- Academic Editor
-
-
-
†These authors contributed equally.
Sepsis-induced myocardial injury (SIMI) represents a major contributor to prolonged hospitalization in intensive care units (ICUs) and is associated with increased mortality rates. Mitochondria serve as the primary energy source for cardiomyocytes and are also essential for various other cell functions. The essential voltage-dependent anion channel 3 (VDAC3) protein located in the outer mitochondrial membrane plays a crucial role in preserving mitochondrial homeostasis by controlling metabolite transport and the shape of cristae. However, the precise mechanism by which VDAC3 is involved in SIMI remains unclear. This study aimed to explore the function and mechanism of VDAC3 in SIMI pathogenesis, with a particular emphasis on its regulatory role in ferroptosis.
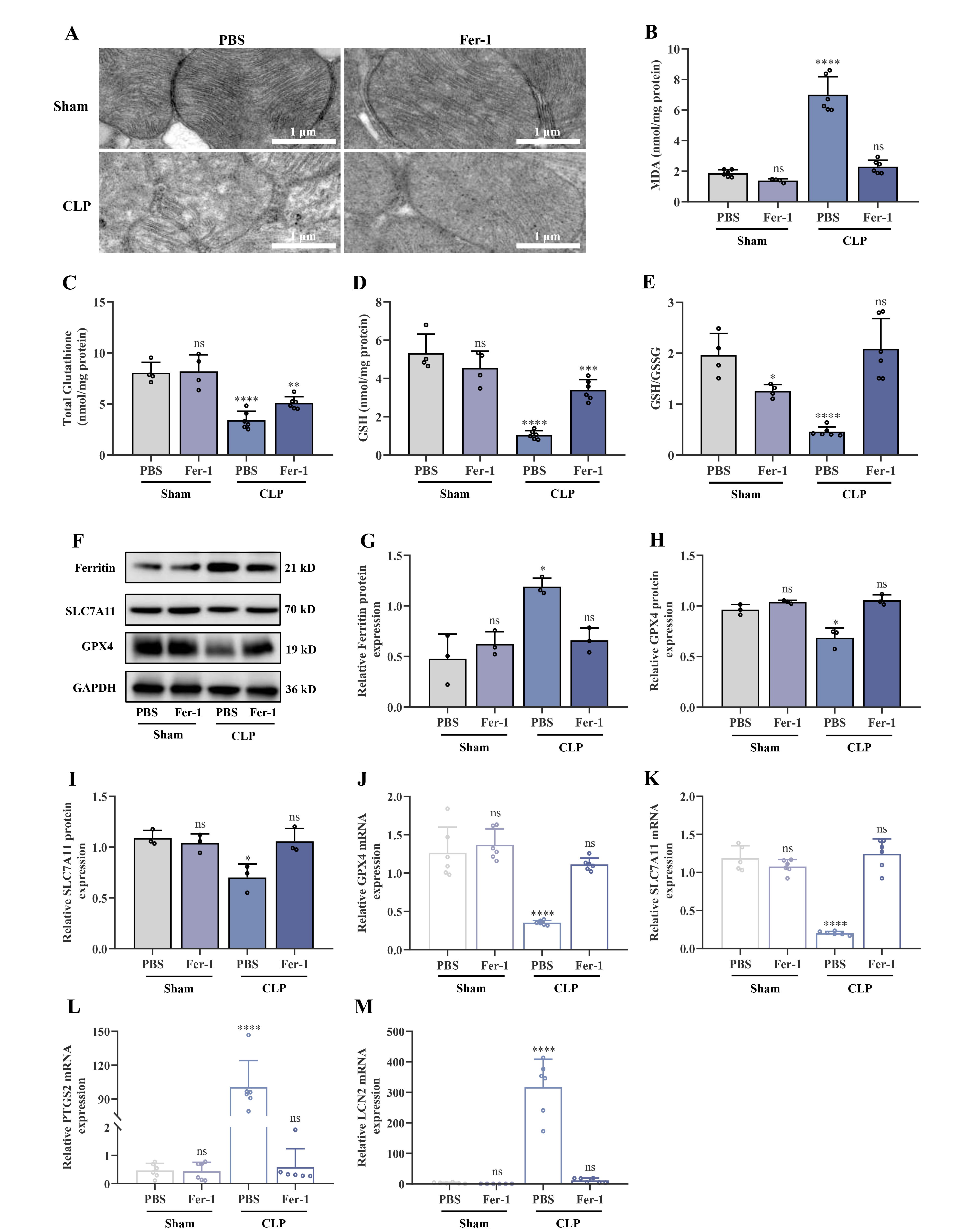
Lipopolysaccharide (LPS)-treated HL-1 cardiomyocytes (a murine cardiomyocyte cell line) were used to construct an in vitro myocardial injury model, and mice were used to establish a cecal ligation and puncture (CLP)-induced in vivo myocardial injury model. Transmission electron microscopy (TEM) was employed to evaluate the mitochondrial ultrastructure in cardiac tissues, while hematoxylin-eosin (H&E) staining was used to assess histopathological alterations. Echocardiography was used to evaluate the structural and functional characteristics of the heart. Integrated transcriptome and proteomic studies were performed to identify differentially expressed genes. VDAC3 expression levels, inflammatory responses, cellular proliferation, and ferroptosis were assessed using colorimetric assays, flow cytometry, enzyme-linked immunosorbent assay (ELISA), Cell Counting Kit-8 (CCK-8) proliferation assay, western blotting, and quantitative reverse transcription PCR (qRT-PCR). The relationship between VDAC3 and ferroptosis was investigated in vitro by transfecting cells with VDAC3 overexpression plasmids.
The injury model group in both the in vitro and in vivo experiments showed a decreased level of the antioxidant glutathione (GSH) and an elevated level of the lipid peroxidation product malondialdehyde (MDA). Moreover, ferroptosis regulation occurred through the modulation of glutathione peroxidase 4 (GPX4), solute carrier family 7 members 11 (SLC7A11), ferritin, prostaglandin-endoperoxide synthase 2 (PTGS2), lipocalin 2 (LCN2), and acyl-coenzyme A (CoA)-synthetase long-chain family member 4 (ACSL4) expression. Administration of ferrostatin-1 (Fer-1), an inhibitor of ferroptosis, markedly reduced the cardiac injury caused by CLP. Additionally, VDAC3 expression was significantly downregulated in experimental models and septic children. In contrast, Fer-1 treatment increased the expression of both VDAC3 and dihydroorotate dehydrogenase (DHODH) and significantly ameliorated cardiac damage. Overexpression of VDAC3 reduced mitochondrial oxidative stress, increased the expression of DHODH, and altered the progression of ferroptosis.
Collectively, this research provides insights into the molecular mechanism behind the VDAC3/DHODH axis in SIMI. This axis mitigates cardiac injury by regulating ferroptosis, thereby suggesting novel therapies for SIMI.