
- Academic Editors
Ionizing radiations (IRs), commonly used in both diagnostic imaging and cancer therapy, generate reactive oxygen species (ROS) and free radicals, causing significant DNA damage that can lead to genetic mutations, cell death, and tissue injury in both normal and tumor tissues. In response to the oxidative stress, the nuclear factor erythroid 2-related factor 2 (NRF2) is activated to induce target genes involved in antioxidant and detoxifying pathways, thereby playing a pivotal role in protecting cells from IR-induced oxidative damage. In clinical diagnostics, IR exposure from imaging techniques can result in DNA damage, inflammation, and increased risk of IR-induced pathologies, including cancer. NRF2 activation in response to these diagnostic exposures can help to protect normal tissues from damage by boosting antioxidant defenses. In radiotherapy, IR induces DNA damage to kill malignant cells, although it may also harm surrounding healthy tissue. Cancer cells exploit NRF2 activation to resist IR-induced cell damage, thereby maintaining redox balance and protecting themselves from oxidative stress. In that case, NRF2 inhibition could sensitize cancer cells to IR effects by disrupting their antioxidant defense, leading to increased ROS accumulation, enhanced DNA damage, and greater cell death. This review will summarize the role of NRF2 in mediating the response to IR in both healthy and cancerous cells, with a focus on its effects in clinical diagnostic and radiotherapy.
Nuclear factor erythroid 2-related factor 2 (NRF2) is a key transcription factor that protects cells from oxidative stress and cell damage caused by environmental stressors, including ionizing radiations (IRs). Under normal conditions, NRF2 is kept inactive in the cytoplasm by its interaction with Kelch-like ECH-associated protein 1 (Keap1), an E3 ubiquitin ligase which drives NRF2 degradation [1]. Under oxidative stress, the interaction between Keap1 and NRF2 is impaired, allowing NRF2 to stabilize and translocate to the nucleus to activate target gene promoters with antioxidant response elements (ARE) whose encoded proteins help protect cells from oxidative stress [2, 3]. In tumour, NRF2 plays a double role. During the first phases of carcinogenesis its activation can be mainly cytoprotective by suppressing oxidative stress and tumor-promoting inflammation, both responsible of increased DNA mutations. On the other hand, NRF2 hyperactivation in many cancers, by different mechanisms, may support tumorigenesis, resistance to chemo- and radiotherapy, and enhanced cancer cell survival by strengthening antioxidant defenses and detoxification pathways that counteract cell death induced by oxidative stress and increase genetic instability [4]. Such dysregulation supports cancer progression, metastasis, and resistance to therapies, making NRF2 a potential attractive therapeutic target to enhance the efficacy of the current cancer treatments [5].
IRs, including those used in medical diagnostic and cancer therapy, can cause oxidative stress and DNA damage [6]. In diagnostic imaging, low doses of IR may increase cancer risk over time. The presence of oxidatively-induced clustered DNA lesions (OCDLs) challenges the linear no-threshold (LNT) model, as even low radiation doses can cause complex, hard-to-repair damage. Due to their high biological impact, OCDLs may disproportionately drive mutations and cancer, suggesting low doses could be more harmful than previously assumed [7, 8, 9, 10]. In radiotherapy, higher doses of IR target cancer cells but can also damage healthy tissues, leading to DNA breaks, inflammation, and eventually secondary cancers [11]. IR can directly interact with DNA causing single-strand or double-strand breaks, or induce indirect DNA damage through water ionization and reactive oxygen species (ROS) generation, which further harm cellular components [11, 12]. Emerging research now shows that NRF2-mediated transcription can protect cells and tissues from the pathogenic consequences of hydroxyl radicals that are directly generated by IR as well as of the hydrogen peroxide and superoxide that are generated as a secondary consequence of irradiation which can have an impact on both normal and cancer tissues.
In this review, we explore the role of NRF2 in the response to the damage caused by IR, with a focus on its effects in clinical diagnostic and radiotherapy.
