

 , Li Guo 2,†, Xianghua Dou 1,†, Wenwen Jiang 2, Bowen Qian 2, Jian Liu 1, Jun Wang 2, Chunling Wang 1, Congmin Xu 1,3,*
, Li Guo 2,†, Xianghua Dou 1,†, Wenwen Jiang 2, Bowen Qian 2, Jian Liu 1, Jun Wang 2, Chunling Wang 1, Congmin Xu 1,3,*1 Shandong Key Laboratory of Biophysics, Institute of Biophysics, Dezhou University, 253023 Dezhou, Shandong, China
2 Department of Bioinformatics, Smart Health Big Data Analysis and Location Services Engineering Lab of Jiangsu Province, School of Geographic and Biologic Information, Nanjing University of Posts and Telecommunications, 210023 Nanjing, Jiangsu, China
3 Department of Biomedical Engineering, Georgia Institute of Technology and Emory University, Atlanta, GA 30332, USA
† These authors contributed equally.
Abstract
Background: Small open reading frames (sORFs) with protein-coding ability present unprecedented challenge for genome annotation because of their short sequence and low expression level. In the past decade, only several prediction methods have been proposed for discovery of protein-coding sORFs and lack of objective and uniform negative datasets has become an important obstacle to sORFs prediction. The prediction efficiency of current sORFs prediction methods needs to be further evaluated to provide better research strategies for protein-coding sORFs discovery. Methods: In this work, nine mainstream existing methods for predicting protein-coding potential of ORFs are comprehensively evaluated based on a random sequence strategy. Results: The results show that the current methods perform poorly on different sORFs datasets. For comparison, a sequence based prediction algorithm trained on prokaryotic sORFs is proposed and its better prediction performance indicates that the random sequence strategy can provide feasible ideas for protein-coding sORFs predictions. Conclusions: As a kind of important functional genomic element, discovery of protein-coding sORFs has shed light on the dark proteomes. This evaluation work indicates that there is an urgent need for developing specialized prediction tools for protein-coding sORFs in both eukaryotes and prokaryotes. It is expected that the present work may provide novel ideas for future sORFs researches.
Keywords
- Small open reading frames
- Small protein
- Gene prediction
- Genome annotation
- Protein-coding gene
Small proteins (shorter than 100 amino acids) encoded by small open reading frames (sORFs) have been ignored in genome annotations during the past decades. Reports of two functional small peptides myoregulin and DWORF encoded by transcripts that had been annotated as long noncoding RNAs [1, 2] aroused unprecedented attention to small open reading frames (sORFs) and their encoded proteins [3, 4, 5, 6, 7]. The discovery of protein-coding sORFs also led to a debate on the definition of noncoding RNA and rethinking of the understanding of genome [8, 9, 10, 11, 12]. Thus, sORFs have gradually become a research hotspot in the past few years. Actually, sORFs have been seriously underestimated because they were believed too short to encode proteins, so that earlier literatures called them evil little fellows (ELFs) [13]. In most cases, traditional methods are not suitable for short sequences [14, 15], therefore identifying protein-coding sORFs is a huge challenge for genome research. Recently, rapid development of versatile omics sequencing technologies such as mass spectrometry and ribosome profiling reveal a large number of protein-coding sORFs with important functions in different genomic regions [12, 16, 17, 18]. Even so, the proteogenomic methods are not sensitive enough and the ribosome profiling sequencing strategies require additional measures to ensure comprehensive and accurate sORF annotation [5], hence there is still lack of efficient technologies for sORF identification [7, 16, 19, 20]. Furthermore, the resolution of ribosome profiling for bacterial cells is lower than that for eukaryotic cells due to technical challenges [18]. Therefore, most sORFs studies mainly focus on several model eukaryotes such as human, mouse, Arabidopsis Thaliana. Thus, a limited number of protein-coding sORFs prediction programs trained by eukaryotic sORFs have been developed recently [21, 22, 23, 24, 25]. Among them, sORF finder [21], MiPepid [25], CPPred-sORF [24] were specially designed for sORFs. Some coding potential prediction programs for normal ORFs, such as CPPred and e also tested on sORFs datasets. These programs provide alternative tools for sORFs detection, but the real efficiency need to be further evaluated. On the other hand, lack of reliable datasets particularly negative samples has become one of the key issues in sORF prediction [5, 14, 16, 19, 24, 25]. Construction of reliable sORFs dataset and annotation platforms have been the foremost challenge in the field [26]. Then, in this work, we perform comprehensive evaluation of nine up-to-date ORF coding potential prediction programs that have been discussed in recent sORFs related studies [16, 27] based on a random sequence strategy. It is expected that the present work may provide novel ideas for future sORFs researches.
