markers | Weight control, corticosteroid, biologics |
| Type VII |
Chemical substances (e.g., NSAIDs) | Allergic rhinitis, asthma, atopic dermatitis, aspirin-exacerbated respiratory disease (AERD), NSAID reactions | Clinical features, drug history | Avoidance of triggers, corticosteroid, leukotriene modifiers, biologics, aspirin desensitisation |
| Cytokine release reaction (CRR) | Cytokines (IL-6) | Fever, chills, malaise, hypotension, elevated mast cell tryptase and IL-6. Drug reactions (biologics, oxaliplatin, beta-lactams) | Serum tryptase, IL-6 levels, C-reactive protein | Tailored approaches, corticosteroid, supportive care |
This table summarises the key immunopathological mechanisms, clinical manifestations, diagnostic approaches, and treatment strategies associated with hypersensitivity reactions, categorised by Gell and Coombs Types I–IV and the extended EAACI Types V–VII. It also includes cytokine release syndrome as an example of a mixed or atypical reaction. This classification aids in the accurate diagnosis and management of allergic and allergy-mimicking conditions across clinical settings.
The
EAACI, European Academy of Allergy and Clinical Immunology; IgE, immunoglobulin E; IgG, immunoglobulin G; IgM, immunoglobulin M; IL-6, interleukin 6; NSAIDs, non-steroidal anti-inflammatory drugs.
This review will focus on Type I reactions and their mimics, their clinical presentations, diagnostic challenges, and management strategies, particularly using mast cell mediators such as serum tryptase for diagnosis. The goal is to offer clarity on how to accurately differentiate between these hypersensitivity types, ensuring that patients receive targeted, safe, and effective care. We begin by briefly outlining the different types of hypersensitivity before focusing on IgE-mediated mechanisms and their clinical expression. The subsequent sections explore diagnostic tools, the role of cofactors and hidden allergens, and emergency and long-term management. The review then turns to common and rare mimics of Type I reactions, including urticaria variants, bradykinin-mediated angioedema, and mast cell disorders. We conclude by highlighting key clinical points and proposing directions for future research and improved clinical pathways.
Type I reactions are immediate and IgE-mediated. In this type, allergen-specific IgE antibodies bind to mast cells and basophils. Upon re-exposure to the allergen, these antibodies cross-link, triggering degranulation and the release of mediators like histamine. Symptoms usually appear within seconds to minutes and include anaphylaxis, urticaria, allergic asthma, rhinitis, diarrhoea and vomiting. Diagnosis often involves skin testing, specific IgE measurement, and assessment of serum tryptase levels. Acute management focuses on adrenaline for anaphylaxis, antihistamines, corticosteroids, and allergen avoidance. Longer-term options for specific cases can include allergen immunotherapy (AIT) and biologics such as omalizumab [3].
Type II reactions are cytotoxic, mediated by IgG or IgM antibodies. These antibodies bind to cell surface antigens, leading to complement activation, phagocytosis, or antibody-dependent cellular cytotoxicity (ADCC), resulting in cell destruction, functional alteration if directed against cell surface receptors, or tissue damage. The onset of these reactions typically occurs within hours to days and includes conditions such as autoimmune haemolytic anaemia, thrombocytopenia, Goodpasture syndrome, and transfusion reactions. Diagnosis involves tests like the Coombs test and detection of autoantibodies. Treatment aims to remove the trigger, using immunosuppressive drugs, transfusion support, or plasmapheresis as necessary [3].
Type III reactions are immune complex-mediated. Soluble antigen-antibody complexes deposit in tissues, activating complement and neutrophils, causing inflammation. These reactions usually appear over days to weeks and are seen in conditions such as serum sickness, lupus nephritis, and hypersensitivity pneumonitis. Diagnosis is based on low complement levels, biopsy to identify immune complexes, and inflammatory markers. Management includes antigen avoidance, corticosteroid, and immunosuppressive treatments [3].
Type IV reactions are delayed and T cell mediated. These reactions involve antigen-specific T lymphocytes that trigger inflammation. There are several subtypes of Type IV reactions. Type IVa involves T helper 1 (Th1) cells and macrophages, causing conditions like contact dermatitis and coeliac disease. Type IVb involves Th2 cells and eosinophils, contributing to chronic asthma, atopic dermatitis, and eosinophilic esophagitis. Type IVc reactions are mediated by cytotoxic T cells and include severe conditions like Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN). Type IVd reactions are neutrophilic, as seen in acute generalized exanthematous pustulosis (AGEP). AGEP reactions have a typical onset 24–72 hours after exposure. Diagnosis is often through patch testing, biopsy, or cautiously conducted drug provocation tests. Treatment strategies include antigen avoidance, corticosteroids, immunosuppressive agents, and biologics in severe cases [3].
Type V hypersensitivity is characterised by epithelial barrier defects that impair immune regulation, leading to chronic inflammation. The breakdown of the skin or mucosal barrier allows allergens and microbes to trigger immune responses, involving T helper cells (Th1, Th2, Th17) and the loss of regulatory cells, resulting in tissue damage. Conditions like asthma, chronic allergic rhinitis, atopic dermatitis, eosinophilic esophagitis, and food protein-induced enterocolitis syndrome are linked to this mechanism. Environmental factors such as pollutants, chemicals, and allergens can disrupt the barrier, exacerbating symptoms. Clinical features include inflammation, hypersensitivity, and tissue damage in affected organs. Management focuses on allergen avoidance, corticosteroid, immunosuppressants, and biologic treatments targeting immune pathways like alarmins [3].
