


 , Yu-Yu Li 2,3, Chia-Hung Yu 1, Kuo-Chuan Hung 1,4,†, Chin-Chen Chu 1, Ping-Hsun Feng 5,*
, Yu-Yu Li 2,3, Chia-Hung Yu 1, Kuo-Chuan Hung 1,4,†, Chin-Chen Chu 1, Ping-Hsun Feng 5,* , Ping-Heng Tan 1,4,*
, Ping-Heng Tan 1,4,*1 Department of Anesthesiology, Chi Mei Medical Center, 701 Tainan, Taiwan, ROC
2 Department of Anesthesiology, Chi-Mei Hospital, Chiali, 722 Tainan, Taiwan, ROC
3 Department of Leisure and Sports Management, CTBC University of Technology, 744 Tainan, Taiwan, ROC
4 School of Medicine, College of Medicine, National Sun Yat-Sen University, 800 Kaohsiung, Taiwan, ROC
5 Department of Anesthesiology, Chi Mei Medical Center, Liouying, 736 Tainan, Taiwan, ROC
†These authors contributed equally.
Abstract
Interferons (IFNs) are cytokines with diverse functions, possessing antiviral, antiproliferative, and immunomodulatory effects. IFN-α and IFN-β, key members of the type I interferon (IFN-I) family, are widely used in the treatment of diseases such as hepatitis and multiple sclerosis. In the nervous system, microglia, astrocytes, and neurons express IFN-I receptors. Beyond their classical transcriptional roles, IFN-Is can suppress neuronal activity and synaptic transmission through nongenomic mechanisms, producing potent analgesic effects. However, IFN-Is are active in signaling pathways such as phosphoinositide 3-kinase (PI3K), mitogen-activated protein kinase (MAPK), and the MAPK-interacting serine/threonine-protein kinase (MNK)-eukaryotic initiation factor 4E (eIF4E) pathway, which can sensitize peripheral nociceptors and contribute to nociceptive responses. This narrative review explores recent advances in understanding the roles of IFN-I and the cyclic-GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) signaling cascade in acute and chronic nociceptive responses, which are increasingly recognized but remain a subject of debate. Recent studies suggest that the STING–IFN-I pathway has complex, stage-dependent effects on nociception. In the middle to late stages of the nociceptive response, this pathway can activate signal transducer and activator of transcription (STAT) signaling, as well as microglial mediated STING pathways and tumor necrosis factor (TNF) receptor-associated factor (TRAF) family member-associated nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB activator) collectively referred to as TANK. These pathways increase pro- and anti-inflammatory cytokine production, promote microglial M1 polarization, and inhibit endoplasmic reticulum-phagy (ER-phagy) in