NRF2 is a transcription factor encoded by the NFE2L2 gene and belonging
to the cap “n” collar subfamily of basic-region leucine zipper proteins [13].
It has a central role in cytoprotection by reducing oxidative stress and
maintaining redox homeostasis. NRF2 regulates the expression of numerous target
genes critical for cellular processes like detoxification and cytoprotection,
particularly those involved in protecting against oxidative stress and supporting
cellular homeostasis including heme oxygenase 1 (HO-1), Aldo-keto reductase
(AKR), NADPH quinone oxidoreductase 1 (NQO-1), superoxide dismutase (SOD),
catalase, multidrug resistance-associated protein (MRP), and ATP-binding cassette
(ABC) transporters [14]. Additionally, it exerts anti-inflammatory activity by
regulating a variety of downstream genes such as Interleukin-6 (IL-6), tumor
necrosis factor alpha (TNF-
| Biological activity | Target genes |
| Antioxidant | HO-1, GPx, Trx, TrxR, PrxR, SOD, catalase, GSR |
| Detoxyfication and Transport | AKR, NQO-1, MRP, ABC transporters, GST, GCLC, GCLM |
| Inflammation | IL-6, TNF- |
| DNA repair | 53BP1, XRCC1, GADD45, OGG1, PARP-1 |
| Cell death | Bcl-2, Caspase-3, SOD1, cIAP1 |
Abbreviations: 53BP1, p53 binding protein-1; ABC, (ATP-binding cassette)
transporters; AKR, Aldo-keto reductase; Bcl-2, B-cell lymphoma
2; cIAP1, cellular inhibitor of apoptosis protein-1; COX2, cyclooxygenase-2;
GADD45, growth-arrest and DNA-damage inducible 45; GCLC,
glutamate-cysteine ligase; GCLM, glutamate-cysteine ligase modifier;
GPx, gluthathione peroxidase; GSR, glutathione reductase;
GST, glutathione S-transferase; HO-1, heme oxygenase-1;
IL-6, interleukin-6; iNOS, inducible nitric oxide synthase;
MCP-1, monocyte chemoattractant protein-1; Mip2, macrophage
inflammatory protein-2; MRP, multidrug resistance-associated protein;
NQO-1, NADPH quinone oxidoreductase-1; NRF-2, nuclear factor erythroid
2-related factor 2; OGG1, 8-Oxoguanine glycosylase; PARP-1,
Poly (ADP-ribose) polymerase-1; PrxR, peroxiredoxin; SOD,
superoxide dismutase; TNF-
Under normal conditions, NRF2 is retained in the cytoplasm through its interaction with two proteins: Keap1, an actin cytoskeleton-associated protein with 25 cysteines that function as “sensors” for the NFR2/Keap1 regulatory system [1], and Cullin 3 (CUL3), a scaffold protein that is part of the E3 ubiquitin ligase complex [21]. This complex mediates NRF2 ubiquitination, which is then targeted for proteasomal degradation (Fig. 1A, left side). As a reaction to oxidative stress, cysteines 273 and 288 on Keap1 are modified by reactive oxygen species and electrophiles leading to protein conformational changes. As a result, the Keap1-CUL3-NRF2 ubiquitination system is impaired, hindering the ubiquitination of NRF2 and causing its accumulation in the cytoplasm [22]. Afterward, NRF2 translocates to the nucleus, where it forms a heterodimer with one of the small musculoaponeurotic fibrosarcoma (MAF) proteins and binds to antioxidant response elements (AREs) in the promoter regions of target genes (Fig. 1A, right side) [4]. Besides this canonical activation, NRF2 can be activated via a non-canonical mechanism by Ser349 phosphorylated p62/sequestome 1 (SQSTM1) (p-STGE), a key autophagyc adaptor that interacts with Keap1, inducing its degradation through autophagy, thereby triggering NRF2 stabilization and activation (Fig. 1B) [23]. Additionally, NRF2 activation can be regulated by a feedback mechanism involving (BTB Domain and CNC Homolog 1) BACH1 [24], a physiological repressor of NRF2 that is itself stabilized by NRF2 (Fig. 1C) [25, 26]. Both NRF2 and BACH1 associate to form a heterodimer with MAF proteins and bind to ARE sites in the promoters of cytoprotective genes. Under basal conditions or low oxidative stress, MAF-BACH1 heterodimers occupy these ARE sites, acting as transcriptional repressors (Fig. 1C, left side). As a reaction to oxidative stress, NRF2 is activated and its increased availability and affinity for AREs displace the BACH1/MAF complex from the ARE sites leading to activation of protective gene expression [27]. Notably, NRF2 activation can also induce BACH1 expression via ARE sites in its promoter, establishing a negative feedback circuit that limits the duration and intensity of the NRF2 response (Fig. 1C, right side) [28].
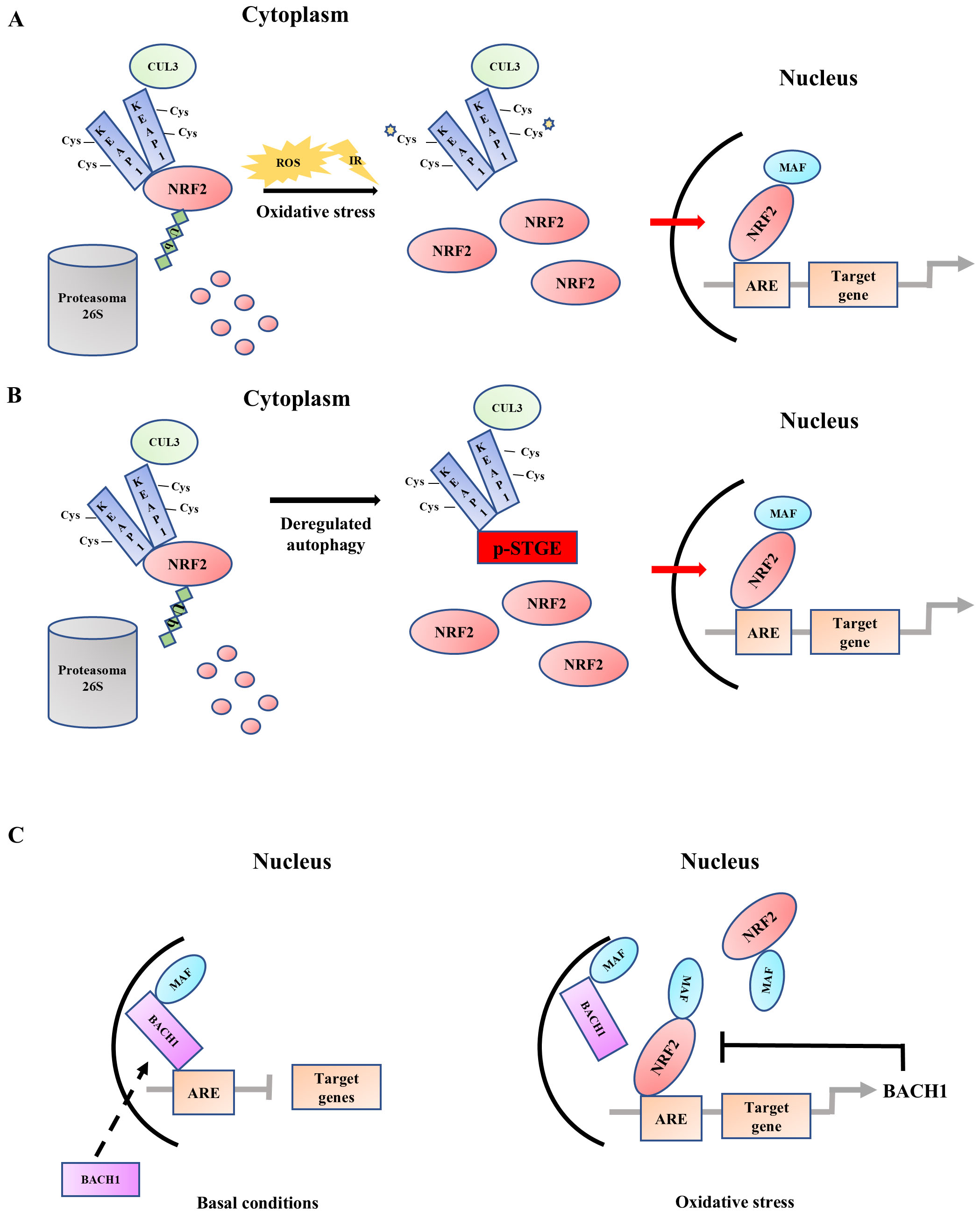 Fig. 1.