Type VI hypersensitivity involves metabolic-induced immune dysregulation, where conditions like obesity and diabetes influence immune responses. Obesity, particularly in asthmatic patients, exacerbates inflammation due to altered inflammatory mediators from adipose tissue and metabolic dysfunction. Increased circulating cytokines, chemokines, and reactive oxygen species contribute to airway inflammation, and gut and lung microbiome alterations are also implicated. This leads to chronic low-grade inflammation, impaired epithelial barriers, and enhanced immune responses. Clinical manifestations include obesity-associated asthma, increased airway remodelling, and systemic inflammation. Management focuses on weight control, addressing metabolic dysfunction, and targeting inflammatory pathways, with potential treatments including corticosteroid and biologics [3].
Type VII hypersensitivity involves direct cellular and inflammatory responses to chemical substances, particularly in conditions like allergic rhinitis, allergic conjunctivitis, asthma, atopic dermatitis, acute urticaria/angioedema, and drug allergies. A key example is aspirin-exacerbated respiratory disease (AERD), where non-steroidal anti-inflammatory drugs (NSAIDs) inhibit cyclooxygenase enzymes, leading to an overproduction of inflammatory mediators like cysteinyl leukotrienes, resulting in asthma, nasal polyps, and hypersensitivity. Another example involves NSAID reactions, where cross-reactivity causes respiratory or cutaneous symptoms. Recent research highlights a possible role of Mas-related G protein-coupled receptor X2 (MRGPRX2) in mediating anaphylactoid reactions to drugs like neuromuscular blockers and fluoroquinolones. A group of cationic peptidergic drugs, including icatibant, a bradykinin B2 receptor antagonist used in the management of hereditary angioedema, has been found to cause mast cell degranulation via MRGPRX2, leading to local injection site reactions [4]. Management includes avoiding triggering substances, managing symptoms with corticosteroid and leukotriene modifiers, and targeting specific immune pathways [3].
Some reactions, particularly cytokine release syndrome or ‘cytokine release reactions’, seen in drug reactions such as those triggered by biologics, exhibit mixed features that do not neatly fit into the established types. These reactions can present with chills, fever, malaise, flushing and hypotension. They can be associated with elevation in mast cell tryptase and interleukin (IL)-6, and require tailored diagnostic and therapeutic approaches [5].
IgE-mediated allergic reactions are Type I hypersensitivity reactions that develop as a consequence of a coordinated adaptive immune response, involving both T and B lymphocytes. For T cell activation to occur, antigens must first be processed and presented by professional antigen-presenting cells (APCs), which display antigenic peptides via major-histocompatibility-complex (MHC) class II molecules to the T cell receptor (TCR). This antigen-specific interaction alone is insufficient; a second, co-stimulatory signal is also required to achieve full T cell activation. Once activated, T cells proliferate and differentiate into effector subsets with specialised immune functions.
In the context of allergic sensitisation, Cluster of differentiation 4-positive
(CD4+) T helper cells—particularly the Th2 subset—produce cytokines such
as interleukin (IL)-4 and IL-13, which together are the first signal to promote
immunoglobulin class switching in B cells, leading to their differentiation into
IgE-producing plasma cells. The second signal is provided by accessory pairs of
molecules, such as
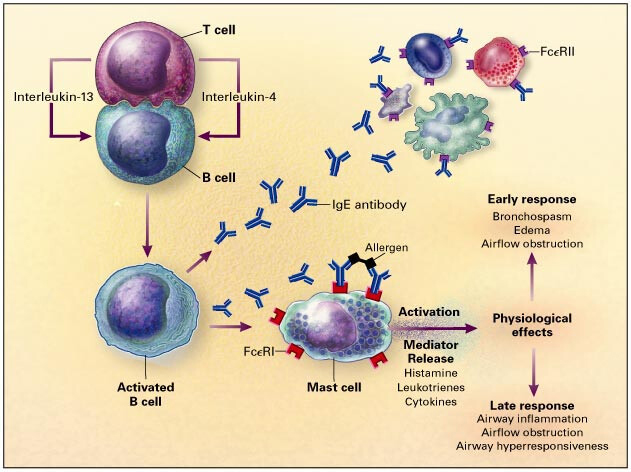 Fig. 1.
Fig. 1.
Interactions between cluster of differentiation 4 (CD4) T cells and B cells that are important
in IgE synthesis. Interleukin-4 and interleukin-13 provide the first signal to B
cells to switch to the production of the IgE isotype. The second signal
is provided by accessory pairs of molecules, such as
This sensitisation phase establishes the immunological basis for immediate hypersensitivity reactions upon subsequent contact with the relevant allergen.
In the absence of allergen, IgE bound to the surface of mast cells, basophils, or other effector cells remains inert and does not cause harm. However, when the corresponding allergen is present and binds to the surface-bound IgE, it induces cross-linking of adjacent IgE molecules. This cross-linking triggers cellular activation and initiates the allergic cascade.
Mast cells contain preformed granules packed with potent mediators, including enzymes such as tryptase and chymase, and vasoactive substances like histamine. Upon activation, these granules are rapidly released, resulting in acute physiological changes such as increased vascular permeability and smooth muscle contraction—hallmarks of the immediate phase of IgE-mediated allergy [1, 6].
In addition to releasing preformed mediators, activated mast cells secrete cytokines, including interleukin-4 (IL-4), IL-5, and IL-13, which contribute to the amplification and maintenance of the allergic response. A more delayed process involves enzymatic degradation of the mast cell lipid membrane, leading to the production of lipid-derived mediators such as leukotrienes. These further enhance vascular permeability and bronchoconstriction, contributing to the late-phase response [1, 6]. Common allergens capable of inducing such reactions include peanut, bee and wasp venom, and certain drugs.