the central nervous system (CNS). These mechanisms contribute to central sensitization while modulating the analgesic effects of IFN-Is. Thus, the STING-IFN-I pathway plays a dual role in nociception, with both pro-nociceptive and analgesic effects that are dependent on the stage of the nociceptive response. Understanding the differential roles of STING–IFN-I signaling in nociceptors under physiological and pathological conditions could pave the way for the development of targeted nociceptive response management therapies.
Graphical Abstract
Keywords
- nociceptive response
- type I interferon
- STING
- IFN-α
- IFN-β
- type I interferon receptor
Interferons (IFNs) were first discovered in 1957 and named for their ability to
“interfere” with viral replication [1]. Over the years, significant progress
has been made in understanding their mechanisms of action and therapeutic
potential. Recent studies have elucidated the pivotal role of IFNs in combating
viral infections with hepatitis B and C, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), and various emerging viral
pathogens [2, 3, 4, 5]. For example, type I IFNs (IFN-Is), including IFN-
Following viral or bacterial infections or tissue injury, pattern recognition
receptors (PRRs) such as Toll-like receptors (TLRs) detect pathogen- and
damage-associated molecular patterns (PAMPs and DAMPs). This activation triggers
the release of cytokines and chemokines, including tIFN-Is, through neuroimmune
interactions, alerting the host immune system to potential threats [10, 11]. TLRs
2, 4, and 5, which are located on the cell surface, primarily detect bacterial
PAMPs. TLR4 can also be found to a lesser extent in endosomes, whereas TLR3 and
TLR7/8 are endosomal receptors that respond mainly to viral and bacterial nucleic
acids, specifically, double-stranded RNA (dsRNA) and single-stranded RNA (ssRNA),
respectively [12, 13]. For example, TLR3 detects viral dsRNA and its synthetic
analog poly (I:C), whereas TLR7/8 can be activated by imidazoquinoline-like
molecules (imiquimod and resiquimod). TLR9, another endosomal TLR, recognizes
dsDNA and unmethylated cytidine-phosphate-guanine deoxyribonucleic acid (CpG)
DNA. The activation of TLRs such as TLR4 by bacterial lipopolysaccharides (LPS)
and viral proteins induces robust IFN-I production and triggers specific
intracellular signaling cascades [10]. When a ligand binds to a TLR, adaptor
proteins such as TIR-domain-containing adapter-inducing IFN-
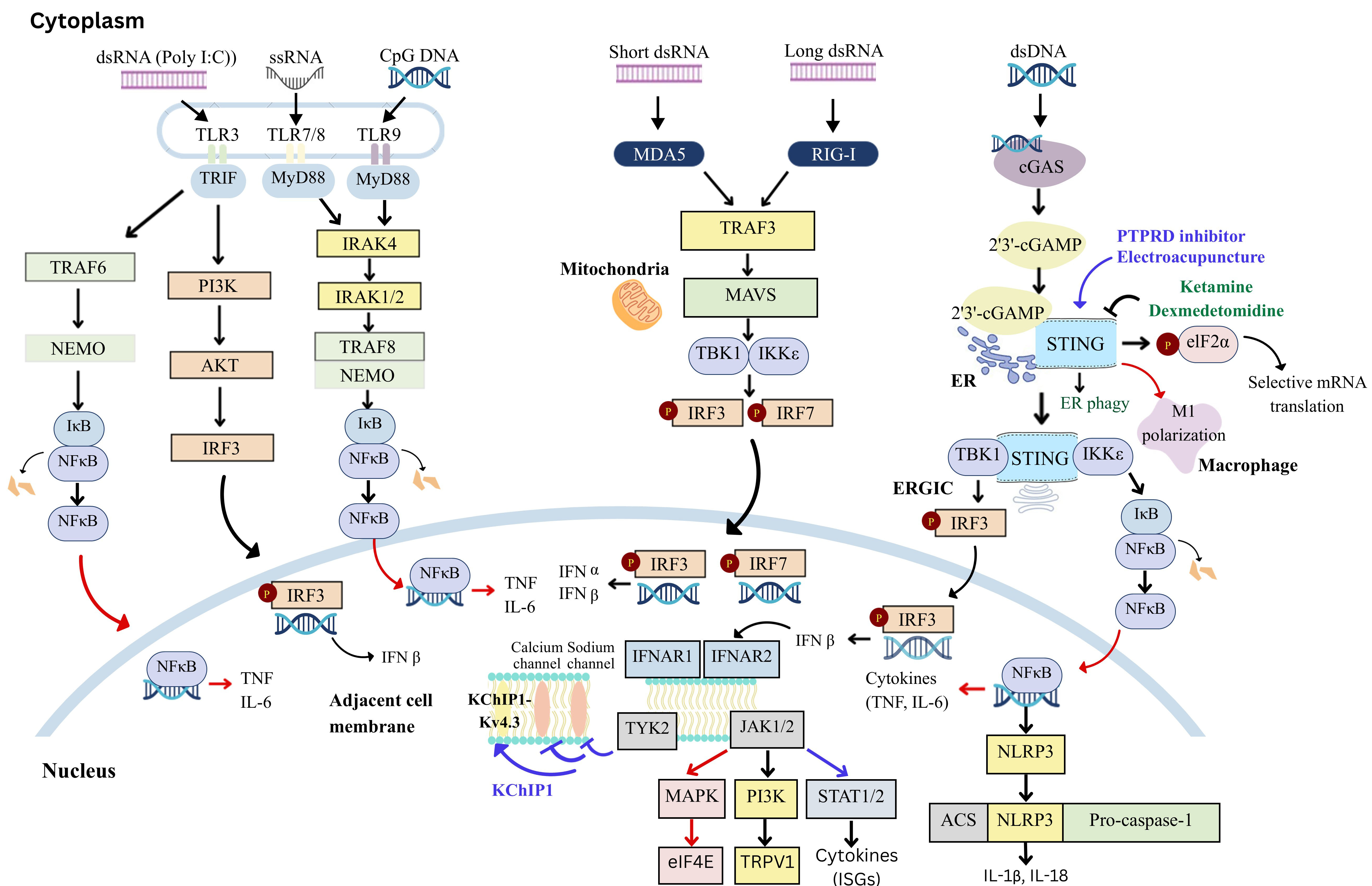 Fig. 1.
Fig. 1.
