



 , Iordanis Pelagiadis 1, Maria Stratigaki 1, Nikolaos Katzilakis 1, Eftichia Stiakaki 1,*
, Iordanis Pelagiadis 1, Maria Stratigaki 1, Nikolaos Katzilakis 1, Eftichia Stiakaki 1,*
1 Department of Pediatric Hematology-Oncology & Autologous Hematopoietic Stem Cell Transplantation Unit, University Hospital of Heraklion & Laboratory of Blood Diseases and Childhood Cancer Biology, School of Medicine, University of Crete, 71003 Heraklion, Greece
Abstract
Myosin heavy chain 9-related disease (MYH9-RD) is a rare inherited disorder characterised by macrothrombocytopenia, often misdiagnosed as immune thrombocytopenia (ITP). Early identification is crucial to prevent unnecessary treatments and to ensure appropriate monitoring. The present case aims to highlight the diagnostic challenges and clinical management of MYH9-RD in a toddler, emphasising the importance of early genetic testing.
We discuss a 13.5-month-old girl with macrothrombocytopenia lacking Döhle bodies, who initially received intravenous immunoglobulin (IVIg) and corticosteroids without any response. Within two months, whole-exome sequencing identified a pathogenic MYH9 mutation (c.287C>T; p.Ser96Leu).
One year later, the patient remains clinically stable without significant bleeding. The occurrence of petechial rash exhibited a more pronounced correlation with platelet mass index (PMI) values compared to platelet count (PLT), underscoring its significance in clinical evaluation.
MYH9-RD should be considered in cases of IVIg-resistant thrombocytopenia accompanied by macrothrombocytes. Timely genetic testing can facilitate accurate diagnosis and may help avoid unnecessary procedures, while routine renal and auditory monitoring is important for managing the S96L variant.
Keywords
- MYH9
- blood platelets
- thrombocytopenia
- case report
- platelet mass index
- Epstein syndrome
Myosin heavy chain 9-related disease (MYH9-RD) is an umbrella term for disorders characterized by MYH9 mutations and macrothrombocytopenia (i.e., low platelet count (PLT) with giant platelets), which can be accompanied by non-syndromic and progressive sensorineural hearing loss, presenile cataracts, elevation of liver enzymes, or progressive nephropathy often leading to end-stage renal disease. Epstein syndrome, Fechtner syndrome (Alport-like syndrome with macrothrombocytopenia), May-Hegglin anomaly, and Sebastian syndrome are pathologic entities comprising the MYH9-RD spectrum. The MYH9 gene encodes a conventional non-muscle myosin (myosin IIA; located in 22q12.3) involved in several essential functions such as cytokinesis, cell motility, and cell shape maintenance [1].
We hereby report the diagnostic challenges of a young female toddler who presented with macrothrombocytopenia and a petechial rash, diagnosed with MHY9-RD two months after admission. One year after the diagnosis, the child remains well without any bleeding events and is undergoing regular monitoring of her renal and auditory functions.
Following the Declaration of Helsinki, and adhering to the CARE guidelines
(Supplementary Table 1)
(https://www.equator-network.org/reporting-guidelines/care/;
accessed on 10 April 2025), we hereby report a well-appearing 13.5-month-old girl
admitted to our department following the results of a routine check-up that
revealed thrombocytopenia with a PLT count of 11
Intravenous immunoglobulin (IVIg) was initially administered as per immune thrombocytopenia (ITP). Two days later, after no significant response in PLT count, prednisolone was administered, adhering to the respective childhood ITP recommendations [2]. New petechiae were halted at this phase, and corticosteroids were discontinued when the next-generation sequencing (NGS) results set the diagnosis, and medication was deemed redundant.