Fig. 1.
Nuclear factor erythroid 2-related factor 2 (NRF2) activation. (A) Under normal conditions, NRF2 is retained in the cytoplasm through its interaction with Keap1 and Cullin3 (CUL3) that allows proteasomal ubiquitination and degradation. During oxidative stress, the Keap1-CUL3-NRF2 ubiquitination system is disrupted, allowing NRF2 to accumulate in the cytoplasm and translocate to the nucleus. In the nucleus, NRF2 forms a heterodimer with MAF proteins and binds to antioxidant response elements (AREs) in the promoter regions of target genes promoting transcription (canonical activation). (B) In a non-canonical activation pathway, p-STGE (Ser349 phosphorylated p62 motif) competes with NRF2 for Keap1 binding, leading to the dissociation of NRF2. This allows NRF2 to accumulate in the cytoplasm, translocate to the nucleus, bind to target genes, and promote transcription. (C) Under basal conditions, BACH1 (BTB Domain and CNC Homolog 1) occupies ARE sites, repressing transcription of target genes. Following oxidative stress, NRF2 is activated and displaces the BACH1/MAF complex from the ARE sites, activating the expression of protective gene, including BACH1, creating a negative feedback loop (dotted arrow). ROS, reactive oxygen species; IR, ionizing radiation.
NRF2 plays a complex and dual role in cancer, influencing different stages of tumorigenesis in either protective or pro-tumorigenic ways and acting on both healthy and tumor cells [4, 29]. Despite the progress in the knowledge of NRF2 in recent years, there is still no clear consensus on its exact role during carcinogenesis [30, 31]. In normal cells, including the very early stages of neoplastic transformation, NRF2, through the expression of antioxidant and detoxifying enzymes, protects the cells against oxidative stress and cellular damage and therefore blocks carcinogenesis [32] (Fig. 2, upper panel). However, a tumor-promoting role for NRF2 in cancer initiation has been reported linked, for instance, to its protection against redox stress in cells with mutations in kirsten rat sarcoma viral oncogene homolog (KRAS) and/or serine/threonine kinase 11 (STK11) [33]. In the subsequent stages of tumor promotion and in the neoplastic progression, the role of NRF2 shifts, as its persistent activation becomes a key driver of cancer cell survival and proliferation and resistance to cancer treatments, often in an interplay with oncogenic pathways such as signal transducer and activator of transcription 3 (STAT3) or mutant p53 [34, 35] (Fig. 2, lower panel). By upregulating a variety of detoxification and antioxidant genes, NRF2 supports the adaptation of tumor cells to the harsh tumor microenvironment, providing resistance to cytotoxic effects of chemo- and radio-therapy in many types of tumors including breast, lung, pancreatic, head and neck, colon, etc., [36, 37, 38, 39, 40, 41, 42, 43, 44]. NRF2 activation also protects cancer cells from ferroptosis, an iron-dependent and lipid peroxidation-driven cell death cascade, occurring when there is an imbalance of redox homeostasis in the cell [45]. NRF2 upregulates multiple genes that block ferroptosis such as SLC7A11 (xCT) or glutamate-cysteine ligase (GCLC)/glutamate-cysteine ligase modifier (GCLM), modulates iron metabolism genes to limit iron-driven ROS, and limit the availability of polyunsaturated fatty acids (PUFAs)—the main substrates for lipid peroxidation [46]. NRF2 also protects cells from apoptosis, allowing cancer cells to escape death especially the one induced by anticancer treatments [20, 47]. Furthermore, NRF2 has been implicated in promoting metastasis, partly by facilitating epithelial-to-mesenchymal transition (EMT), which enhances the invasive capacity of tumor cells [26, 48, 49, 50]. Thus, while NRF2 initially serves as a protective response to cellular oxidative stress to block carcinogenesis, its chronic activation in established cancers contributes to tumor growth, metastasis, and therapy resistance. In summary, this dual role makes NRF2 a challenging molecular target for therapeutic intervention in cancer [30], as its inhibition may on one hand improve cancer cell response to cytotoxic cancer treatments [51, 52, 53], but, on the other hand, may impair its normal protective functions in healthy cells contributing to carcinogenesis. Therefore, more precise targeting strategies need to be taken in consideration [31, 54].