Although IgE cross-linking is a classical activation pathway, mast cells can
also be triggered via alternative mechanisms. These include engagement of Fc
gamma (Fc
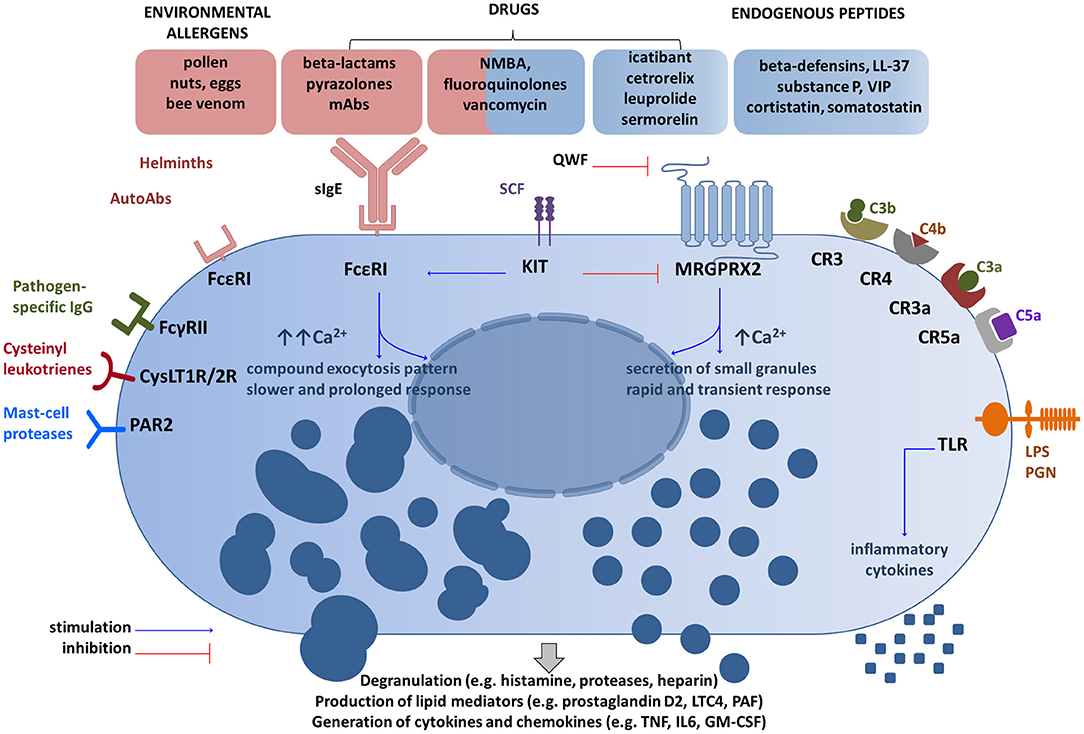 Fig. 2.
Fig. 2.
Main receptor systems and examples of ligands involved in mast
cell activation. Mast cells can be activated by a
number of receptor—ligand interactions, including allergen binding to IgE as
depicted in red, complement components binding to complement receptors as
depicted on the right of the figure, pathogen specific IgG binding to IgG
receptors and binding of drugs or endogenous peptides to the transmembrane
MRGPRX2 receptor, depicted in blue at the top of the figure. Some drugs may
activate mast cells by more than 1 mechanism. Reprinted from Porebski et al. [8], available under the terms of the CC BY 4.0
(https://creativecommons.org/licenses/by/4.0/).
Fc
The clinical history is of paramount importance in determining what is and what is not allergy (Type I hypersensitivity). When taking an allergy history, there are a few key features that should be included:
Symptoms: Are symptoms consistent with acute mast cell degranulation? Typical manifestations include urticaria, angioedema, flushing, nasal congestion, bronchospasm, gastrointestinal symptoms (e.g., nausea, vomiting, diarrhoea), and hypotension.
Timing: IgE-mediated reactions are typically rapid in onset from allergen exposure, often within minutes, and not usually delayed by more than 1–2 hours.
Attributable allergen: Are symptoms attributable to a possible allergen?
Reproducible: Symptoms should be reproducible on repeat exposure, however, this can be modified by co-factors, see below.
Biomarkers can be of help in determining whether acute symptoms are a result of
mast cell activation. Measurement of mast cell tryptase may confirm an acute mast
cell degranulating event and may identify those rare patients with an underlying
mast cell disorder [9]. Unlike histamine, which has a short
half-life, making it an impractical biomarker in the context of acute allergy,
mast cell tryptase has a half-life of approximately 2.5 hours. It is stable in
serum at room temperature and does not require any special sample handling. An
isolated normal mast cell tryptase result does not exclude allergy. A mast cell
tryptase series is most helpful, with the first sample taken after any necessary
resuscitation, with further samples at 1–2 and 24 hours or in convalescence
[10]. A dynamic change, defined as the baseline
mast cell tryptase
Cofactors can exacerbate allergy, and in some circumstances, are necessary for the development of allergic symptoms. A good example is wheat-dependent, exercise induced allergy/anaphylaxis, where the combination of exercise and wheat consumption are required to elicit an allergic response [11]. Such patients are often seen in the emergency department in advance of allergy referral having presented with systemic symptoms. Additional cofactors such as NSAID use, or alcohol consumption may modify reactions. Another example of where cofactors, including exercise and NSAID use, can exacerbate allergic symptoms is in patients with lipid transfer protein (LTP) allergy, where patients have hypersensitivity to lipid transfer proteins of plant-derived foods [12]. Unlike the oral allergy syndrome, where patients usually have mild oral allergic symptoms on exposure to certain raw fruits, raw vegetables, and some nuts in the context of pollen allergy, LTP hypersensitivity can lead to anaphylaxis. Other cofactors modifying an allergic response include stress and infection. These types of food reactions could also lead to confusion about possible NSAID hypersensitivity, if NSAID is not recognised as a cofactor.