Pattern recognition and type I interferon (IFN-I) signaling
pathways. IRF3, interferon regulatory factor 3; IRF7, interferon regulatory
factor 7; RIG-I, retinoic acid-inducible gene I; MDA5, melanoma
differentiation-associated protein 5; MAVS, mitochondrial antiviral signaling
protein; MYD88, myeloid differentiation primary response 88; NF-
The signaling pathways involved in the production of IFN-Is through the
activation of toll-like receptors (TLRs), melanoma differentiation-associated
protein 5 (MDA5) and retinoic acid-inducible gene I (RIG-I)-like receptors are
shown. TLRs, which are key pattern recognition receptors, detect pathogens and
are located on the cell surface or within intracellular compartments such as
endosomes (e.g., TLR3 and TLR7-9) in immune and glial cells. The induction of
IFN-Is occurs via distinct intracellular signaling molecules: TRIF mediates
signaling for TLR3 and TLR4, while MyD88 facilitates signaling for TLR7-9.
Additionally, IFN-Is are produced through the activation of the stimulator of
interferon genes (STING) signaling cascade. STING, an adaptor protein in the
endoplasmic reticulum, responds to viruses and intracellular DNA by activating
TBK1. This, in turn, stimulates the transcription factors NF-
In addition to TLRs, cytoplasmic sensors such as Asp-Glu-x-Asp (DExD)/H-box RNA
helicases and STING detect microbial RNA and DNA to induce IFN-I production [6].
This pathway includes receptors such as retinoic acid-inducible gene I
(RIG-I)-like helicases, melanoma differentiation-associated protein 5 (MDA5) and
cGAS, which synthesizes cyclic guanosine monophosphate–adenosine monophosphate
(cGAMP) upon the detection of cytosolic DNA [14, 15, 16]. STING is a transmembrane
protein in the endoplasmic reticulum (ER) that responds to cGAMP or other
bacterial cyclic dinucleotides (CDNs). In the Golgi, STING interacts with TBK1
once it is translocated from the ER to the ER-Golgi intermediate compartment
(ERGIC). STING and IRF3 are phosphorylated by TBK1, promoting IRF3 nuclear
translocation and inducing IFN-I production [12, 17, 18, 19] (Fig. 1). IFN-I
production supports immune responses by activating cells such as macrophages and
natural killer (NK) cells. Plasmacytoid dendritic cells (pDCs) are key producers
of IFN-Is, particularly during viral infections [20, 21, 22]. Additionally, STING can
activate NF-
IFN-
Upon ligand binding, IFNAR1 associates with tyrosine kinase (TYK)2, and IFNAR2 interacts with janus kinase (JAK)1, triggering receptor subunit rearrangement and dimerization. This conformational change activates receptor-associated JAKs, leading to the phosphorylation of tyrosine residues on IFNAR, which serve as docking sites for signal transducer and activator of transcription (STAT) proteins. Activated JAKs phosphorylate STAT1 and STAT2, initiating the JAK-STAT signaling cascade [27] (Fig. 1). Phosphorylated STAT1 and STAT2 dimerize and recruit IRF9 to form the interferon-stimulated gene factor 3 (ISGF3) complex. This complex translocates to the nucleus and binds interferon-stimulated response elements (ISREs) in the promoters of ISGs, driving their transcription. ISGs encode proteins with antiviral, immunomodulatory, and apoptotic functions that are essential for effective host defense. In parallel, phosphorylated STAT1 homodimers bind gamma-activated sequence (GAS) elements in ISG promoters, providing an additional layer of regulation. The ISGF3 complex and STAT1 homodimers act synergistically to amplify ISG expression, ensuring a robust and tailored response to type I IFN signaling. This dual mechanism allows fine-tuning of gene expression depending on the cellular context and external stimuli. Notably, some ISGs further enhance IFN signaling, establishing positive feedback loops that amplify the antiviral response [28]. These ISGs inhibit viral replication in infected cells and provide protection to adjacent uninfected cells [29]. ISG15, viperin, ribonuclease L (RNase L), myxovirus resistance (Mx), and 2′-5′-oligoadenylate synthetase (OAS) are key antiviral ISGs. Many of the biological effects of IFN-I are mediated by the JAK-STAT signaling cascade. Studies [27, 28] have shown that IFN-Is can regulate hundreds to thousands of genes via this canonical pathway (Fig. 1). In addition to the JAK-STAT pathway, IFNAR1/2 activation also engages noncanonical signaling cascades, such as the phosphoinositide 3-kinase (PI3K) and mitogen-activated protein kinase (MAPK) pathways, expanding the biological effects of IFN-Is [6] (Fig. 1).