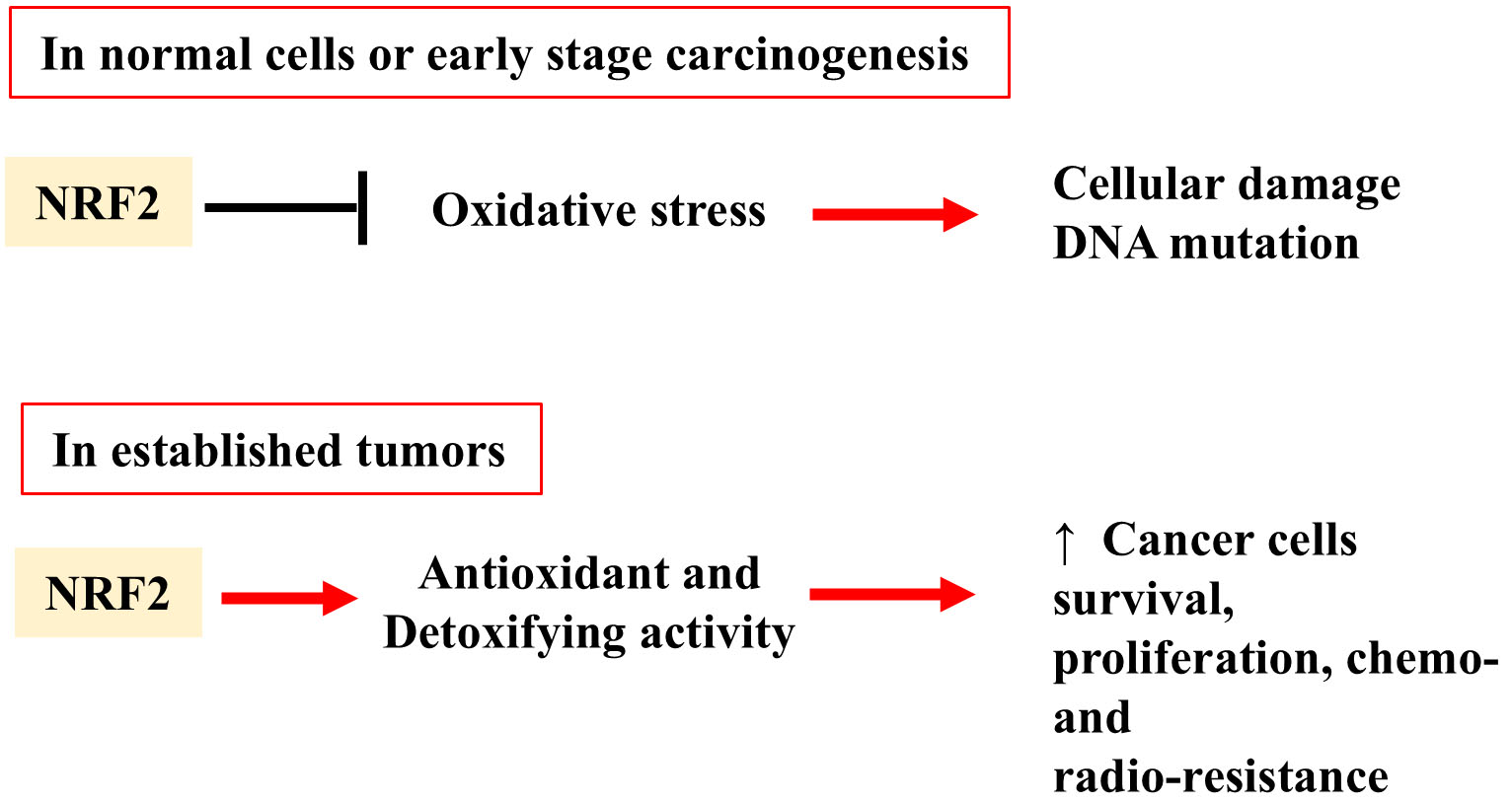 Fig. 2.
Fig. 2.
Dual role of NRF2 in both normal and cancer cells. In normal
cells or early stage carcinogenesis, NRF2 acts as a protector against oxidative
stress-induced cellular damage. In established tumors, NRF2 can acquire a
pro-tumorigenic function. Its persistent activation can support tumor growth and
proliferation by inducing the expression of antioxidant and detoxifying enzymes.
“
IR interacts with biological tissues generating either direct or indirect effects [12, 55]. The extent of DNA damage caused by IR depends primarily on the ionization density, absorbed dose, dose rate, and linear energy transfer (LET) [56]. The high linear energy transfer (LET) IRs, i.e., alpha-particles and heavy ions, induce damage mostly by direct action, whereas the low LET IR, i.e., X-Rays and gamma-rays, by indirect one. Direct effects occur by the direct ionization and excitation of DNA molecules, which disrupt the molecular structure. The direct interaction can result in DNA single-strand breaks (SSBs) or double-strand breaks (DSBs) which, if not properly repaired, can result in mutations, genomic instability, or cellular death [57]. Indirect effects, in contrast, occur through the radiolysis of water, leading to the formation of highly reactive species like hydroxyl radicals (OH•), hydrogen peroxide (H2O2), and superoxide anion (O2•-). These reactive oxygen species (ROS) can cause damage to cellular components, including DNA, proteins, and lipids. In addition, reactive nitrogen species (RNS) such as nitric oxide (NO•) and peroxynitrite (ONOO-) may form, contributing to further cellular damage. This cascade of oxidative and nitrosative stress can result in DNA mutations, cell death, and in a broad range of biological effects, from immediate tissue damage to long-term outcomes such as cancer development [58].
The most severe damage resulting from IR exposure is DNA DSBs, which is mainly repaired through non-homologous end joining (NHEJ) and homologous recombination (HR) mechanisms [59, 60, 61, 62]. These repair processes rely on a network of factors that detect, repair, and maintain genomic stability, ensuring the cell can recover from IR-induced DNA injury or, if necessary, trigger appropriate cell death to prevent genomic instability. Key sensors like ataxia telangiectasia mutated (ATM) and ATM and Rad3-related (ATR) detect DSBs and SSBs, respectively, and initiate a signaling cascade involving checkpoint proteins like checkpoint kinase 1 (CHK1) and CHK2 to arrest the cell cycle and allow DNA damage repair [63, 64]. If the damage is irreparable, oncosuppressor p53 triggers permanent cell cycle arrest by replicative senescence or cell death by apoptosis [65]. Additionally, proteins such as breast cancer gene 1 (BRCA1) and BRCA2 play crucial roles in DNA repair and checkpoint regulation, particularly in the HR pathway [66].
IR used in clinical setting have distinct effects depending on whether they are employed for diagnosis purposes or radiotherapy and also present risks because of their ability to induce molecular and cellular damage, as seen above [67]. In diagnostic applications, IR are primarily used to create detailed scans of internal bodily organs. Techniques such as X-rays, computed tomography (CT), mammography and nuclear medicine aid in the diagnosis of various medical conditions. X-rays are commonly employed to obtain images of bones, detect fractures, and diagnose lung conditions like tuberculosis [68]. CT scans, which involve a series of X-ray images combined to create detailed cross-sectional views, provide comprehensive information on complex issues such as tumors, internal bleeding, and organ diseases [69]. Mammography, is a critical imaging modality in the screening of breast carcinoma, utilizing IR to obtain detailed breast tissue images that can identify nodules, calcifications, or other abnormalities indicative of breast cancer [70]. In nuclear medicine, small amounts of radioactive isotopes are used to create images of organs and tissues, providing functional information about the body’s physiology through diagnostic procedures like positron emission tomography (PET) and single-photon emission computed tomography (SPECT) [71] (Fig. 3). These procedures are generally quick, non-invasive, and offer valuable diagnostic insights. However, despite the relatively low doses used, this exposure can potentially cause cellular damage or increase the risk of cancer over time. It is therefore essential to carefully assess the frequency of diagnostic tests and IR doses used to manage and minimize the risk of potential harm [72, 73]. At the moment, unlike CT or X-Ray exposure, PET and SPECT do not have large-scale epidemiological studies linking them directly to increase cancer incidence.
