






1 Department of Nephrology, The Third Affiliated Hospital of Jinzhou Medical University, 121017 Jinzhou, Liaoning, China
2 Department of Cardiovascular, The Affiliated Hospital (The First Clinical College/Liaoning Hospital of (Traditional Chinese Medicine) TCM), 110000 Shenyang, Liaoning, China
Abstract
Type 2 cardiorenal syndrome (CRS) is a complex disease characterized by the interplay between the heart and kidneys. The pathophysiology of type 2 CRS involves multiple molecular signaling pathways. Transient receptor potential melastatin 2 (TRPM2) is a reactive oxygen species (ROS)-sensitive and non-selective calcium-permeable cation channel, which plays a regulatory role in intracellular Ca2+ homeostasis. Thus, this study aimed to explore the biological functions and mechanisms of the ROS–TRPM2 signaling axis in type 2 CRS.
Type 2 CRS model rats (a rat model of type 2 CRS induced through left anterior descending coronary artery ligation combined with 5/6 total nephrectomy) and lipopolysaccharide (LPS)-induced CRS cell lines, human kidney-2 (HK-2), were transfected with small interfering RNA (siRNA) to knock down TRPM2 or a calcium ion channel activator Yoda1 to evaluate the involvement of the ROS–TRPM2 signaling axis on type 2 CRS. Changes in kidney tissue morphology were observed using H&E staining; cell viability and apoptosis were monitored using CCK-8, Annexin V-FITC/PI, and TUNEL kits, alongside quantitative real-time polymerase chain reaction (qRT-PCR), Western blot, ELISA, and immunofluorescence assays to confirm the interaction between ROS, TRPM2, and Ca2+.
TRPM2 is highly expressed in HK-2 cells after LPS stimulation and renal tissues of type 2 CRS rats. Intervention via TRPM2 improves injured cell viability, mitigates apoptosis, inhibits the inflammatory cytokines interleukin 10 (IL-10) and tumor necrosis factor-α (TNF-α), as well as indices of oxidative stress—malondialdehyde (MDA) and ROS—promotes total antioxidant capacity (T-AOC) expression, and alleviates pathological changes in CRS; Yoda1 promoted a contrasting effect to the biological effect induced by TRPM2 deletion.
TRPM2 is abnormally highly expressed in damaged kidneys during the pathogenesis of type 2 CRS. Silencing TRPM2 can inhibit inflammatory and oxidative stress responses, reduce cell apoptosis, promote survival, and alleviate pathological loss; this may be related to the inhibition of Ca2+ influx. This suggests that the ROS–TRPM2 signaling pathway is significant for CRS development, and TRPM2 may be an effective therapeutic target for type 2 CRS.
Keywords
- type 2 cardiorenal syndrome
- ROS
- TRPM2
- Ca2+-sensitive channel
Cardiorenal syndrome (CRS) is a multi-organ dysfunction with a complex pathogenesis involving the interplay of a variety of signaling pathways [1, 2]. It is a clinical syndrome characterized by the simultaneous dysfunction of the heart and kidneys, leading to pathophysiological changes with high morbidity and mortality rates. In recent years, with the increasing aging population, the incidence and mortality rates of CRS have been rising. Epidemiological studies indicate that 24% to 45% of patients with acute decompensated heart failure develop acute kidney injury during their illness, and among these patients, 40% to 60% eventually suffer from renal insufficiency [3, 4]. Additionally, nearly 50% of chronic kidney disease patients have cardiovascular disease as the cause of death [5, 6]. Currently, the clinical treatment of CRS mainly involves correcting hemodynamic disturbances and renal replacement therapy. Therefore, a large number of diuretics and vasodilators are needed in the early treatment stage. However, the use of a large number of diuretics will aggravate the occurrence of CRS, which is contradictory. Resolving this contradiction is crucial for improving the treatment outcomes of CRS and alleviating the patient’s condition [7]. In CRS, kidney injury is closely related to calcium ion regulation. Kidney damage can lead to an imbalance in calcium ion regulation, affecting physiological functions such as renal filtration and reabsorption, which exacerbates kidney injury [8]. Additionally, calcium ion imbalance also impacts cardiac contraction and relaxation functions, further worsening cardiac failure [9, 10]. Therefore, maintaining calcium ion balance is of great significance in the treatment of cardiorenal syndrome. As the main form of CRS disease, type 2 CRS is characterized by the gradual progression of chronic kidney disease caused by chronic cardiac insufficiency [11]. The pathology of type 2 CRS is considered to be complex and multifaceted, and its pathological mechanism is still not fully understood. Therefore, it is necessary to explore more possible mechanisms to provide ideas for the treatment of type 2 CRS.