Regulatory mechanisms involve suppressor of cytokine signaling (SOCS) proteins,
such as SOCS1 and SOCS3. SOCS1 dampens IFN-I responses by directly inhibiting
JAK2 and STAT1
IFN-Is have been successfully used to treat various viral infections, including
hepatitis B and C, as well as autoimmune diseases such as multiple sclerosis
(MS), where they reduce inflammation and disease activity [31]. In cancer
therapy, IFN-Is enhance cytotoxic T lymphocyte and natural killer cell
activation, promote tumor cell apoptosis, and inhibit angiogenesis, with evidence
demonstrating improved disease-free survival in melanoma patients [32]. In
addition to these therapeutic effects, IFN-Is modulate immune responses by
inducing anti-inflammatory cytokines such as interleukin-10 (IL-10) [19] and
programmed cell death-ligand 1 (PD-L1) [33] while simultaneously suppressing
proinflammatory mediators such as matrix metalloproteinase-9 (MMP-9) and tumor
necrosis factor-
IFN-Is, particularly IFN-
Neuroinflammation, a hallmark of various CNS disorders such as strokes, injuries, neurodegenerative diseases, and chronic pain syndromes, is characterized by blood-brain barrier (BBB) disruption, immune cell infiltration, and the activation of glial cells. Activated microglia and astrocytes release proinflammatory mediators, including cytokines, chemokines, and matrix metalloproteinases (e.g., MMP-9), which exacerbate inflammation and tissue damage [41, 42, 43, 44, 45, 46, 47]. IFN-Is play dual roles in these pathological processes. They stabilize the BBB by modulating endothelial cell function and reducing inflammatory mediator-induced permeability. Additionally, they attenuate the proinflammatory responses of glial cells while promoting anti-inflammatory pathways, fostering a neuroprotective environment. By orchestrating the expression of chemokines such as CCL5 and CXCL10, IFN-Is regulate immune cell trafficking and activation, facilitating the resolution of neuroinflammation and tissue repair. These multifaceted actions highlight the pivotal role of IFN-Is in balancing protective and pathological responses during neuroinflammatory events.
Extensive preclinical research highlights the pivotal role of neuroinflammation,
which is characterized by glial activation and the release of proinflammatory
mediators, in the development and maintenance of the chronic nociceptive response
[48, 49, 50]. This neuroinflammatory response is also evident in clinical chronic
nociceptive response conditions, supporting the findings from preclinical
studies. IFN-
| Agent | Actions | IFN doses, STING treatment agent | Routes | Species | Conditions | Mechanisms | References |
| IFN- |
Antinociception | 500 U | Intracerebral | Mouse | Naïve | Opioid receptor dependent | Blalock et al., 1980 [51] |
| 4, 8, 16 pmole | Intracranial ventricle | Rat | Naïve | Mu-opioid receptor dependent | Jiang et al., 2000 [52] | ||
| 100 ng | Intrathecal | Rat | Naïve, CFA | Opioid receptor dependent | Tan et al., 2012 [9] | ||
| 100 ng | Intrathecal | Rat | Naïve, CFA | Inhibits EPSC and capsaicin-induced p-ERK | Liu et al., 2016 [53] | ||
| 100 U | Intrathecal | Mouse | Naïve | IFNAR-mediated actions; inhibition of NaV1.7 and calcium channel activities | Donnelly et al., 2021 [8] | ||
| Hyperalgesia | 300 U | Intraplantar | Mouse | Naïve | Activation of MAPK and MNK-elF4e translation | Barragán-Iglesias et al., 2020 [55] | |
| IFN- |
Antinociception | 100 ng | Intrathecal | Mouse | LPS | IFNAR1-mediated and TLR-mediated actions | Stokes et al., 2013 [56] |
| 3600 U | Intrathecal, coinjection with TNF antibody | Mouse | Arthritis | Induction of IL-10 expression | Woller et al., 2019 [57] | ||
| 1000, 5000, 10,000 U | Intrathecal | Mouse | Spared nerve injury (SNI) | Induction of ISG-15 and inhibition of MAPK | Liu et al., 2020 [54] | ||
| 100 U | Intrathecal | Mouse | Naïve | IFNAR-mediated actions; inhibition of NaV1.7 and calcium channel activities | Donnelly et al., 2021 [8] | ||
| Hyperalgesia | 300 U | Intraplantar | Mouse | Naïve | Activation of MAPK and MNK-elF4e translation | Barragán-Iglesias et al., 2020 [55] | |
| STING | Antinociception | STING agonist (ADU-S100) | Intrathecal (Spinal cord) | Mouse | SNI, CCI | Microglia STING-IFN-I activation | Silveira Prudente et al., 2024 [58] |
| STING agonist (ADU-S100) | Intrathecal (DRG) | Mouse | Inflammatory pain | KChIP1-Kv4.3 regulation | Defaye et al., 2024 [59] | ||
| STING agonist (ADU-S100) | Intrathecal (DRG) | Rat | Incision pain | Activation of the STING-IFN-I pathway | Ma et al., 2023 [60] | ||
| STING agonists (DMXAA, ADU-S100) | Intrathecal (Spinal cord, DRG) | Mouse, Macaca mulatta | CIPN, CCI | Activation of the STING-IFN-I pathway | Donnelly et al., 2021 [8] | ||
| STING agonists (DMXAA, ADU-S100) | Intrathecal (Spinal cord, DRG) | Mouse | Bone cancer pain | Activation of the STING-IFN-I pathway | Wang et al., 2021 [61] | ||
| PTPRD inhibitor (7-BIA) | DRG injection | Mouse | CCI | Upregulation of STING and IFN- |
Sun et al. 2022 [62] | ||
| Electroacupuncture (EA) | Bilateral ST36 (Zusanli) and SP6 (Sanyinjiao) acupoints | Rat | Acute postoperative pain (APP) | STING/IFN-1 pathway activation, mitigation of neuroinflammatory response | Ding et al., 2023 [63] | ||