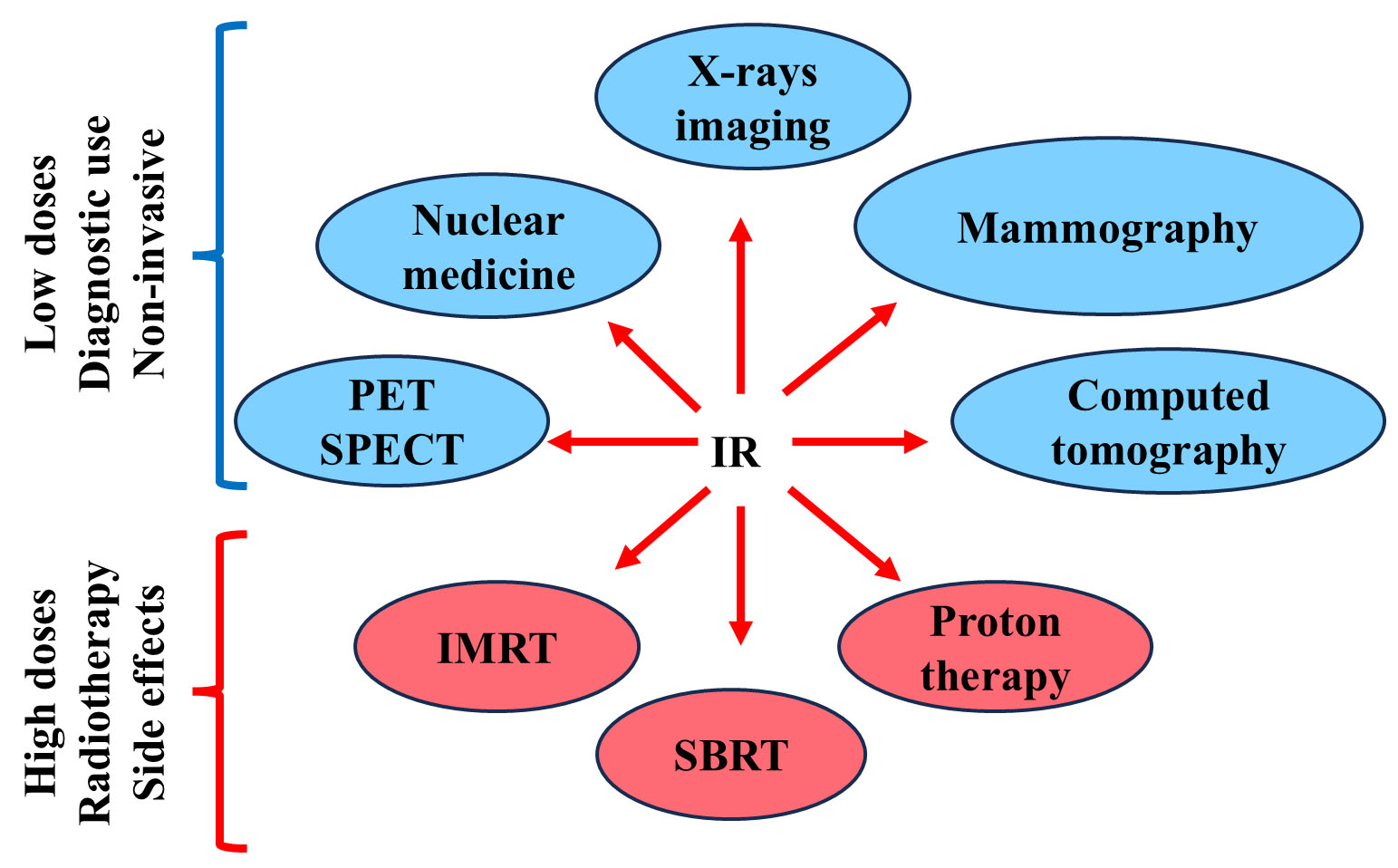 Fig. 3.
Fig. 3.
IR in diagnostic imaging and therapy. Diagnostic and therapeutic application of Low and High doses of IR in clinical applications. PET, positron emission tomography; SPECT, single-photon emission computed tomography; IMRT, intensity-modulated IR therapy; SBRT, stereotactic body radiotherapy.
On the other hand, radiotherapy involves higher doses of IR aimed at destroying cancerous cells or shrinking tumors. Indeed, radiotherapy is a cornerstone in the treatment of various cancers, including lung [74, 75], breast [76], cervical [77], prostate [78] and colorectal cancers [79], often as part of a multi-modal approach, combined with surgery, chemotherapy, or immunotherapy to improve outcomes [80]. It is used in curative treatment, adjuvant therapy post-surgery, or in palliative care to alleviate symptoms in advanced cancer stages. In Europe, the cancers with the highest number of patients in radiotherapy departments are breast cancer, lung cancer, prostate cancer, head and neck cancer, and rectal cancer [81]. Advances in IR technology, such as intensity-modulated IR therapy (IMRT), stereotactic body radiotherapy (SBRT), and proton therapy, have significantly improved precision, enabling targeted IR delivery to tumors while minimizing exposure to surrounding healthy tissues [82]. However, despite the precision of modern radiotherapy techniques, the higher doses used can cause side effects, including damage to normal tissues, skin reactions, fatigue, or long-term effects such as secondary cancers [83, 84]. While the therapeutic benefits of radiotherapy are substantial, the potential risks necessitate careful monitoring and planning to balance efficacy with patient safety.
NRF2 is involved in cellular response to IR and promotes a pro-survival response
in irradiated cells and tissues, both healthy and cancer ones, through ROS
detoxification, supporting DNA repair and modulating cytokine responses [85].