A number of hidden allergens can lead to more atypical presentations of
IgE-mediated allergy. For example, in the medical setting, chlorhexidine allergy
is a consideration in patients having otherwise unexplained allergic reactions in
the operating theatre or following vascular or urinary catheterisation
[13]. Alpha-gal syndrome refers to hypersensitivity
to non-primate mammalian meat proteins in patients with a history of tick bite
[14]. Tick bites can sensitise the human host to the
oligosaccharide galactose-
Anaphylaxis is a severe, potentially life-threatening allergic reaction that occurs rapidly after exposure to an allergen. It is characterised by systemic mast cell and basophil activation, leading to the release of histamine and other mediators. Anaphylaxis is difficult to define, but common symptoms include hypotension, airway obstruction, urticaria, angioedema, and bronchospasm, with rapid onset within minutes to hours of exposure to the trigger. Anaphylaxis can progress quickly to cardiovascular collapse and respiratory failure, necessitating urgent treatment to prevent fatal outcomes [10].
The UK Resuscitation Council’s 2021 guidelines emphasise the rapid and systematic approach required to manage anaphylaxis, and are the recommended management pathway in the UK. The first-line treatment is intramuscular adrenaline, administered as soon as anaphylaxis is suspected. Adrenaline acts on alpha- and beta-receptors, leading to vasoconstriction, improved myocardial contractility, bronchodilation, and reduced mediator release, thus reversing the most dangerous effects of anaphylaxis. The recommended dose is 500 micrograms for adults, with a repeat dose after 5 minutes if necessary. Once adrenaline has been administered, the patient should be monitored closely, ideally in a hospital setting, as further reactions may occur. Patients must be placed in a comfortable position, which may vary depending on specific factors like pregnancy, and must not walk or stand during acute reactions [10]. See Fig. 3 (Ref. [10]).
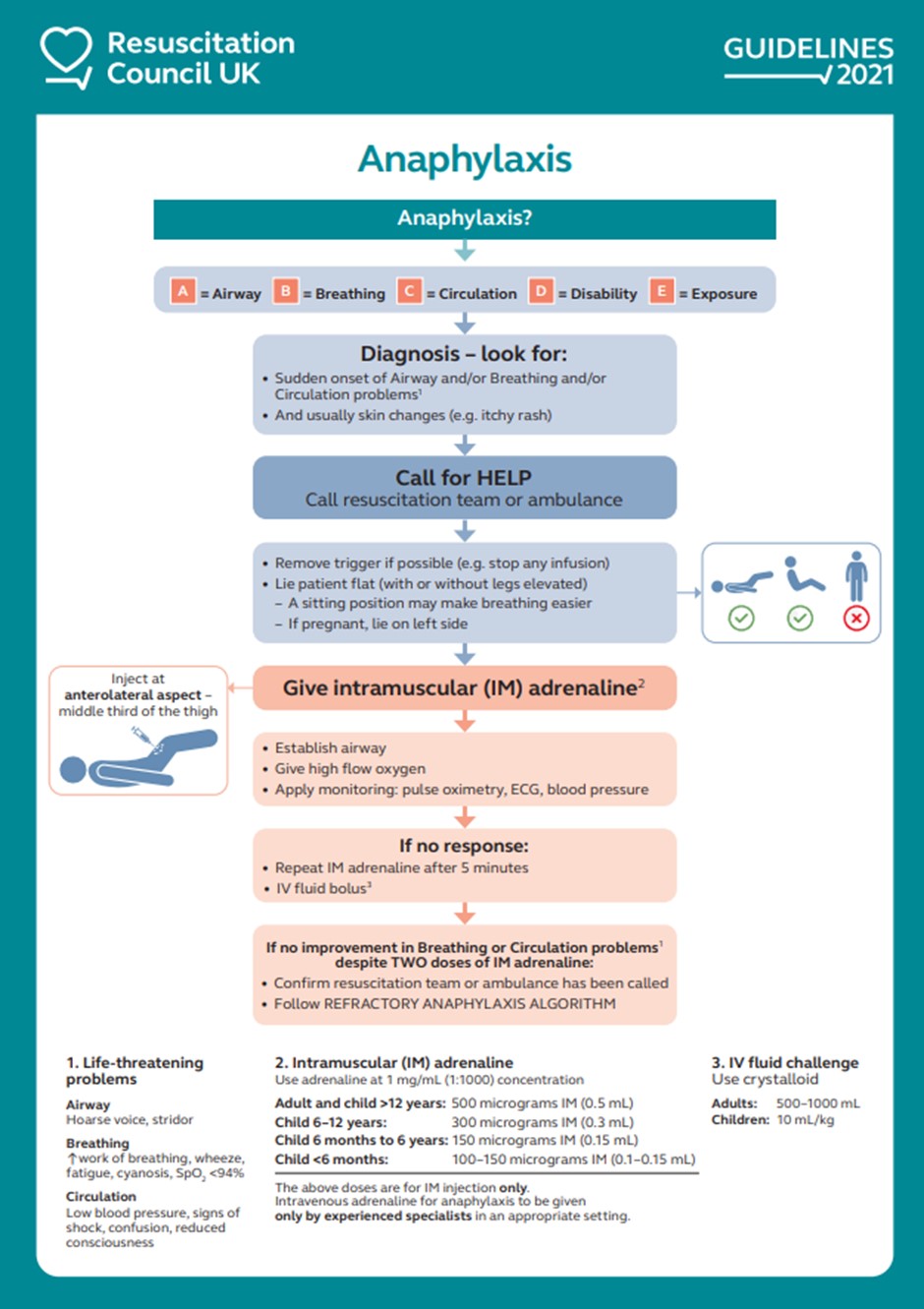 Fig. 3.