| Hyperalgesia | STING antagonist (C-176) | Intrathecal (Spinal cord) | Mouse | SNI | Activation of the STING/NF-κB/IL-6 | Sun et al., 2022 [64] | |
| STING antagonist (C-176) | Spinal dorsal nerve (Spinal cord) | Rat | SNI, CCI | Suppression of the microglial M1-polarization | Wu et al., 2022 [65] | ||
| Dexmedetomidine, ketamine | Intraperitoneal (Spinal cord) | Rat | Spinal nerve ligation | Inhibition of the STING/TBK1 pathway to increase ER-phagy | Liu et al., 2022 [66] | ||
| STING antagonist (C-176) | Intrathecal (DRG) | Rat | Bone cancer pain | Activation of the STING-TBK1-NF-κB pathway | Zhang et al., 2023 [67] |
IFN, Interferon; SNI, Spared nerve
injury; CCI, Chronic constriction injury; CIPN, chemotherapy-induced peripheral
neuropathy; DRG, dorsal root ganglion; RTPRD, Protein tyrosine phosphatase
receptor type D; elF4e, eukaryotic initiation factor 4E; CFA, complete freund’s
adjuvant; EPSC, excitatory postsynaptic current; ERK, extracellular
signal-regulated kinase; IFNAR, interferon-
We reported in 2012 that high-dose intrathecal injections of short interfering
RNAs (siRNAs) (10 or 20 µg) produced IFN-
In the rat spinal cord, IFN-
In vivo, intrathecal administration of a neutralizing antibody to
endogenous IFN-
In a murine arthritis model, the intrathecal administration of IFN-
Emerging evidence suggests that TLRs modulate the nociceptive response by
regulating IFN-Is. TLR3, which detects dsRNA and activates IFN-I responses
through TRIF signaling, plays a role in managing nociceptive and itch responses
and is expressed primarily in immune and glial cells [13, 70, 74]. Interestingly,
in addition to being expressed by immune and glial cells, TLR3 is also expressed
in nociceptive sensory neurons, where it influences nociceptive synaptic
transmission within the spinal cord [11, 75, 76]. However, whether TLR3
contributes to the antinociceptive effects of double-stranded RNAs remains
unexplored. Von Frey testing has shown that intrathecal IFN-
Recent reports indicate that IFN-Is can exert pronociceptive effects [66, 67, 68]
(Table 1). Intraplantar injection of IFN-
Similar to host immunity, the nociceptive system serves as an alert mechanism to
detect and respond to “danger signals”. Pathogens can exploit these signals to
disrupt sensory responses such as smell and taste. Nociceptors recognize
pathogen- and damage-associated molecular patterns (PAMPs and DAMPs) through
PRRs, including TLRs, RIG-I-like receptors (RLRs), NOD-like receptors (NLRs), and
cytosolic DNA sensors (CDSs). In DRG neurons, STING acts as a cytosolic DNA
sensor, detecting self-DNA, viral DNA, and bacterial CDNs to trigger the
production of type I interferons (IFN-
This process drives the expression of IFN-Is and other immune response genes,
facilitating pathogen clearance and the removal of damaged cells during
inflammation. By binding to the IFN-
STING has recently emerged as a key regulator of nociception. STING-deficient
mice exhibit mechanical allodynia, while in neuropathic nociceptive response
models, STING agonists have antinociceptive effects. IFN-Is have been shown to
suppress nociceptor excitability in mice, monkeys, and humans [82]. In
naïve, neuropathic, and cancer nociceptive response models, the intrathecal
administration of STING agonists results in sustained antinociception lasting
24–48 hours without motor impairment [8, 58] (Table 1). This effect is
correlated with elevated IFN-
Defaye et al. [59] investigated the transcriptional changes in
sensitized nociceptive neurons to identify genes involved in nociceptor
plasticity during inflammation-induced sensitization and its resolution (Table 1). Their study revealed that the activation of STING in nociceptors plays a
crucial role in resolving the inflammatory nociceptive response. Inflammation
upregulated STING expression in DRG nociceptors, where its activation initiated
TBK1-mediated signaling, leading to the production of IFN-I, predominantly
IFN-
The neuropathic nociceptive response is a challenging condition to manage with
currently available analgesics. Spinal microglia are central to the development
and persistence of this response, largely through PRR signaling and the release
of proinflammatory cytokines [49]. Silveira Prudente et al. [58] investigated the
role of the STING pathway in spinal microglia and its modulation of the
neuropathic nociceptive response, revealing significant sex-specific effects
(Table 1). STING, which is expressed predominantly in spinal microglia, was
upregulated following peripheral nerve injury. However, microglial STING
expression was not required for the development of nerve injury-induced
mechanical allodynia. Activation of STING with agonists such as 2′3′-c-di-AM (PS)2 (Rp,Rp) (ADU-S100)
alleviated the neuropathic nociceptive response in male mice by reducing
mechanical and cold allodynia as well as pinprick hyperalgesia. This analgesic
effect was absent in female mice, likely due to increased production of
proinflammatory cytokines in females, which counteracted the beneficial effects.