The prokaryotic sORFs were filtered from the RefSeq database [28]. The human and mouse sORFs were downloaded from sORFs.org database [29], and the sORFs of Arabidopsis thaliana were downloaded from the TAIR database [30].
Four non-redundant positive datasets (Hum-7111 dataset, Mou-7385 dataset, Ara-2125 dataset, Pro-6318) are constructed in this work. To construct the Hum-7111 dataset and the Mou-7385 dataset, 10000 human sORFs and 10000 mouse sORFs were downloaded from the sORF.org database respectively, and 2888 Arabidopsis thaliana sORFs were downloaded from the TAIR database to construct the Ara-2125 dataset. To construct the Pro-6318 dataset, the sORFs with definite functions were derived from 56 prokaryotic genomes (Supplementary Table 1), the genomic GC contents of the 56 selected prokaryotic genomes have a wide range from 20% to 70%. Thus a total of 6578 prokaryotics sORFs were obtained. These candidate sORFs were further filtered as follows:
(i) Excluding the redundant sequences in each dataset using the CD-Hit program [31] with the similarity threshold of 80% at DNA level;
(ii) Excluding the sORF
(iii) Excluding the sORFs whose sequence length cannot be divisible by 3;
(iv) Excluding the sORFs that do not end with a stop codon;
(v) Excluding the sORFs with stop codon in its sequence;
(vi) Excluding the sORFs that start with stop codon.
In this way, 7111 human sORFs, 7385 mouse sORFs, 2125 Arabidopsis thaliana sORFs and 6318 prokaryotic sORFs are obtained. These datasets are as the positive testing sets. In Supplementary file 1, the four datasets are provided in fasta format.
It is difficult to construct negative sORFs datasets. The sORFs from intergenic region and noncoding region were usually extracted as negative samples, but there is great possibility of the existence of protein-coding sORFs in these regions. For prokaryotes, there are few noncoding and intergenic regions, therefore constructing negative samples is a challenging task for sORF prediction. Negative ORFs generated based on random sequencestrategies have been used in gene prediction works [32, 33]. Then, in this work, a strict negative sORFs generating strategy is proposed by following steps:
(i) Randomly shuffling each positive sORF sequence to get a corresponding negative sequence without any stop codons before the stop codon at the end of sequence;
(ii) Ensuering that the negative sequence shares the same start codon and stop codon with its original positive sORF and there is no pre-mature stop codon in the sequence.
(iii) Excluding the redundant sequences with the abovementioned standard.
Furthermore, an experimentally verified dataset (Eexp-150-53) released by Hemm et al. [18] is also employed as test set. This dataset includes 150 positive sORFs and 53 negative sORFs detected from E. coli genome.
At present, there are only a few sORF prediction programs. Some prediction programs reviewed in recent works are proposed for long ORF, but several of them are also applied to sORFs [16, 27]. Then, in total of nine ORF coding potential prediction programs [22, 23, 24, 25, 34, 35, 36, 37, 38] with source codes available are evaluated based on the test sets constructed above. The operating system and parameters used to run these programs are listed in Table 1. It is noted that MiPepid, CPPred-sORF are specially proposed for sORFs. On the other hand, although the abovementioned sORFs prediction programs are trained on the ORFs (sORFs) derived from different eukaryotic species, some of them were declared to have cross species prediction ability. Even so, no uniform standard has been proposed to measure their real efficiency. Therefore, different sORFs prediction programs are evaluated in this work.
| Programs | Parameters | Type | Operating system |
| CPC2 | - | - | online |
| CPPred | Integrated | - | Linux |
| DeepCPP | Human | sORF | Windows |
| CPPred-sORF | Integrated | - | Linux |
| CPAT | - | - | online |
| MiPiped | - | - | Windows |
| CNCI | Vertebrate | - | Linux |
| PLEK | - | - | Linux |
| LGC | - | - | online |
Currently, most sORFs studies mainly focus on several model eukaryotes such as human and mouse. To verify the efficiency of the random sequence strategy, we propose an alternative protein-coding sORFs prediction algorithm (PsORFs) based on the presented random sequence strategy. This algorithm uses the frequency of the 64 kinds of codons as numerical parameters, and the random forest is adopted as classifier. Detailed description of this prediction algorithm is provided in Supplementary file 2. Our earlier studies indicated that some prokaryotic genomes exhibit properties of universal protein-coding genes regardless of their genome sizes and genomic GC contents [32]. The protein-coding genes in these genomes can be used as training set to accurately predict the protein-coding genes in other prokaryotic genomes. Then, the protein-coding sORFs with known functions derived from five prokaryotic genomes (NC_009089, NC_003103, NC_012962, NC_000913, NC_008380) are adopted as the positive training set, and the negative sORFs in the training set are generated according to the random sequence generation procedure mentioned above. Furthermore, the training set is processed according to the abovementioned filtering steps, and finally 1228 positive sORFs and 1327 negative sORFs are obtained. In Supplementary file 3, we provide the training sets in fasta format.