Fig. 3.
Resuscitation Council UK anaphylaxis algorithm. Reproduced from Resuscitation Council UK [10], with the kind permission of Resuscitation Council UK. IV, intravenous; ECG, electrocardiogram; SpO; Peripheral Capillary Oxygen Saturation.
The 2023 guideline for the management of anaphylaxis in the peri-operative setting emphasises early intravenous adrenaline administration in this context [15], but this is beyond the scope of this review.
Following initial resuscitation, the management focuses on stabilisation and preventing recurrence. Oxygen should be provided if necessary, and intravenous fluids may be required to manage hypotension. Antihistamines and corticosteroids are now considered second-line treatments, as they do not provide immediate benefit in reversing the acute symptoms of anaphylaxis. These medications are used to reduce symptoms like urticaria or angioedema and prevent delayed-phase reactions, but they are not life-saving in the acute setting. Steroids, while helpful for inflammation (especially in the context of asthma exacerbation and anaphylaxis), take several hours to take effect and should not delay the administration of adrenaline [10].
In the emergency department (A&E), after initial treatment with adrenaline, patients should be observed for at least 4–6 hours. Fast-tracked discharge after 2 hours can be considered for patients with good response (within 5–10 minutes) to a single dose of adrenaline given within 30 minutes of onset of reaction, who have experienced complete resolution of symptoms, have access to unused adrenaline auto-injectors and appropriate training, and have adequate supervision following discharge. The patient’s clinical history, the severity of the reaction, and the allergen involved should be reviewed to guide further management, including specialist referral to an allergy department. Ensuring the patient has an adrenaline autoinjector for future use and providing appropriate education are crucial for long-term management [10].
All patients with systemic allergy should be issued with an allergy action plan and adrenaline autoinjectors with appropriate training. Patients should be advised regarding appropriate avoidance measures, to record their allergy within their mobile medical information, carry a medical emblem confirming the allergy, and electronic patient records should be updated at the time of allergy diagnosis [10].
Patients often present for medical attention with clinical features that resemble allergy but are not allergy-related. Depending on clinical context, common pathologies such as sepsis, haemorrhage, embolus, arrhythmia, or airway manipulation need to be considered in the differential diagnosis of hypotension and/or bronchospasm [16].
Other allergy mimics presenting to the emergency department include acute exacerbation of asthma or anxiety disorder, although other features of acute systemic allergy will be absent [16]. Vocal cord dysfunction, with involuntary paradoxical adduction of the vocal cords during inspiration, can present with dyspnoea, cough, wheeze and or inspiratory stridor [17]. A careful clinical history may differentiate this from anaphylaxis. The diagnosis is confirmed by direct observation of the vocal cords by laryngoscopy.
Flushing with medication or alcohol may be diagnosed from the clinical history.
Urticaria and angioedema without additional features of IgE-mediated hypersensitivity—such as rapid onset, reproducibility, and systemic symptoms—are frequently misattributed to allergy. True acute allergic urticaria typically occurs within one hour of exposure to a specific allergen, resolves within hours (especially with appropriate treatment), and does not recur outside of allergen exposure. These IgE-mediated cases account for a minority. In contrast, most acute urticaria is non-specific, tends to persist for several days, lacks a clearly identifiable immediate trigger, and is most commonly associated with intercurrent infection.
Chronic spontaneous urticaria (CSU) is a common condition defined by the spontaneous occurrence of wheals, with or without angioedema, on most days for over six weeks. Most patients present with urticaria alone; approximately 40% also experience angioedema, and 10% present with angioedema in the absence of urticaria [18]. See the section below for further discussion on isolated angioedema. As CSU is not typically allergy-mediated, extensive allergen testing is rarely useful. In some cases, CSU is associated with autoimmunity, particularly thyroid autoimmunity.
Recognised physical triggers include temperature changes, sweating, and cutaneous pressure. Some patients with CSU experience symptom exacerbation with NSAID use and should be specifically asked about tolerance. Patients intolerant to cyclooxygenase (COX)-1 inhibitors may tolerate them once urticaria is well controlled; specialist assessment can help guide safe reintroduction. Alternatively, paracetamol or a selective COX-2 inhibitor may be considered if anti-inflammatory treatment is needed [19].
In patients with residual bruising at urticarial sites, urticarial vasculitis should be suspected. Complement levels should be measured to evaluate for hypocomplementaemic urticarial vasculitis [20]. These patients should be referred to dermatology or rheumatology for further evaluation.
Autoimmune CSU is recognised in a subset of patients, with functional IgG
autoantibodies directed against the high-affinity IgE receptors alpha subunit
(Fc
Management:
Second-generation non-sedating H1 antihistamines are the first-line treatment for CSU. According to British Society for Allergy and Clinical Immunology (BSACI) guidelines, doses may be escalated up to fourfold if standard doses are ineffective, with stepwise increases every 2–4 weeks while monitoring symptom control and side effects (see Fig. 4) [21]. Patients who remain symptomatic despite high-dose antihistamines should be referred to a specialist for consideration of add-on therapies such as omalizumab or ciclosporin.