In male mice, STING agonists reduced the neuropathic nociceptive response through
the activation of TBK1 and IFN-
In the leukocyte common antigen-related receptor family, protein tyrosine phosphatase receptor type D (PTPRD) is located on human chromosome 9. It consists of extracellular immunoglobulin and fibronectin domains and plays an important role in adhesion and synaptic differentiation [84]. Inhibiting PTPRD activity reduces cocaine addiction [85], which suggests that PTPRD might be a potential therapeutic target for addiction disorders. Furthermore, chronic constriction injury (CCI) significantly increases PTPRD expression in the DRGs of rats [86, 87]. Given its role in addiction treatment, findings by Sun et al. [62] suggest that targeting PTPRD may offer a safe, low-addiction-risk analgesic approach for managing the neuropathic nociceptive response. The CCI-induced neuropathic nociceptive response was alleviated through PTPRD knockdown and PTPRD inhibitor 7-butoxy-3-hydroxy-6-methoxy-1-oxoisochromane-5-carbaldehyde (7-BIA) treatment. Additionally, H-151, a STING inhibitor, reversed the analgesic effects of PTPRD knockdown (Table 1). Overall, the study by Sun et al. [62] indicated that elevated PTPRD levels in the DRG following CCI may contribute to the development of a neuropathic nociceptive response through the STING-IFN-I pathway (Fig. 1).
Electroacupuncture (EA) stimulation at 2/15 Hz has been shown to alleviate the
postlaparotomy nociceptive response in rats [88], although the underlying
mechanisms remain unclear. Ding et al. (2023) [63] investigated the
analgesic effects of EA on the acute postoperative nociceptive response (APP) in
rats, focusing on the role of the STING and IFN-1 signaling pathway (Table 1)
(Fig. 1). APP was induced through abdominal surgery, and EA was applied at
acupoints ST36 and SP6. This study revealed that APP caused mechanical and
thermal hypersensitivities, reduced EEG rhythmic power, and increased
neuroinflammation in the DRG and spinal dorsal horn. EA significantly mitigated
these hypersensitivities, restored electroencephalography (EEG) rhythmic power, and reduced
neuroinflammation. APP suppressed the STING/IFN-1 pathway, dampening
anti-inflammatory signaling in the DRG and spinal cord dorsal horn (SDH), whereas
EA reversed this suppression, activating the pathway and enhancing
anti-inflammatory responses. Intrathecal administration of the STING inhibitor
C-176 abolished the analgesic and anti-inflammatory effects of EA, confirming the
pivotal role of this pathway in EA-mediated analgesia. The STING/IFN-1 pathway is
expressed predominantly in C-type nociceptive DRG neurons (CGRP+ and IB4+), which
are involved in mechanical and thermal nociceptive response detection. EA also
shifted astrocytes and microglia toward an anti-inflammatory profile, further
contributing to its analgesic effects. These findings highlight the STING/IFN-1
pathway as a promising therapeutic target and support the potential clinical
application of EA as a nonpharmacological treatment for APP. Similarly, in an
incision nociceptive response model, Ma et al. [60] demonstrated that
activating the STING-IFN-I pathway alleviated the acute postoperative nociceptive
response by inhibiting the activation of satellite glial cells and macrophages,
thereby reducing neuroinflammation in the DRG. This pathway activation also
downregulated the expression of IL-6, TNF-
Peripheral nerve injury in the neuropathic nociceptive response triggers M1 polarization of spinal microglia, contributing to neuronal hyperexcitability and central sensitization [49]. Suppressing M1 polarization has been shown to alleviate the neuropathic nociceptive response. In a mouse model of middle cerebral artery occlusion, cGAS knockdown shifted microglial polarization toward the M2 phenotype, suggesting that the cGAS-STING signaling cascade may modulate microglial polarization [89]. Wu et al. [65] demonstrated that spared nerve injury (SNI) promoted M1 polarization and activated the cGAS-STING signaling cascade in spinal microglia and neurons, as confirmed by double-label immunofluorescence (Table 1) (Fig. 1). In vitro, LPS-induced M1 polarization in BV-2 microglia activated the cGAS-STING signaling cascade, which was suppressed by cGAS-STING antagonists. In vivo, cGAS and STING antagonists reduced microglial M1 polarization and improved SNI-induced mechanical allodynia.