For evaluation purpose, the sensitivity (s
In addition, the Matthew’s correlation coefficient (MCC) is also used to describe the agreement of prediction and annotation with a single value in the range of [–1, 1], i.e.,
Where, TP and TN denote the number of coding sORFs and
non-coding sORFs that have been correctly predicted respectively, FP and
FN denote the number of coding sORFs and non-coding sORFs that have been
falsely predicted respectively. Then, s
Computational methods can provide quick and convenient tool for sORFs prediction. Several of the nine computational algorithms evaluated in this work have been tested in sORFs in their original literatures [22, 23, 24, 25], and we summarize their reported performances in Supplementary Table 2. The CPPred program got its best performance with ACC 0.8788 and MCC 0.7650 on their integrated test set. As its improved version, CPPred-sORF got its best performance with ACC 0.8849 and MCC 0.7680 on an integrated eukaryotic sORFs dataset. Mipepid got prediction accuracy 0.9576 on integrated dataset and 0.96 on human sORFs dataset, but it only predicts the sORFs start with ATG. DeepCPP is a deep neural network-based method for RNA coding potential prediction, and its reported prediction accuracy and MCC on a human sORFs test set are 0.8858 and 0.7740, respectively. The sORF finder program seems to achieve the lowest performance. Lack of reliable negative samples is one of the key challenges for sORFs prediction. As two specially designed programs for protein-coding sORFs prediction, Mipepid and CPPred-sORF defined ORFs derived from miRNA and lncRNA as negative samples in their training and test datasets respectively. In Fig. 1, we analyzed the length distribution of the protein coding datasets in Mipepid and CPPred-sORF. Obviously, there is apparent length bias between the positive dataset and negative dataset in both programs. It is noted that more than 90% negative samples from Mipepid are shorter than 20 aa, while more than 80% negative samples from CPPred-sORF are longer than 100 aa. It means that one can discriminate negative samples from positive samples only based on their length. However, sORFs generally exist in different parts of the genome, so the length of sORFs cannot be different between positive and negative samples. Therefore, in order to develop the prediction method of sORFs better, it is necessary to evaluate these procedures based on the third party datasets objectively.
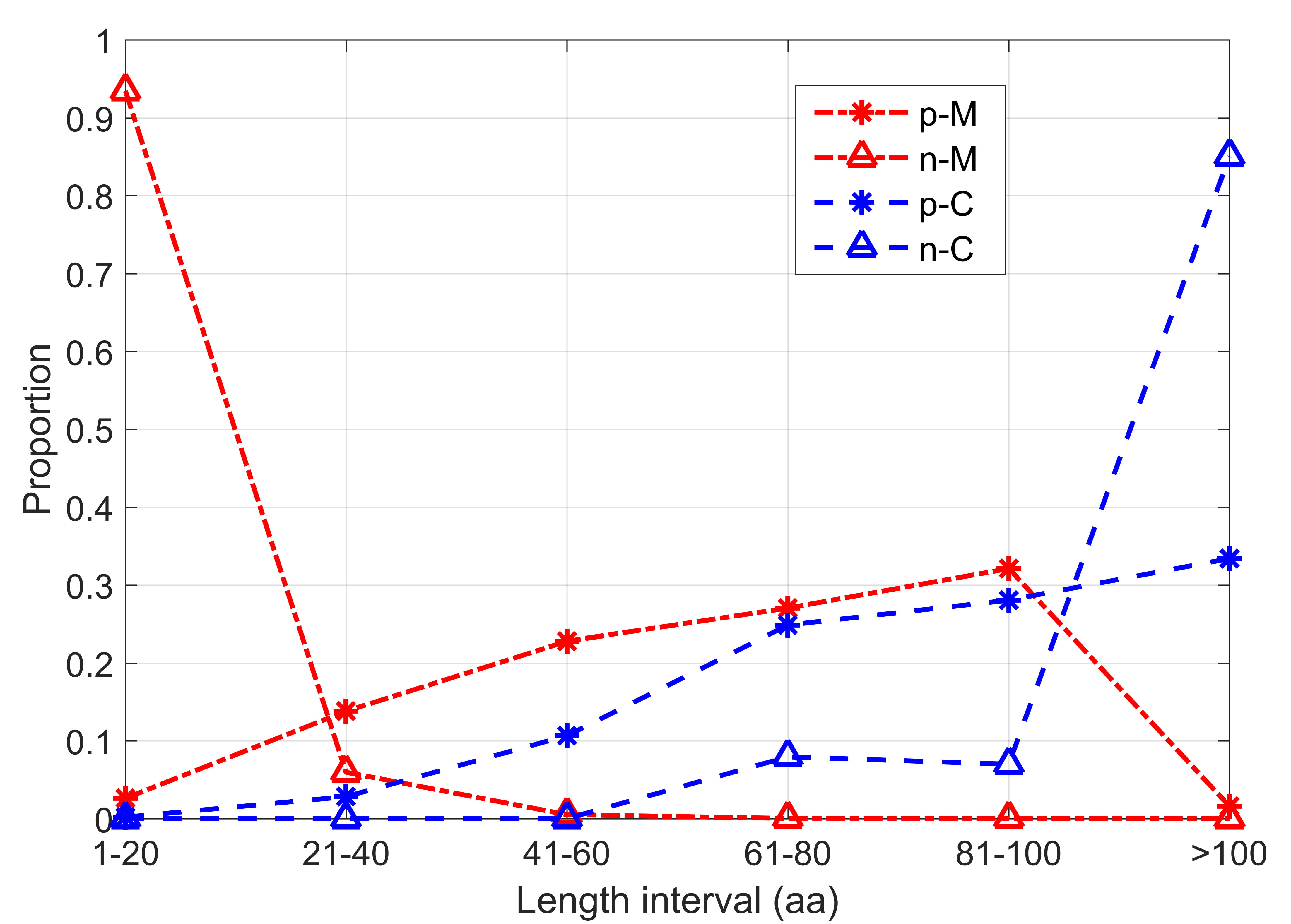 Fig. 1.
Fig. 1.Length distribution intervals of each sORFs dataset. Where, p-M and n-M represent the positive and negative samples of Mipepid, p-C and n-C represent the positive and negative samples of CPPred-sORF.