Transient receptor potential melastatin 2 (TRPM2) is a non-selective cation channel mediating calcium influx and ubiquitously expressed endogenously in tissues and cells, such as heart and kidney. During various physiological and pathological conditions, reactive oxygen species (ROS) activates Ca2+ signaling which regulates cell metabolism, bioenergetics, signaling pathways and apoptosis. Then TRPM2 serves as a key executor that sensor oxidative stress (OS) and keep intracellular calcium homeostasis [12, 13]. Yu et al. [14] found that the inhibition of TRPM2 decreases the production of ROS in acute kidney injury (AKI) and protects against acute kidney injury through a Ca2+-dependent autophagy mechanism. During oxidative cardiomyopathy, TRPM2 converts ROS into Ca2+ signal and acts on its downstream pro-survival targets in cardiac myocytes [15]. But dysregulation of TRPM2 also trigger cytosolic Ca2+ dyshomeostasis that form a redundant feedback cycle to pathologically promote each other’s function. For instance, aberrant ROS production induces the massive influx of Ca2+ into the cell upon open TRPM2 channel, resulting in caspase cascade activation and ultimately cardiomyocyte cell death [16]. In renal ischemia-reperfusion injury (IR), blockage of TRPM2 obviously reduced histopathological harm, expressions of caspase-3 and OS index [17]. In summary, the TRPM2 channel may play diverse roles and regulatory mechanisms to exacerbate and mitigate CRS injury, and in-depth research on the ROS-TRPM2 signaling pathway is all-important for not only elucidating the pathogeny of CRS but also seeking new therapy targets and strategies for related diseases.
This study showed that the expression of TRPM2 was high in HK-2 cells stimulated by lipopolysaccharide (LPS) and the kidney tissue of CRS rats. After silencing TRPM2, LPS-induced human kidney-2 (HK -2) cell viability increased, apoptosis rate decreased, and calcium channel activation was inhibited, which inhibiting inflammatory response and oxidative stress response. This study identifies the influence of TRPM2 on CRS and uncovers its molecular mechanisms.
CRS cell lines HK-2 was obtained from Hanpu Nuosai Life Science and Technology Co., Ltd. (CL-0109, Wuhan, Hubei, China). The cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM, 11965118, Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FSD500, Excell Bio, Shanghai, China) and 1% cyanine/streptomycin (C0222, Beyotime, Shanghai, China) at 37 °C with 5% CO2.
An in vitro cell damage model was established by treating HK-2 cells with 1 µg/mL LPS (MFCD00131520, Merck, Kenilworth, NJ, USA) for 48 hours [18]. Subsequently, the calcium ion channel activator Yoda1 (0.2 µM, HY-18723, Medchem Express, Elizabeth, NJ, USA) was used to activate the calcium ion channels in the cells for an additional 24 hours [19]. All cell lines were validated by short tandem repeat (STR) profiling and tested negative for mycoplasma.
The small interfering RNAs (siRNAs) targeting TRPM2 and the nonsense control (si-NC) were obtained via chemical synthesis (Gemma Genetics, Suzhou, China). Subsequently, transfections were carried out with Lipofectamine 3000 reagent (L3000015, ThermoFisher, Boston, MA, USA) in HK-2 cells following the manufacturer’s instructions. Stable cell lines were generated through with puromycin (10 µg/mL, HY-B1743A, MedChem Express, NJ, USA) selection for 7–14 days. The specific sequences of si-TRPM2: 5′-GGACAAGCTCTGTCTGCAAA -3′; si-NC: 5′- GTTCTCCGAACGTGTCACGT -3′.