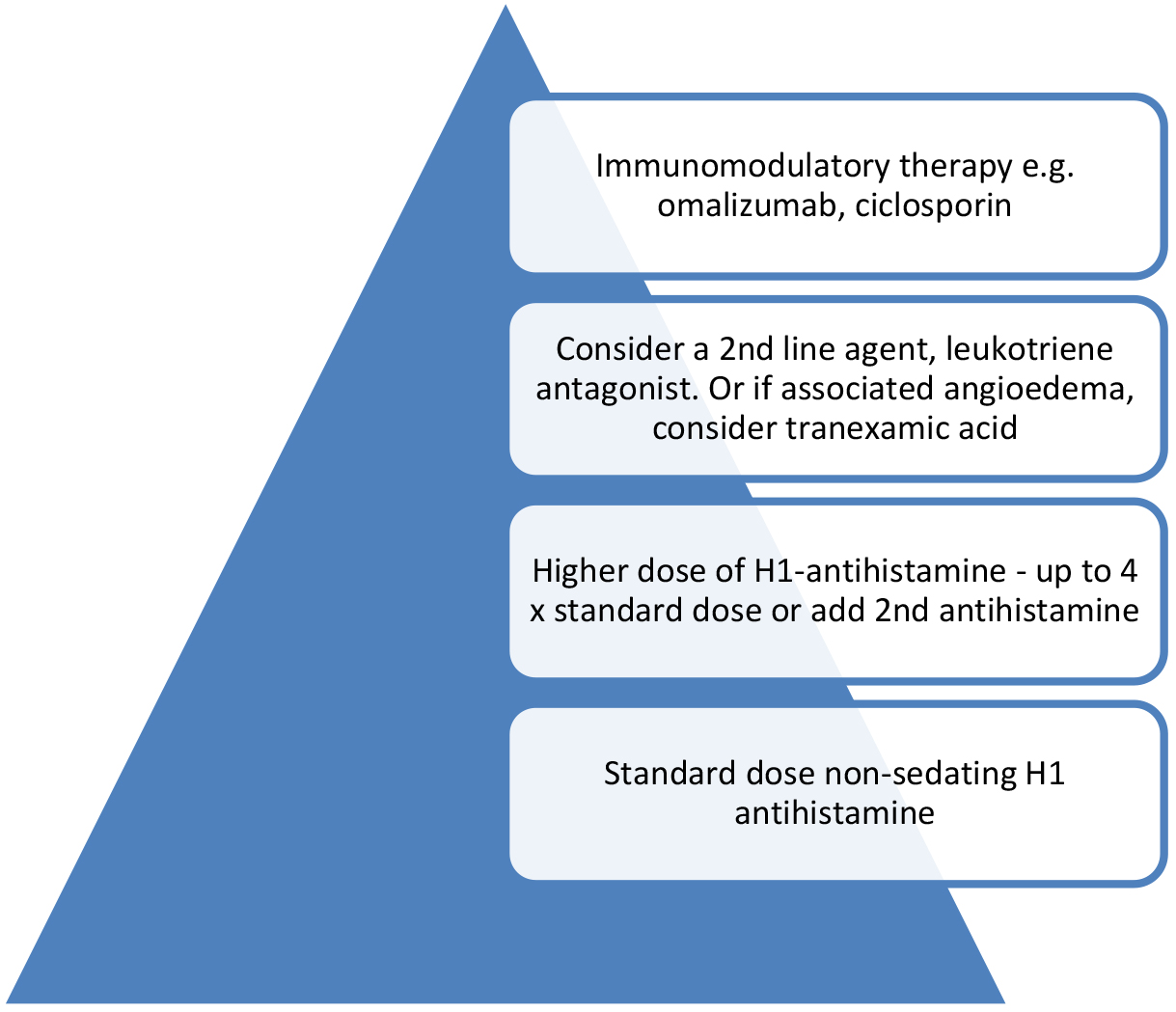 Fig. 4.
Fig. 4.
Management plan for patients with chronic urticaria. If triggers are identified, e.g., NSAID use, physical stimuli, these should be avoided. The figure was created using Microsoft PowerPoint 2021 (Microsoft Corporation, Redmond, WA, USA).
Omalizumab is a humanised monoclonal antibody that binds free IgE, preventing its interaction with high- and low-affinity receptors on mast cells and basophils. The resulting IgE–antibody complexes are cleared by the reticuloendothelial system. Omalizumab does not bind to cell-bound IgE or IgE receptors and therefore does not activate mast cells or basophils. The clinical efficacy of omalizumab in CSU, even among non-atopic individuals, supports a role for IgE- or IgE-receptor-related autoimmunity in the disease pathogenesis.
Chronic Inducible Urticarias (CIndUs) are a group of physical urticarias triggered by specific and reproducible stimuli. Common subtypes include symptomatic dermographism (the most frequent form), cold urticaria, delayed pressure urticaria, cholinergic urticaria, solar urticaria, and vibratory angioedema. These may occur in isolation or coexist with CSU, and diagnosis relies heavily on clinical history supported by provocation testing where available.
Cold urticaria, in particular, is important to identify due to its potential for systemic involvement, including syncope and anaphylaxis [22]. Patients with moderate to severe cold urticaria should be referred for specialist allergy evaluation, issued with an adrenaline autoinjector, and counselled on avoidance of high-risk exposures such as cold water immersion or anaesthesia. Cold stimulation testing (e.g., ice cube test or TempTest) can assist with diagnosis and risk stratification (see Fig. 5).