Whole-exome sequencing (WES) was performed using the Twist Human Core EF Multiplex
Complete Kit (Twist Bioscience, San Francisco, CA, USA) and the NextSeq 500
System (Illumina, Inc., San Diego, CA, USA). Bioinformatic analysis was conducted
using the SOPHiA DDM™ platform (Sophia Genetics SA, Boston, MA, USA). WES
revealed a de novo pathogenic c.287C
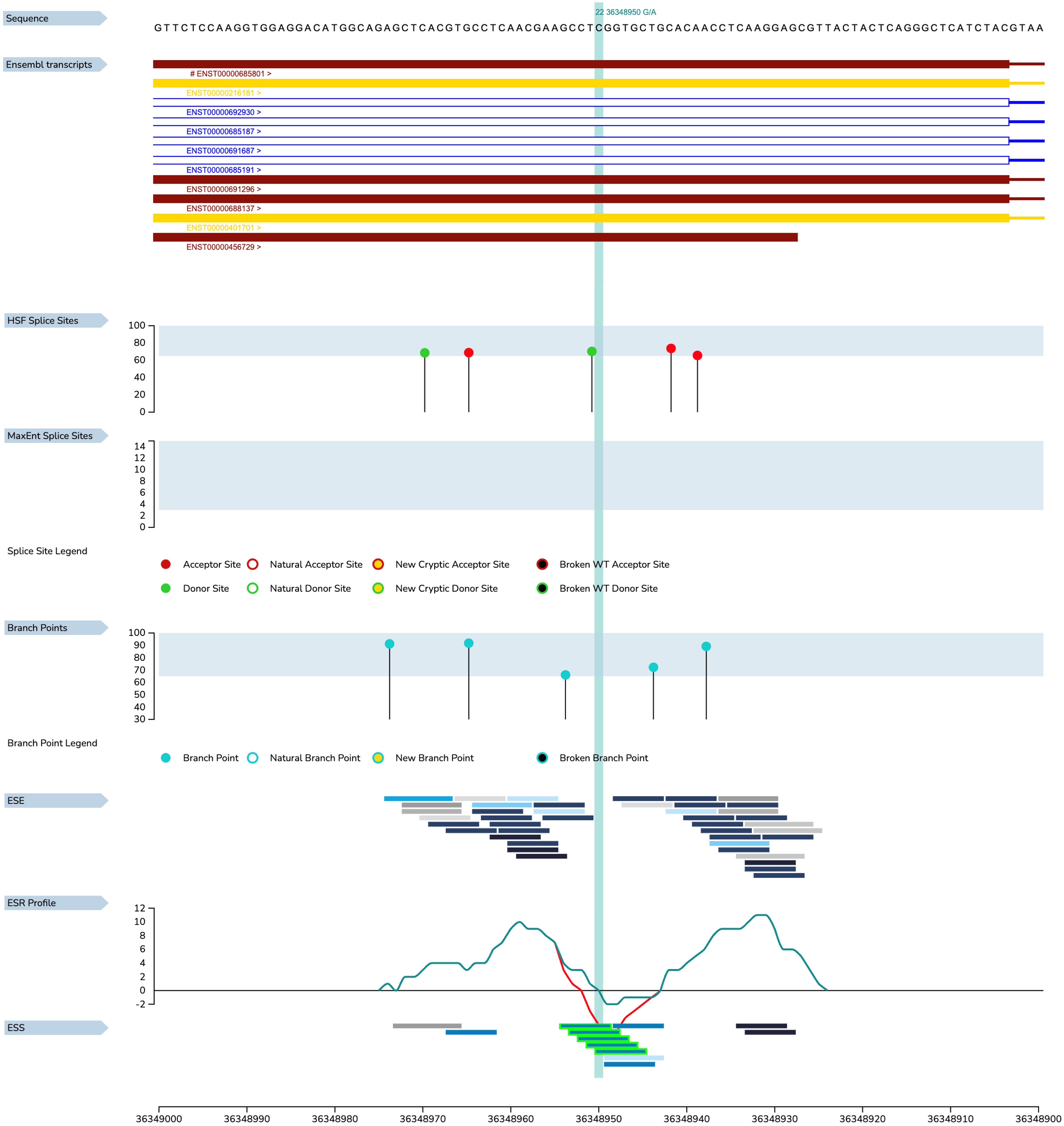 Fig. 1.
Fig. 1.
The predicted impact of myosin heavy chain (MYH9) c.287C
Regarding the patient’s follow-up period, the median PLT count was 10
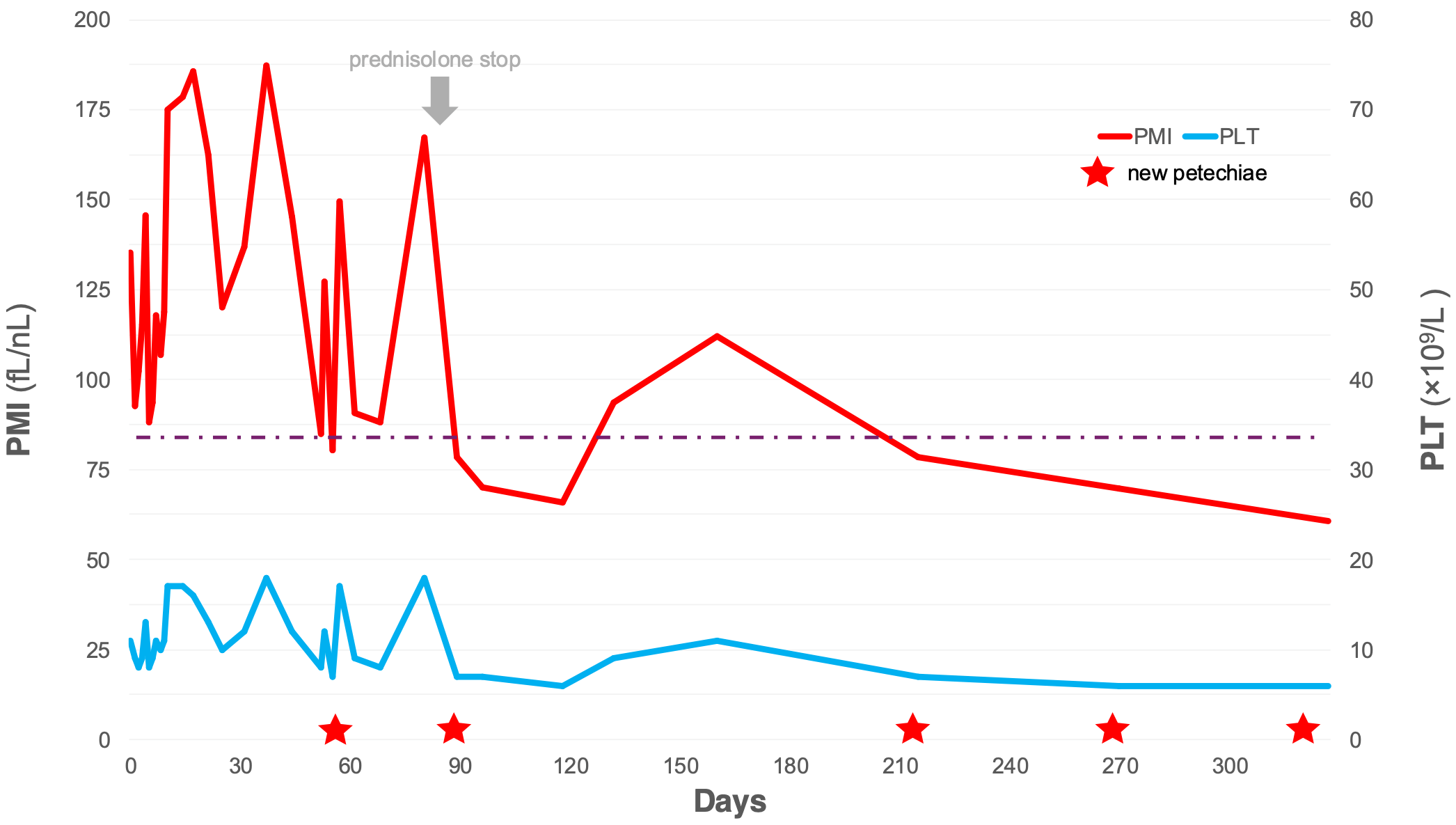 Fig. 2.
Fig. 2.
PMI drops below 85 fL
One year later, the patient is generally well without bleeding incidents and is regularly monitored, including kidney function and hearing acuity, according to published relevant schedules.
MYH9-RD is a rare genetic disorder with a reported prevalence of 1–9/1,000,000 (https://www.orpha.net/en/disease/detail/182050; accessed on 10 April 2025). Recent studies, however, suggest a significantly elevated prevalence rate of approximately 1 in 20,000. This discrepancy is attributed to advancements in the molecular characterisation of thrombocytopenia cases [3]. Notably, MYH9-RD is commonly misdiagnosed as immune thrombocytopenia in over 90% of cases, even in individuals with markedly elevated MPV [4]. Promptly identifying the pathogenic MYH9 variant can provide valuable insights into our patient’s clinical course and prevent unnecessary treatments [5, 6, 7].
The localisation pattern of neutrophil MYH9 provides critical insights for
documenting Döhle inclusions. In the present case, neutrophil inclusions were
not observed, which aligns with the localisation pattern associated with the S96L
mutant [8]. The clinical course of known mutations (such
as S96L) can be predicted based on published genotype-phenotype correlations
[9]. In this case, although not present, hearing loss and
nephritis are anticipated (each with up to a 75% chance), while cataracts are
not expected; therefore, the corresponding follow-up schedule was approved.