All animal experiments were carried out with approval by the Institutional Laboratory Animals Care and Use Committee of Liaoning University of Traditional Chinese Medicine (Approval No. 21000042024002). SPF-grade SD rats (male, 6–7 weeks old, weighing 220–250 g) were obtained from Beijing Vital River Laboratory Animal Technology Co., Ltd (China). Before the experiment, all animals were acclimated for one week.
The rats were randomly divided into Sham group (n = 6) and CRS group (n = 24). Rat model of cardiorenal syndrome (CRS) group: according to the previous method, the rat model of CRS was established by ligating the left anterior descending coronary artery and inducing acute renal ischemia-reperfusion [20]. In simple terms, after ligation with 2% isoflurane, the left anterior descending coronary artery was permanently ligated to induce myocardial infarction. Two weeks after myocardial infarction, the left kidney was removed and subtotal nephrectomy was performed. The rats underwent right subcapsular nephrectomy and about 2/3 renal infarction (because 2/3 of the right renal artery was ligated). The heart-kidney rat model was successfully established 4 weeks after myocardial infarction.
Then, CRS rats were randomly divided into CRS+KD-TRPM2, CRS+Yoda1, and CRS+KD-NC groups, with 6 rats in each group. CRS+KD-TRPM2 group: After the CRS model was constructed by the same operation method as the CRS group, AAV2/2-U6-sh-TRPM2 (TRPM2 knockdown lentiviral expression vector) recombinant vector was injected into the tail vein on the 7th, 14th, 21th and 28th days. CRS+Yoda1 group: After surgery, Yoda1 was alternately injected into the left and right arms at 5 mg/kg/d for 10 days. CRS+KD-NC group: AAV2/2-U6-sh-NC (empty vectors) recombinant vector was injected into the tail vein on the 7th, 14th, 21st and 28th day after surgery. After two operations, rats need intramuscular injection of ampicillin to prevent infection. Following the observation period, the rats were sacrificed by exsanguination following intraperitoneal injection of pentobarbital sodium (50 mg/kg), and their blood samples, and kidney tissues were collected for further study.
Total RNA was extracted from treated cells using the Trizol reagent (RC112, Vazyme, Nanjing, China) and reverse-transcribed using the HiScript lll 1st Strand cDNA Synthesis Kit (R312, Vazyme, China). Subsequently, qRT-PCR was performed on an applied biosystems Inc. (ABI) (Foster,CA,USA) real-time PCR system following the manufacturer’s recommended cycling parameters. The specific cycle parameters are shown in Table 1. Relative fold changes in gene expression were analyzed by normalizing to the endogenous controls GADPH or U6 to account for loading variation. The primer sequences are as followed: TRPM2 (Rat), Forward: CGACGAGCCAGATGCTGAG; Reverse: ATCAGGGTAGAGGAGGTGCC; GAPDH (Rat), Forward: GAAGCTGGTCATCAACGGGA; Reverse: ACGACATACTCAGCACCAGC; TRPM2 (Human), Forward: CCGAGCAGAAGATCGAGGAC; Reverse: GGGTGGTTACTGGAGCCTTC; GAPDH (Human), Forward: AATGGGCAGCCGTTAGGAAA; Reverse: GCGCCCAATACGACCAAATC.
| Stage | Response | Cycle index | Temperature | Time |
| Stage 1 | pre-denaturation | Reps: 1 | 95 °C | 30 sec |
| Stage 2 | circular reaction | Reps: 40 | 95 °C | 10 sec |
| 60 °C | 10 sec | |||
| Stage 3 | melting curve | Reps: 1 | 95 °C | 15 sec |
| 65 °C | 60 sec | |||
| 95 °C | 15 sec |
PCR, polymerase chain reaction.