 Fig. 5.
Fig. 5.
The figure demonstrates induction of cold urticaria following
ice provocation to the volar aspect of the patient’s forearm. The
Management of CIndUs involves trigger avoidance, use of non-sedating H1 antihistamines (with dose escalation if needed), and in refractory cases, consideration of omalizumab. As with CSU, a detailed history is essential to distinguish spontaneous from inducible patterns of urticaria.
Isolated angioedema is a common reason for emergency department attendance [23]. While chronic spontaneous urticaria (CSU) accounts for most cases, it is essential to exclude angiotensin-converting enzyme inhibitor (ACEi)-induced angioedema—a well-recognised class effect that can be life-threatening and usually occurs within the first week of treatment. All patients presenting with angioedema should undergo a thorough medication review, with particular attention to current or past ACEi use.
Some reports suggest that ACEi-induced angioedema can arise after months or even years of previously well-tolerated treatment and may continue intermittently for several months after drug discontinuation. However, these observations have not consistently accounted for the background prevalence of CSU, which may act as a confounding factor in such retrospective assessments. Therefore, the claim that ACEi-induced angioedema can present after long-term use or persist well after discontinuation should be interpreted with caution and weighed against alternative diagnoses, including CSU.
Clinically, ACEi-induced angioedema tends to involve the tongue or upper airway, occurs without urticaria or pruritus, and typically affects deeper submucosal tissues with significant danger of airway obstruction. ACEi-induced angioedema is caused by bradykinin accumulation due to ACE inhibition and may need bradykinin-targeted therapies (e.g., icatibant, a selective bradykinin B2 receptor antagonist). In contrast, angioedema related to CSU often coexists with urticaria (although not necessarily), involves superficial cutaneous tissues of the face, limbs, or genitalia (without deep mucosal involvement), responds well to high doses of antihistamines (as with CSU, these may need fourfold up-dosing), and usually resolves within 24–48 hours. However, some cases may be ambiguous, as factors such as anxiety or inducible laryngeal obstruction can create confusion.
Angiotensin II receptor blockers (ARBs) are implicated in angioedema less frequently than ACEi. While earlier reviews suggested a cross-reactivity rate of up to 10% [24], more robust data from a Danish nationwide registry-based cohort study found no significant increase in angioedema risk with ARBs among patients who had experienced ACEi-related swelling [25]. Therefore, if renin–angiotensin system blockade remains clinically indicated, an ARB may be cautiously considered.
NSAID-related angioedema is another important differential, particularly when urticaria is also present, as explained above in patients with CSU or as a co-factor. Overall, careful clinical assessment and a detailed medication history are crucial in patients presenting with isolated angioedema.
Hereditary or acquired angioedema (HAE/AAE, respectively) presents with recurrent swelling of deep cutaneous tissue and mucosae. These conditions are rare but important to recognise, as management follows a different pathway from idiopathic angioedema.
Hereditary angioedema is an autosomal dominant condition, resulting from quantitative or qualitative deficiency of complement component 1 (C1) inhibitor [26]. This leads to intermittent activation of the kinin pathway, with increased bradykinin production resulting in increased vascular permeability and angioedema. Patients with HAE can have subtle cutaneous erythema, but do not usually have urticaria. Patients typically present with intermittent attacks of swelling, particularly affecting the skin, gastrointestinal tract and airway, the latter may lead to life-threatening airway compromise. Certain triggers are recognised, such as stress, infection, and mechanical trauma. As this is an autosomal dominant condition, the family history should be considered.
Acquired angioedema is also rare. The most common associated conditions are autoimmunity and B cell lymphoproliferative disease [27]. AAE results from consumption due to complement activation, and less commonly due to an autoantibody directed against C1 inhibitor.
A low C4 at the time of an acute attack of isolated angioedema should raise the suspicion of HAE or AAE. Testing of C1 inhibitor levels and function requires fresh serum, as sample decay is a common cause of falsely low functional levels. A low complement component 1q (C1q) can help distinguish hereditary from acquired angioedema, following specialist assessment.
Management:
Acute attacks can be managed by administration of plasma-derived or recombinant C1 inhibitor concentrate, or the subcutaneous bradykinin 2 receptor blocker Icatibant. If patients have regular attacks, then prophylaxis should be considered, targeting the kallikrein pathway [26]. Kallikrein is a key player in the generation of bradykinin. Targeting of kallikrein is the basis of several new treatment approaches, including gene editing and transcriptional interference [28]. Such patients should be referred to immunology services to ensure long-term access to emergency treatment, that an action plan is in place to manage elective procedures, and to consider prophylaxis as appropriate. Patients with HAE/AAE should also be advised to carry an appropriate medical emblem.
Food protein induced enterocolitis syndrome (FPIES) is a non-IgE-mediated food allergy that presents with often severe, delayed, and reproducible gastrointestinal upset following specific food ingestion, with projectile vomiting, diarrhoea lasting for extended periods, and associated lethargy, and in some cases, hypotension and electrolyte disturbance [29]. Culprit foods vary geographically; limited data suggest that FPIES in adults is most commonly triggered by shellfish. The diagnosis is primarily based on history, with improvement following withdrawal of the trigger food [30]. Fluid and electrolyte replacement, with anti-emetic therapy, are the mainstay of acute treatment, with avoidance of the offending food as the longer-term strategy. However, the syndrome can improve over time so that not all patients have to avoid the eliciting food lifelong. Oral food challenge can be considered, but this is often declined in adult practice, given the discomfort associated with this syndrome. Drug-induced enterocolitis syndrome is rare but recognised, with amoxicillin-based antibiotics as the main culprit agent [31].