The cGAS-STING signaling pathway also plays a role in pain regulation in neurons, macrophages, and T cells, which is mediated by regulating the excitability of nociceptive neurons and neuroinflammatory responses. Recent studies [61, 67, 83] have provided compelling evidence that cGAS-STING signaling in nociceptive neurons has dual roles, contributing to both pronociceptive and antinociceptive processes. The activation of STING in sensory neurons has been shown to regulate nociceptor excitability via the modulation of ion channels, including NaV1.7, and to suppress synaptic transmission in spinal nociceptive circuits, as demonstrated by Donnelly et al. [8]. Zhang et al. [83] reported the activation of the downstream inflammatory pathway of STING in DRG neurons in a cancer-induced bone pain model, which was alleviated by administering a STING inhibitor during the middle to late stages of bone cancer. These findings highlight its biphasic function in pain modulation, suggesting potential therapeutic implications depending on the context of activation. Additionally, macrophages and T cells are central to neuroinflammatory processes, with the cGAS-STING pathway playing a critical role in their polarization and recruitment. For example, this pathway drives M1 macrophage polarization, exacerbating inflammation and pain, while also regulating T-cell differentiation, including proinflammatory Th1 and Th9 responses and anti-inflammatory Tregs [90, 91]. These immune cell interactions emphasize the complexity of pain modulation by cGAS-STING signaling in different phases of pain progression. Emerging evidence also implicates astrocytes as significant contributors, with studies showing STING expression and activity in these glial cells. Astrocytic STING activation has been associated with inflammation in pathological states, and recent data suggest that STING agonists may attenuate astrogliosis and reduce pain signaling, although further investigation is needed to fully delineate these mechanisms [92, 93]. Together, these findings underscore the multifaceted and cell type-specific roles of the cGAS-STING pathway in pain regulation.
The STING/TBK1 pathway plays a critical role in ER stress, where STING acts as an ER adaptor necessary under elevated ER stress conditions [94]. In a study by Liu et al. [66], dexmedetomidine and ketamine were shown to have analgesic and antianxiety effects in SNL rats, where the STING/TBK1 signaling pathway was found to be activated (Table 1) (Fig. 1). Dexmedetomidine and ketamine both appear to enhance ER-phagy by inhibiting the STING signaling cascade, thus reducing ER stress in spinal nerve ligation (SNL) rats. This antinociceptive mechanism differs from previous findings suggesting that STING/TBK1 signaling is a direct mechanism for nociceptive response relief. Variations in dosing and timing may impact STING modulation outcomes. Unlike Donnelly et al.’s study [8], which primarily collected behavioral data within hours post-injection, Liu’s study [66] assessed nociceptive and anxiety-related behaviors over days and weeks following surgery.
STING is activated by endogenous and exogenous dsDNA, initiating immune and
inflammatory responses [95]. Nerve injury induces the release of DNA from dying,
dead, or injured cells into the extracellular environment through necrosis or
apoptosis. Typically, this self-DNA is nonimmunogenic. However, emerging evidence
indicates that extracellular self-dsDNA can enter the cytosol via Fc receptors.
Additionally, the antimicrobial peptide LL37 facilitates the transfer of
extracellular self-dsDNA into monocytes. Other cytoplasmic DNA receptors in
macrophages may also contribute to DNA detection. mtDNA
represents another significant source of cytosolic DNA for STING activation.