Protein samples were extracted from cells or tissues using RIPA lysis buffer. Protein quantification was performed by bicinchoninic acid (BCA) method. Total protein extracts (30 µg) from each sample were subjected to SDS-PAGE gels and transferred onto PVDF membranes for western blotting. At room temperature, the PVDF membrane was blocked with a rapid blocking solution for 30 min, and then incubated with primary antibody overnight at 4 °C Then the secondary antibody was added and incubated at room temperature for 2 h. The primary antibody: anti-TRPM2 (1:1000, ab11168, Abcam, Cambridge, UK) and anti-GAPDH (1:10,000, bsm-33033M, Bioss, Beijing, China); The second antibody: Goat Anti-Rabbit IgG H&L (bs-0295G-HRP), Goat Anti-Mouse IgG H&L (1:20,000, bs-0296G-HRP, Bioss, Beijing, China). Finally, JP-K6000 chemiluminescence imager (Jiapeng, Shanghai, China) was used to develop and Image J software 1.8.0 (NIH, Bethesda, MD, USA) was used for quantitative analysis.
HK-2 cells were plated in 96-well plates at a suitable density of 2000 cells/well. After the cells adhered, the old complete medium was removed and replaced with fresh serum-free medium for 12 hours. Subsequently, the cells were incubated with a 10% CCK8 solution, protected from light, for 1 hour. Finally, cell viability was assessed by measuring absorbance at 450 nm by an RT-6000 enzyme microplate reader from Rayto (Santa Cru, CA, USA).
The BD Pharmingen FITC Annexin V Apoptosis Detection Kit (C1062L, Beyotime, China) was used for evaluating cell apoptosis. Briefly, the cells were washed, collected, and resuspended in binding buffer. Next, the cells were stained with 5 µL Annexin V-FITC and 5 µL PropidiumIodide (PI) in the dark for 15 minutes, and positive cells were analyzed on an Attune NxT (Themo Fisher, Boston, MA, USA).
The pre-cultured cells were taken out, the medium was removed, and the cells were washed with buffer 3 times. Then the Rhod-2/Acetoxymethyl ester (AM) working solution was added to the cells, and the amount of addition was based on the coverage of the cells, and the cells were cultured at 37 °C for 30 min; after the Rhod-2/AM working solution was poured out, the buffer was added and the cells were washed three times. Next, the cells were covered with buffer and incubated at 37 °C for about 30 min. Finally, the cells were detected under a microscope (KEYENCE, Osaka, Japan).