Literature data show that the activation of NRF2 signaling, either through
electrophilic modification of Keap1 or through deficiency in Keap1 expression,
leads to a reduction in intracellular ROS levels, increased cell viability and
resistance to treatments including radiotherapy, in almost healthy and tumor
cells [30, 86, 87, 88] The involvement of NRF2 in protecting from IR damage was first
demonstrated in a preclinical study using a lung cancer cell line carrying Keap1
gene mutation with consequent NRF2 hyperactivation. The authors showed that the
constitutive overexpression of NRF2 leads to low intracellular ROS levels and
IR-resistant cancer cell phenotype [89]. Importantly, shRNA-mediated reduction of
NRF2 expression induces ROS generation and increases protein oxidation resulting
in enhanced sensitivity to radiation-induced cell death [89]. Similar results
were obtained in mouse embryo fibroblasts (MEF) with gain of NRF2 function
(Keap1-/-) as well loss of NRF2 function (NRF2-/-).
The Keap1-/- MEF cells with high antioxidant capacity and low
endogenous ROS levels were shown to be resistant to ionizing radiation-induced
cell death. On the contrary, NRF2-/- MEF with low antioxidant
capacity and high endogenous ROS levels resulted in enhanced sensitivity to
Repeated pretreatment of human fibroblasts exposed to IR with sulforaphane (SFN), a natural substance present in cruciferous vegetables that activates NRF2 [90], has been demonstrated to protect fibroblasts from IR. This leads to reduced ROS levels, decreased DNA damage and increased cell survival [91]. In agreement, SFN treatment was unable to protect NRF2 knockout mouse embryonic fibroblasts, indicating that the sulforaphane-induced radioprotection was NRF2-dependent. Conversely, the use of RNA interference or pharmacological inhibition of NRF2 in these same cell types results in increased ROS production and a radiosensitive phenotype [91]. Similar results were obtained in colon cancer cells, with or without NRF2 knockdown with the CRISPR-Cas9 technology, treated with sulforaphane and chemotherapy. The results showed that SFN reduces cisplatin-induced cell death only in NRF2-proficient cells compared to NRF2-Cas9 cells. Mechanistically, the authors found that NRF2 activation protects NRF2-proficient cells from the drug-induced DNA damage and the apoptotic function of the unfolded protein response (UPR) [92], suggesting a common function of NRF2 in response to chemo- and radiotherapy.
In the study by McDonald and colleagues [88], the authors report that IR activates NRF2-ARE pathway but this occurs only after a significant delay of five days. This activation was detected in breast cancer cells, both with single doses of IR (2–8 Gy) and with clinically relevant daily dose fractions (0.5–4 Gy), and it was shown to be dose-dependent [88]. An important aspect is that no ARE activation was found in the 24 hours following IR exposure, even at high doses of up to 10 Gy. This suggests that the NRF2-ARE response to IR exposure is a second-tier antioxidant response, which is activated only a little later after exposure. Moreover, they observed that NRF2 activation prior to IR exposure does not improve the survival of cells or animals after irradiation, indicating that early activation of the pathway does not offer protection against IR damage under normal conditions. Conversely, the loss of NRF2 makes the cells significantly more radiosensitive, both in vitro and in vivo [88].
IR was also shown to activate NRF2 in a dose-dependent manner in non-small cell lung cancer (NSCLC) cells. RNAi-mediated reduction of NRF2 significantly increases endogenous ROS levels, and decreases the expression of NRF2 target genes, dampening Notch1 expression and inducing IR-induced cellular apoptosis. These authors conclude that the NRF2-mediated Notch signaling is an important determinant in radio-resistance of lung cancer cells [93].
Knockdown of NRF2 shows potential DNA damage after X-rays irradiation in lung cancers. The authors showed that NRF2 knockdown disrupts damaged DNA repair by inhibiting DNA-dependent protein kinase catalytic subunit and interfering with Rad51 expression, suggesting that NRF2 plays a critical role in the development of radio-resistance by upregulating DNA damage response via the mitogen-activated protein kinase (MAPK) pathway [94].
In a more recent study, the authors showed that NRF2-/- MEFs are
more sensitive to spontaneous and to IR-induced transformation and that NRF2
deficiency increases IR-induced NF-
Altogether, these data highlight the key role of NRF2 in cellular protection from the IR-induced damage and in pro-survival and radio-resistance in tumors.
In this paragraph the different mechanisms of NRF2-mediated response to IR will be analysed.
Exposure to IR induces oxidative stress leading to the production of ROS responsible for DNA damage. The activation of NRF2 following IR exposure induces the increased expression of antioxidant enzymes that neutralize ROS, thereby preventing cellular damage caused by IR [88] as above described. IR-induced damage can activate also Sirtuins such as Sirtuin 1 (SIRT1), that induces DNA repair and antioxidant defense. NRF2 and SIRT1 are therefore important players in the defense of normal tissues from IR damage, working through antioxidant and DNA repair mechanisms. On the other hand, NRF2 and SIRT1 overactivation in tumors induces radio-resistance. Their interconnected pathways highlight the complexity of cellular responses to radiation and offer potential targets for therapeutic interventions to protect against radiation-induced damage [4, 96].
In addition to its antioxidant activity, NRF2 plays a crucial role in repairing the DNA damage induced by IR through the HR pathway. Jayakumar and colleagues [97] investigated NRF2 role in DNA repair using two cancer cell models, where they inhibited NRF2 both pharmacologically and genetically. Their study confirmed that the inhibition of NRF2 results in increased radiosensitization in tumor cells, and consequent reduced survival fraction after IR exposure. Notably, NRF2 inhibition caused a significant delay in DNA repair, as evidenced by the persistence of residual DNA damage. This effect was not directly linked to NRF2-mediated antioxidant function. Even when ROS levels were reduced with N-acetyl cysteine (NAC), NRF2 inhibition still impaired DNA repair, suggesting that NRF2 involvement in DNA repair is independent of its activity in redox homeostasis. The authors found that NRF2 primarily affects the repair of DNA through the HR pathway, a critical pathway for DNA DSB repair. In support of this, NRF2 inhibition resulted in a significant reduction in IR-induced Rad51 foci formation, with Rad51 being a key protein in HR and its foci serving as a marker of HR pathway activation. Additionally, NRF2 inhibition decreased the mRNA levels of Rad51, indicating that NRF2 regulates Rad51 expression. However, NRF2 did not appear to affect the NHEJ repair pathway. When both NRF2 and DNA-dependent protein kinase (DNA-PK) (a key enzyme in NHEJ) were inhibited, there was a synergistic reduction in cell survival, suggesting that NRF2 does not act through this pathway [97].