Scombroid poisoning may present with acute gastrointestinal upset and rash following ingestion of poorly stored oily fish, because of bacterial decomposition of fish muscle L-histidine, into histamine, and hence a high histamine load on consumption [32]. A good clinical history is essential, with patients often reporting flushing, headache, nausea, vomiting, diarrhoea, and abdominal pain. If others who consumed the same food were similarly affected, that points towards a food poisoning episode rather than allergy. Allergy testing will be negative, with oral food challenge with fresh ingredient being the gold standard to exclude food allergy.
A minority of patients presenting with allergic symptoms will have an underlying mast cell disorder, identified by mast cell tryptase assessment at the time of an acute event. Mast cell activation disorders is an umbrella term that describes typical clinical signs of severe, recurrent, episodic acute systemic mast cell activation, with objective evidence of mast cell mediator release on biochemical testing, with a dynamic rise in serum tryptase [9], and symptoms that respond to mast cell targeted therapy [33]. Mast cell activation disorders can be further subdivided into primary disorders, with KIT(KIT receptor tyrosine kinase) D816V mutated, clonal mast cells, with or without an underlying diagnosis of mastocytosis; secondary, where clonal disease is absent, but an IgE dependent allergy or other hypersensitivity reaction is detected; and idiopathic, where no KIT mutation or hypersensitivity trigger can be found [33]. Patients can have more than one type, for example, mastocytosis and IgE-mediated venom allergy. Patients with combined primary and secondary forms are at high risk for life-threatening anaphylaxis. Patients with a persistently elevated baseline mast cell tryptase with additional clinical features, such as skin lesions, musculoskeletal abnormalities or organomegaly, may have underlying mastocytosis. The classification of systemic mastocytosis has recently been updated [34], and there are a number of scoring systems that can be used to determine a patient’s risk, such as the Red Española de Mastocitosis score (REMA) score [35]. These patients can be complex and require expert, multidisciplinary assessment, and management. Other haematological malignancies, particularly myeloid malignancies, can also be associated with mastocytosis [36].
A recently recognised cause of a raised baseline tryptase is an increased copy number of the alpha tryptase gene, known as hereditary alpha tryptasaemia (HAT). Whilst this was initially thought to represent a distinct clinical phenotype [37], HAT has a reported prevalence of in the United Kingdom and France of 5–6%. Symptomatic patients can present with a constellation of symptoms, including those associated with mast cell activity, such as flushing, urticaria and pruritus. HAT is more common in patients with systemic mastocytosis [38] and can be a risk factor for anaphylaxis.
Neuroendocrine conditions may present with flushing and systemic symptoms. Carcinoid syndrome, for example, may present with flushing and gastrointestinal symptoms. Other gastrointestinal tumours that produce vasoactive components may mimic clinical features of allergy. Phaeochromocytomas more commonly present with intermittent headache, sweating and tachycardia. Intermittent hypertension rather than hypotension occurs in approximately 50% of patients, but orthostatic hypotension is recognised. Such differentials are considered in the allergy clinic when patients present with unusual, episodic reactions, in the absence of a reproducible trigger. Appropriate assessment, including 24-hour urine testing, may need to be considered, with referral to endocrinology for investigation.
This review highlights the importance of recognising Type I hypersensitivity reactions and differentiating them from a range of mimicking conditions. Accurate diagnosis depends on a thorough clinical history, timely recognition of key symptoms, and appropriate use of biomarkers such as serum tryptase. While IgE-mediated reactions follow well-characterised immunological pathways, overlapping symptoms with non-IgE and non-immune mechanisms—such as bradykinin-mediated angioedema or neuroendocrine tumours—can complicate clinical assessment.
Key messages include the early use of intramuscular adrenaline for anaphylaxis, the utility of the Symptoms, Timing, Attributable allergen, Reproducibility (STAR) approach in history taking, and the necessity of specialist referral in complex or atypical cases. Raising awareness of cofactors, hidden allergens, and pseudoallergic mechanisms also helps to avoid mislabelling and guide safe, individualised care.
Future research should focus on improving diagnostic accuracy through novel biomarkers and point-of-care testing. Clinical practice would benefit from greater interdisciplinary collaboration, particularly in perioperative and emergency contexts. Further guidance is needed on standardised pathways for evaluating suspected allergy and managing patients with overlapping or unclear phenotypes. Ultimately, advancing diagnostic precision will reduce harm, minimise unnecessary avoidance strategies, and improve outcomes for patients presenting with suspected allergic reactions.
All the data of this study are included in this article.
SLJ and RMB designed the study. SLJ and RMB contributed equally to the writing of the manuscript. SLJ prepared the figures and obtained the necessary reprint permissions. Both authors contributed to important editorial revisions of the manuscript. Both authors read and approved the final version of the manuscript. Both authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Patient consent was confirmed for inclusion of medical imaging (Fig. 5). Ethical approval was not required for this review.
We gratefully acknowledge the patient for consenting to the use of clinical images in this publication.
This research received no external funding.
Neither author has any conflict of interest with respect to this publication. Both authors are members of the Perioperative Allergy Network Steering Committee, a UK-wide network created jointly by the British Society for Allergy and Clinical Immunology, the Association of Anaesthetists and the British Society for Immunology Clinical Immunology Professional Network. Neither author has any financial interests or other biases related to these institutions.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.