The expression levels of interleukin 10 (IL-10), tumor necrosis
factor-
The samples from each experimental group were exposed to 4% paraformaldehyde at ambient temperature overnight. For the TUNEL assay, the sections were incubated with proteinase K solution and labeled with TdT reaction solution following the instructions. Cell nuclei were counterstained with DAPI. Then images for apoptotic cells were captured using a Nikon Eclipse 80i light microscopy (BZ-H4XD, Keyence Corporation, Osaka, Japan) with identical parameters.
Briefly, collected rat kidneys were routinely dehydrated, embedded in paraffin and sliced into 4 µm-thick sections. Then, Hematoxylin and eosin (H&E) staining was carried out and representative photographs of the stained sections were captured to observe the histomorphological and structural changes as previously described [21].
Statistical analysis was conducted via Graphpad Prism 9 software (Graphpad,
Boston, MA, USA). Continuous variables were first tested for normal distribution
using the Shapiro-Wilk method. For normally distributed continuous data, the
format (mean
We initially investigated the role of TRPM2 in LPS-injured HK-2 cells
and in cells with Yoda1-activated calcium ion channels. HK-2 cells were treated
with 1 µg/mL LPS for 48 hours, after which TRPM2 expression was
quantified using Western blot and qRT-PCR. Our findings revealed significantly
elevated TRPM2 expression in LPS-injured HK-2 cells, with further
increases upon Yoda1-induced activation of calcium ion channels (p
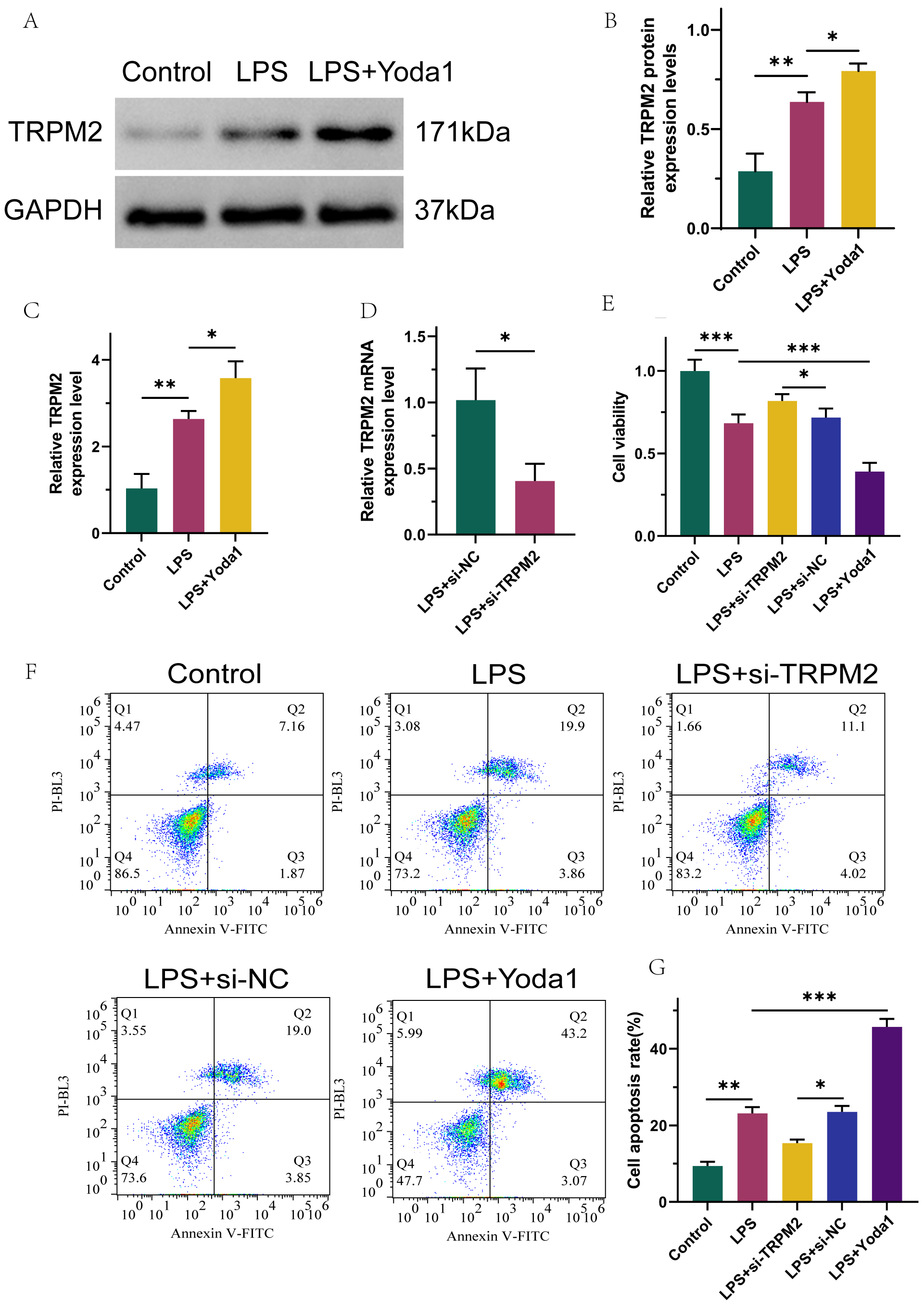 Fig. 1.
Fig. 1.
Transient receptor potential melastatin 2 (TRPM2) is highly
expressed in LPS-induced renal tubular epithelial cells, and TRPM2 promotes
proliferation and inhibits apoptosis of LPS-stimulated HK-2 by inhibiting calcium
ion channel activation. (A,B) Western blot results of TRPM2 protein exhibition.
(C) qRT-PCR analysis of TRPM2 mRNA expression. (D) qRT-PCR analysis of
TRPM2 transfection efficiency. (E) CCK-8 assay measuring cell viability.