The original prediction results of each program are provided in
Supplementary Tables 3.1–3.5, where coding and noncoding represent the positive
or negative sORFs that are predicted as coding sORFs or noncoding sORFs, and
unknown represents the sORFs cannot be predicted (among these programs, MiPiped
can only predict the sORFs start with start codon of ATG, therefore the sORFs
with other start codons cannot be predicted). In Table 2, we provide the
prediction efficiency of different programs based on the four random
sequence-based test datasets of Hum-7115, Ara-2142, Mou-7385, Pro-6578 and the
experimental verified dataset of Eexp-150-53. It can be seen from the results
that the prediction performances of different programs for the two data types are
consistent. For comparison, we mark the lower s
| Dataset | Index | Programs | |||||||||
| CPC2 | CPPred | DeepCPP | CPAT | CNCI | PLEK | LGC | CPPred-sORF | MiPiped* | PsORFs | ||
| Hum-7111 | s |
0.0014 | 0.0634 | 0.0416 | 0.0678 | 0.0553 | 0 | 0.0018 | 0.8187 | 0.9474 | 0.6840 |
| s |
0.9993 | 0.9535 | 0.9854 | 0.9941 | 0.9684 | 1 | 0.9977 | 0.2313 | 0.0860 | 0.5022 | |
| ACC | 0.5004 | 0.5084 | 0.5135 | 0.5309 | 0.5118 | 0.5000 | 0.4998 | 0.5250 | 0.5167 | 0.5928 | |
| MCC | 0.0108 | 0.0370 | 0.0817 | 0.1642 | 0.0579 | - | 0.0047 | 0.0619 | 0.0656 | 0.1887 | |
| Ara-2125 | s |
0.0918 | 0.3842 | 0.3802 | 0.0847 | 0.0513 | 0 | 0.2221 | 0.9939 | 0.9876 | 0.3934 |
| s |
0.9139 | 0.7741 | 0.7152 | 0.9986 | 0.9506 | 1 | 0.7699 | 0.0028 | 0.0338 | 0.9487 | |
| ACC | 0.5028 | 0.5778 | 0.5478 | 0.5416 | 0.5009 | 0.5000 | 0.4960 | 0.4984 | 0.5107 | 0.6656 | |
| MCC | 0.0099 | 0.1719 | 0.1014 | 0.2052 | 0.0043 | - | 0.0096 | –0.0247 | 0.0713 | 0.3970 | |
| Mou-7385 | s |
0.0027 | 0.0726 | 0.0450 | 0.0269 | 0.0489 | 0 | 0.0018 | 0.8079 | 0.9677 | 0.3885 |
| s |
0.9995 | 0.9541 | 0.9874 | 0.9981 | 0.9671 | 1 | 0.9992 | 0.2265 | 0.0495 | 0.7607 | |
| ACC | 0.5011 | 0.5213 | 0.5162 | 0.5125 | 0.5080 | 0.5000 | 0.5005 | 0.5172 | 0.5086 | 0.5853 | |
| MCC | 0.0269 | 0.0567 | 0.0968 | 0.1051 | 0.0403 | - | 0.0132 | 0.0423 | 0.0433 | 0.1758 | |
| Pro-6318 | s |
0.1009 | 0.5426 | 0.4635 | 0.3118 | 0.1610 | 0 | 0.1364 | 0.9737 | 0.8763 | 0.9888 |
| s |
0.9805 | 0.8048 | 0.7134 | 0.9926 | 0.9649 | 1 | 0.8627 | 0.0635 | 0.1123 | 0.8845 | |
| ACC | 0.5407 | 0.6737 | 0.5885 | 0.6522 | 0.5629 | 0.5000 | 0.4995 | 0.5186 | 0.4931 | 0.8996 | |
| MCC | 0.1713 | 0.3600 | 0.1828 | 0.4155 | 0.2116 | - | 0.0013 | 0.0899 | –0.0177 | 0.8032 | |
| Eexp-150-53 | s |
0 | 0.2333 | 0.0200 | 0.0133 | 0.0933 | 0 | 0 | 1 | 0.8593 | 0.9434 |
| s |
1 | 0.8679 | 0.9808 | 1 | 0.9245 | 1 | 1 | 0.0755 | 0.1556 | 0.4133 | |
| ACC | 0.2611 | 0.3990 | 0.2673 | 0.2709 | 0.3103 | 0.2611 | 0.2611 | 0.7586 | 0.6833 | 0.5517 | |