According to Kim and colleagues [98], NRF2 repairs the broken DNA duplexes primarily by regulating p53 binding protein-1 (53BP1), a key component of DNA damage response. They found that NRF2 binds to the three ARE core sequences present in the 53BP1 promoter, resulting in an increase in 53BP1 expression. As described by Sekhar and Freeman [85], NRF2 plays a role in IR-induced DNA repair, also through base excision repair (BER). Chromatin immunoprecipitation (ChIP) assays have shown that NRF2 binds to the promoter of 8-Oxoguanine glycosylase 1 (OGG1), a glycosylase involved in base excision repair [99], and RNA interference experiments have demonstrated that NRF2 deficiencies suppress OGG1 expression [100]. The authors showed that OGG1 deficiency increases IR sensitivity in human cells, thus supporting the hypothesis of a mechanistic link between NRF2, DNA base damage repair, and IR sensitivity [100].
In addition, it was shown that NRF2 preserves genomic integrity by facilitating ATR activation and G2 cell cycle arrest [101]. Consistent with previous reports [97, 102], NRF2 influenced the protein levels of BRCA1 and Rad51. The inhibition of NRF2 by brusatol increases the radio-sensitivity of tumor cells in xenografts by perturbing ATR and CHK1 activation [101].
IR-induced damage that results from both indirect and direct interactions
between IR and biological molecules leads to tissue response including
inflammation, fibrosis, ferroptosis, and endothelial dysfunction [103, 104, 105, 106, 107, 108]. NRF2
activation in response to IR-induced oxidative stress may also modulate
inflammation, either by stimulating or suppressing inflammatory signals [103].
NRF2 achieves the anti-inflammatory effects by modulating antioxidant and
anti-inflammatory genes, such as for instance the HO-1 axis, which is a potent
anti-inflammatory target, by modulating macrophages, and by interacting with
other key inflammatory pathways like NF-
Several studies suggest a complex interaction between NRF2 and
NF-
IR-induced tissue fibrosis (RIF) is a common and delayed detrimental outcome of
delayed IR exposure [104, 113]. It is characterized by the abnormal activation of
myofibroblasts and excessive deposition of extracellular matrix components.
Differentiation of fibroblasts into myofibroblasts plays a critical role in the
development of fibrosis [114]. This process is promoted by increased levels of
ROS in fibroblasts, which result from oxidative stress induced by IR exposure
[115]. RIF can impact various organs, including lung, skin, liver, and kidney.
Extensive research has elucidated that RIF involves a complex interplay of
extracellular signals, such as immune cell activation and dysregulated cytokine
release, as well as intracellular signaling pathways, including cyclic GMP-AMP
synthase/stimulator of interferon genes (cGAS/STING), oxidative stress responses,
metabolic reprogramming, and proteasomal pathway activation, all contributing to
myofibroblast activation [105]. Clinical studies have highlighted the association
between NRF2 and IR-induced fibrosis [116]. Traver and colleagues [117]
demonstrated that upon IR exposure, NRF2 deficiency inhibits the mobilization of
NRF2 exerts a protective role in fibrosis by inhibiting
fibroblast-to-myofibroblast differentiation. Artaud-Macari and colleagues [118]
found that reduced NRF2 expression correlates with a myofibroblastic phenotype in
idiopathic pulmonary fibrosis, and NRF2 activation by sulforaphane led to the
dedifferentiation of myofibroblasts. Han and colleagues [119] observed that NRF2
activation by dimethyl itaconate protects against pulmonary fibrosis by
inhibiting Thioredoxin- interacting protein (TXNIP), an
Ferroptosis is a programmed, oxidative stress- dependent cell death process,
important in IR-induced cell death [122]. It presents with iron accumulation,
lipid peroxidation [45], and excess of ROS, as it kills cells by amplifying
oxidative stress or inhibiting the antioxidant system [123]. Ling and colleagues
[124] have suggested that ferroptosis plays a fundamental role in regulating EMT
in pulmonary fibrosis. Studies have shown that treatment of acute IR damage with
ferroptosis inhibitors results in a significant decrease in ROS levels and serum
inflammatory cytokines (TNF-
According to recent research, NRF2 is closely involved in inhibiting ferroptosis
by interfering with iron metabolism, inhibiting glutathione synthesis, and
enhancing DNA repair [126, 127, 128]. NRF2 positively regulates the transcription of
heme oxygenase 1 (HMOX1), increases iron storage, and reduces intracellular free
iron by rapidly upregulating the transcription of ferritin heavy (FTH) and light
(FTL) chains, thereby controlling the FTL/FTH ratio [129]. On the other hand,
NRF2 directly promotes the expression of glutathione peroxidase 4 (GPX4), a
central regulator in ferroptosis, by protecting cells against membrane lipid
peroxidation and regulates the GTP cyclohydrolase-1/tetrahydrobiopterin
(GCH1/BH4) pathway to mediate cellular redox reactions and inhibit ferroptosis
[130, 131]. Studies have revealed that NRF2 can also mediate glutathione synthesis
by promoting the expression of solute carrier family 7 member 11 (SLC7A11),
glutamate-cysteine ligase (GCLC), and glutathione synthetase (GSS), which play
crucial roles in preventing ferroptosis [132, 133, 134]. Hypoxia-inducible
factor-1
These data indicate that NRF2 may prevent ferroptosis by modulating proteins involved in iron metabolism and ROS detoxification pathways, ultimately reducing oxidative stress and inflammation caused by IR and enhancing cellular radio-sensitivity.
Modulation of NRF2 activity through genetic or pharmacologic approaches, as described above [54, 92] significantly affects cellular and tissue injury caused by IR, including radiotherapy. Below are described the main NRF2 activating or inhibiting molecules that are used in pre-clinical studies given that at the moment there are not molecules approved for clinical use. NRF2 activators enhance resistance to IR-induced damage by increasing antioxidants and detoxifying enzymes, reducing oxidative stress and DNA damage, and improving cell survival [137]. NRF2 activation is a key mechanism for many chemopreventive agents, helping to prevent tumorigenesis, although an excessive NRF2 activation may protect tumor cells from radiotherapy, reducing treatment efficacy [138]. NRF2 activators include a variety of natural and synthetic compounds, including sulforaphane, curcumin, and seleno-hormetic molecules.