Despite the high penetrance of S96L, the variation in the age of onset of
nephritis and hearing loss among the affected patients depends on epigenetic
modifications, environmental influences, and genetic epistasis. Therefore,
longitudinal monitoring is necessary to detect any future changes [8, 9]. PLT counts may exhibit a slight increase after
treatment with prednisolone; however, the presence of giant PLTs in this case,
along with the initial lack of response to IVIg administration, prompted the
investigation using WES when the coagulation and PLT function tests yielded
negative results. Notably, a mean PLT diameter of
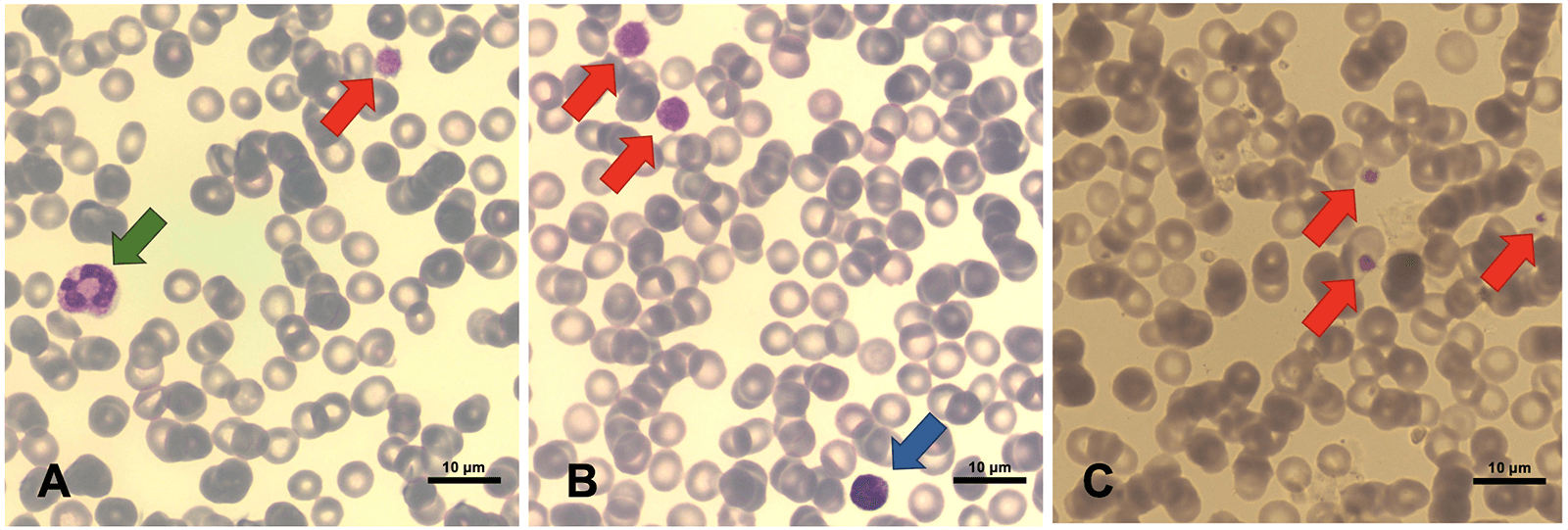 Fig. 3.
Fig. 3.
The patient’s latest peripheral blood smear (magnification: 100
No clinical practice guidelines for MYH9-RD have been published. Annual hematologic evaluations are recommended, and immediate medical attention is essential for each bleeding episode. Hemostatic challenges (menorrhagia, surgery, delivery, and other invasive procedures) require careful planning. Screening with an audiogram every three years (with immediate reporting of any worsening of hearing function) and for nephropathy every 6–12 months (depending on the variant) is also recommended. Aside from supportive care, no specific therapy exists to alleviate the systemic effects of the mutated MYH9. However, some patients have undergone long-term treatment with a thrombopoietin receptor agonist (TPO-RA) and achieved positive outcomes regarding thrombocytopenia [15]. The efficacy and safety of long-term TPO-RA administration for treating MYH9-RD have yet to be determined. A watch-and-wait strategy was chosen for our patient, involving regular follow-up assessments, with interventions considered only occasionally in the event of hemostatic challenges.
Our findings indicate that prednisolone may increase PLT counts, yet it does not prevent the occurrence of new petechiae. Furthermore, PMI may serve as a useful predictor of bleeding episodes. The strength of this case lies in the early identification of the MYH9 mutation using WES, which directed appropriate clinical management and averted unnecessary treatments. However, a limitation of the study is the short follow-up period of one year, which may not adequately capture the long-term progression of nephropathy or hearing loss, both of which are part of the expected phenotype associated with S96L mutations. Additionally, all limitations relevant to case reports apply here as well.
The present case reinforces the importance of considering MYH9-RD in the differential diagnosis of thrombocytopenia with large platelets. Early genetic testing is crucial for an accurate diagnosis and appropriate management, preventing unnecessary treatments and enabling tailored follow-up strategies. Additionally, PMI may be a valuable tool in assessing bleeding risk and an accessory to the established PLT count.
All data included in this study are available from the corresponding author upon reasonable request.
IP & ES: conceptualization; IK & IP: methodology; IP, MS, NK & ES: validation; IP, MS & NK: investigation; IK, NK & ES: resources; IK: writing - original draft preparation; IP, MS, NK & ES: writing - review and editing; IK: visualization; IP, NK and ES: supervision; IP, NK and ES: project administration. All authors contributed to the important editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Written informed consent was obtained from the patient’s parents. The research was conducted in strict accordance with the ethical principles outlined in the Declaration of Helsinki.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/BJHM50376.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.