| MCC | - | 0.1098 | 0.0024 | 0.0593 | 0.0276 | - | - | 0.2385 | 0.0182 | 0.3358 | |
| * The indexes of MiPiped are evaluated by the sORFs start with ATG. The lower
s | |||||||||||
Protein-coding sORFs have a widespread occurrence in diverse species and can be of high functional importance. However, no single identification method developed to date is sufficient to identify all sORFs, hence sORFs detection is a multidisciplinary strategy [16]. The evaluation results of CPPred-sORF and MiPiped indicate that protein-coding sORFs prediction is still in its infancy. There are few noncoding regions in prokaryotic genomes, so it is very difficult to construct prokaryotic negative sORFs datasets. Then, we propose the PsORFs model based on the random sequence strategy. The random forest is employed as the core algorithm to train the PsORFs model. The number of bags was set as 200 according the evaluation result during K-fold cross validation. The five-fold cross validation was used to evaluate the model performance, the accuracy and MCC (threshold set as 0.5) of which are 0.8925 and 0.7852, respectively. To compare with other programs, PsORFs is evaluated by the five independent test datasets and its prediction results are also provided in Table 2. It can be found that the prediction efficiency of PsORFs is better than other methods in each test dataset. Although PsORFs is trained based on the prokaryotic sORFs, its prediction efficiency in eukaryotic sORFs is superior to other programs, which indicates the random sequence can provide robust data sources for sORFs prediction. The source code of PsORFs algorithm in Matlab format can be downloaded from http://211.64.32.111:8888/.
The important roles of protein-coding sORFs in biological activities have been confirmed by a large number of studies in recent years. As a kind of important functional genomic element, discovery of protein-coding sORFs has shed light on the dark proteomes [39]. In this work, we evaluated different types of prediction programs, and the results showed that our evaluation study can provide important theoretical basis and novel ideas for sORFs discoveries.
JY and CX designed the experiments; JY, LG, XD perform the experiments; XD, BQ, WJ and CW wrote the codes, JY, CX, WJ, BQ developed the prediction method, LG, JW, CW analyzed the data; JY, LG, JW and CX wrote the manuscript.
Not applicable.
Thanks to all the peer reviewers for their opinions and suggestions.
This work was supported by the National Natural Science Foundation of China (61771093, 62011530044 and 61671107), the Youth Science and technology innovation plan of universities in Shandong (2019KJE007).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://www.fbscience.com/Landmark/articles/10.52586/4943.