Sulforaphane is one of the most extensively studied natural compounds that
targets the NRF2-Keap1 signaling pathway, recognized for its antioxidant,
chemopreventive and antiproliferative properties [139]. Mathew and colleagues
[91] demonstrated the radioprotective effect of sulforaphane in human skin
fibroblasts. Through site-directed mutagenesis and mass spectrometry analysis, it
was shown that sulforaphane can directly modify the critical cysteine residue at
position 151 of Keap1, causing the activation of the NRF2-Keap1-ARE signaling
pathway [140]. Curcumin is another extensively studied natural compound with many
therapeutic properties, including antioxidant and anticancer activities
[52, 53, 141, 142]. Notably, it was shown to act as a sensitizer for IR and
chemotherapy in several human cancers, including prostate cancer, by oncogene
mouse double minute2 (MDM2) downregulation [143] and colorectal cancer, by
suppressing NF-
Seleno-hormetic compounds exhibit a broad spectrum of biological activities including anticancer and antiproliferative effects and modulate hormetic genes and antioxidant enzyme functions. Studies have demonstrated the redox-modulating and IR-protective properties of seleno-hormetic compounds showing that seleno-compounds protect against IR-induced toxicity activating NRF2 transcription factor and, consequently, upregulating the adaptive stress response to IR [147].
While several compounds that activate NRF2 have been identified, there are only
a few agents recognized as NRF2 inhibitors with suitable pharmacokinetic
properties for in vivo use [137]. Given the aberrant activation and the
complex and varied role of NRF2 in cancer, these inhibitors are gaining interest
as potential anticancer agents. Indeed, they might enhance the responsiveness of
cancer cells to radio and chemotherapy and reduce chemo- and radio-resistance by
downregulating the activity of enzymes responsible for detoxification and drug
elimination through the inhibition of NRF2. However, NRF2 inhibitors might also
increase damage to healthy tissues, causing side effects like inflammation and
fibrosis [103, 115, 120, 121]. NRF2 inhibitors exert their activity at different
levels depending on their characteristics: they can regulate NRF2 at the
post-transcriptional level by degrading its mRNA or affecting NRF2 translation
and post-translational regulation [147, 148]. They can block the translocation of
NRF2 to the nucleus, or alter the binding between NRF2 and DNA [140, 149, 150].
NRF2 inhibitors can be identified in various classes of compounds, including
flavonoids, natural compounds found in fruits, vegetables, and plants, with
antioxidant, anti-inflammatory, and anticancer properties. While many flavonoids
have been identified as NRF2 activators, some have exhibited inhibitory effects.
For instance, in vivo studies have shown that luteolin reduces NRF2 mRNA
and protein levels, sensitizing cells to anticancer drugs [151]. However, the
effects of flavonoids on NRF2 activity are cell-type specific and
concentration-dependent, and their application in cancer therapy requires further
investigation. A known NRF2 inhibitor is brusatol, a natural compound extracted
from the plant Brucea javanica [148]. Brusatol has been shown to reduce
NRF2 protein levels by affecting NRF2 translation and post-translational
regulation in various cell lines, and sensitizes A549 lung tumor cells to the
anticancer drug cisplatin, both in vitro and in vivo [148, 152, 153]. However, the application of brusatol in humans has encountered
challenges due to unresolved issues such as toxicity, transient nature of the
reduction in NRF2 protein levels, and difficulties in drug delivery [152, 154].
Another interesting inhibitor of NRF2 is metformin (1,1-dimethylbiguanide
hydrochloride), a drug used to treat type 2 diabetes and approved as an
anticancer agent for certain tumors [155]. Metformin was shown to increase
sensitivity to anticancer treatments by reducing NRF2 mRNA and protein levels
through suppression of the rapidly accelerated fibrosarcoma (RAF)-ERK-NRF2
signaling pathway or induction of the microRNA-34a, which decreases NRF2 protein
expression via the SIRT1/peroxisome proliferator-activated
receptor gamma coactivator-1 alpha (PGC-1
In conclusion, NRF2 modulation presents a promising yet complex therapeutic strategy, requiring a careful balance between protecting normal tissues and enhancing tumor response to treatment, particularly radiotherapy. Moreover, the double role of NRF2 in both carcinogenesis and tumor progression adds further complexity to its therapeutic targeting [157]. Therefore, the use of specific NRF2 activators or inhibitors in cancer therapy must be approached with prudence. It is also important to note that, at present, no NRF2-targeting molecules have been approved for medical use.
NRF2 plays a crucial role in the cellular response to IR used both in clinical diagnostic imaging and radiotherapy. While NRF2 activation offers protection against IR-induced oxidative damage in healthy tissues and helps prevent the onset of long-term damages, including malignant transformation, it may also promote tumor viability and resistance to radiation therapy. Inhibiting NRF2 represents an encouraging strategy to improve sensitivity of tumor cells to IR and the development of NRF2 inhibitors for clinical use as adjuncts to radiotherapy holds great potential. However, several challenges remain. One of the main obstacles is the selective targeting of NRF2 pathways in tumor cells without affecting normal tissues, which could increase the risk of side effects. Moreover, the potential for compensatory mechanisms in the cell, where other stress response pathways may take over when NRF2 is inhibited, must be carefully considered in the design of combination therapies. Further research is needed to better understand the precise role of NRF2 in IR responses and to identify the optimal timing and dosing strategies for NRF2 inhibition. Clinical trials assessing the safety and efficacy of NRF2 inhibitors, either alone or in combination with existing treatments, are essential to translating these findings into therapeutic practice.
AV and GD designed the outline, wrote the original draft, and reviewed the literature. Both authors contributed to editorial changes in the manuscript. Both authors read and approved the final manuscript. Both authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
The authors wish to thank S. Soddu for critical reading of the manuscript.
This work was supported by the institutional “Ricerca Corrente” granted by the Italian Ministry of Health.
The authors declare no conflict of interest. Given her role as the Guest Editor, Gabriella D’Orazi had no involvement in the peer-review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Alexandros G. Georgakilas and Graham Pawelec.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.