Mechanisms for mtDNA release include B-cell lymphoma 2 (BCL-2)-like protein 4
(BAX)- and BCL-2 homologous killer (BAK)-mediated apoptosis, mitochondrial
permeability transition pore (mPTP) activation, and deficiencies in transcription
factor A mitochondrial (TFAM) or aberrant mtDNA packaging. These processes
collectively contribute to increased cytosolic dsDNA levels in SNI models and underscore the role of the cGAS-STING pathway in driving
inflammatory and nociceptive responses [64]. Sun et al. (2022) [64]
reported that SNI significantly elevates dsDNA levels, activating the
STING/TBK1/NF-
There are ongoing clinical trials investigating STING agonists, which activate the production of IFN-I, primarily in the context of cancer immunotherapy. These trials aim to harness the ability of the STING pathway to stimulate innate immunity and enhance antitumor responses [96]. For example, several phase I and phase II trials are evaluating the safety and efficacy of STING agonists in patients with various cancers. These studies are exploring the use of STING agonists alone or in combination with other treatments, such as immune checkpoint inhibitors, to enhance antitumor immunity [96]. While these trials focus primarily on oncology, the role of the cGAS-STING pathway in the production of IFN-Is suggests potential therapeutic applications beyond cancer, including in the treatment of conditions involving immune dysregulation. However, clinical trials specifically targeting the STING-IFN-I pathway for pain management are currently lacking, and their analgesic effects could be assessed as secondary endpoints, particularly in cancer-related pain conditions. This highlights a gap in translational research that could explore the analgesic potential of this pathway, given its demonstrated effects on nociception in preclinical studies [4]. Importantly, therapeutic modulation of the STING pathway must be approached with caution, as inappropriate activation can lead to excessive IFN-I responses and associated immunopathology [97]. STING agonists are being actively investigated in clinical trials for cancer treatment, and their application in pain management remains an area for future exploration.
IFNs are well-known cytokines involved in antiviral defense, with emerging
evidence highlighting IFN-I regulation by the STING signaling cascade, which
influences nociceptive response modulation and is active in the DRG under normal
conditions. The role of IFN-Is in the nociceptive response remains controversial,
as studies have reported both pronociceptive [58] and antinociceptive [8, 9, 51, 52, 53, 54, 55, 56, 57, 71] actions of IFN-
In the CNS, IFN-Is generally exhibit antinociceptive effects. The cGAS-STING
signaling cascade, a primary driver of IFN-I production in immune cells following
infection or injury, has been shown to mediate analgesic outcomes. Donnelly
et al. (2021) [8] demonstrated that STING activation confers significant
nociceptive response relief, independent of opioid pathways, across models of
chemotherapy-induced nociceptive responses, nerve injuries, and bone cancer
nociceptive responses. These findings suggest the potential of STING-based
therapies in refractory nociceptive response conditions, as evidenced by the
small molecule 7-BIA, which reduces the neuropathic nociceptive response through
STING activation and subsequent IFN-
Nevertheless, the influence of the cGAS-STING signaling cascade on the chronic
nociceptive response is complex. While some studies link STING activation to
proinflammatory cytokine release and worsened neuroinflammation, others highlight
the nociceptive response-relieving effects of enhanced IFN-I signaling in sensory
neurons. Consecutive and repeated administration of STING agonists likely caused
central sensitization and nociception [84]. The STING–IFN-I pathway may have
different effects on different parts of the peripheral nervous system (PNS) and
CNS. These opposing roles underscore the need for further research to delineate
the mechanisms underlying the dual influence of STING on the nociceptive
response, including potential interactions with TLRs and the
NF-
To advance the understanding of the role of the cGAS-STING pathway in pain
processing, several critical areas warrant further exploration. While this
pathway is activated in various central nervous system diseases [64, 98], its
specific contributions to pain modulation in distinct brain regions, such as the
anterior cingulate cortex and amygdala, remain largely unexplored. These regions
are central to pain perception and pain-related mood disorders, underscoring the
need for focused research on the cGAS-STING pathway at the brain level. Another
key area involves investigating the mechanisms underlying pain-induced aberrant
DNA accumulation, which activates the cGAS-STING pathway. Elucidating how these
DNA changes contribute to pain phenotypes could reveal novel targets for
intervention. Furthermore, the pathway regulating pain through immune cells,
particularly astrocytes, macrophages, and T cells, requires detailed
investigation, as these cells play pivotal roles in pain modulation.
Additionally, understanding the relationship between the cGAS-STING pathway’s
downstream regulatory mechanisms (e.g., TBK1, STATs, and NF-
PHT and TCC contributed to the sections of Type 1 interferon, STING, nociceptive response modulation, and conclusions as well as Fig. 1. TCC, KCH and PHF wrote the draft manuscript and Table 1 of this review. YYL and CHY edited the manuscript and Table 1. CCC contributed the conception and design of the article. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We would like to express our gratitude to Li-Li Wu and Se-Ming Liu for her technical support in creating the figure and table.
The manuscript was partly supported by grants from the National Science and Technology Council (NSTC 112-2314-B-384-008-MY3), Chi-Mei Hospital Grants (CMNDMC11106, 11103).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.