(F,G) Flow cytometry assay detecting cell apoptosis. n = 3, *p
To elucidate the role of TRPM2 in modulating cellular inflammatory responses and
OS through the activation of calcium ion channels, this study involved the
transfection of LPS-injured HK-2 cells with a TRPM2-targeting small
interfering RNA (siRNA) expression vector (p
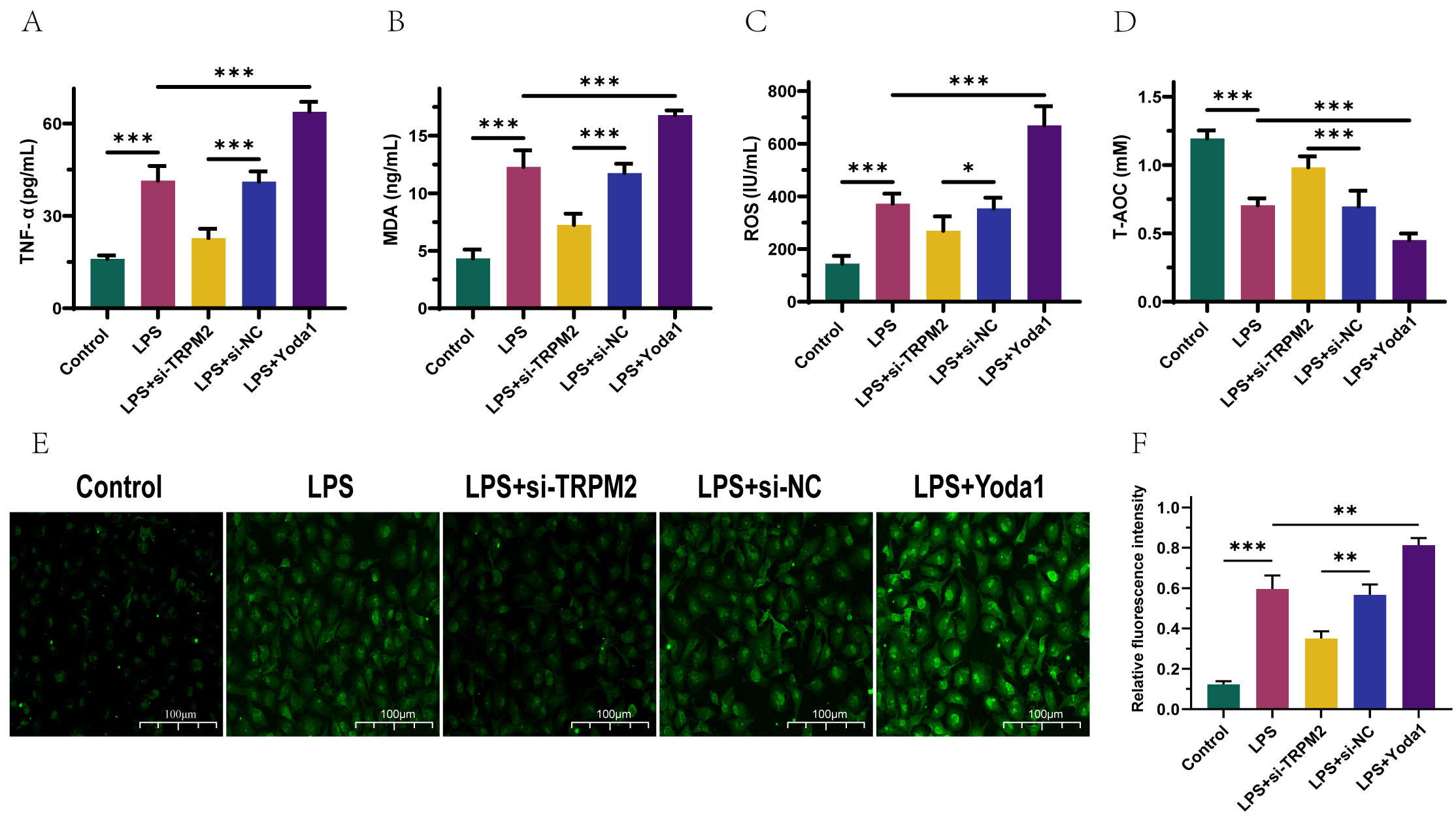 Fig. 2.
Fig. 2.
TRPM2 inhibits the activation of calcium ion channels, thereby
suppressing cellular inflammatory and oxidative stress (OS) reactions. (A–D)
ELISA detection of TNF-
The expression of biomarkers such as IL-10 and TNF-
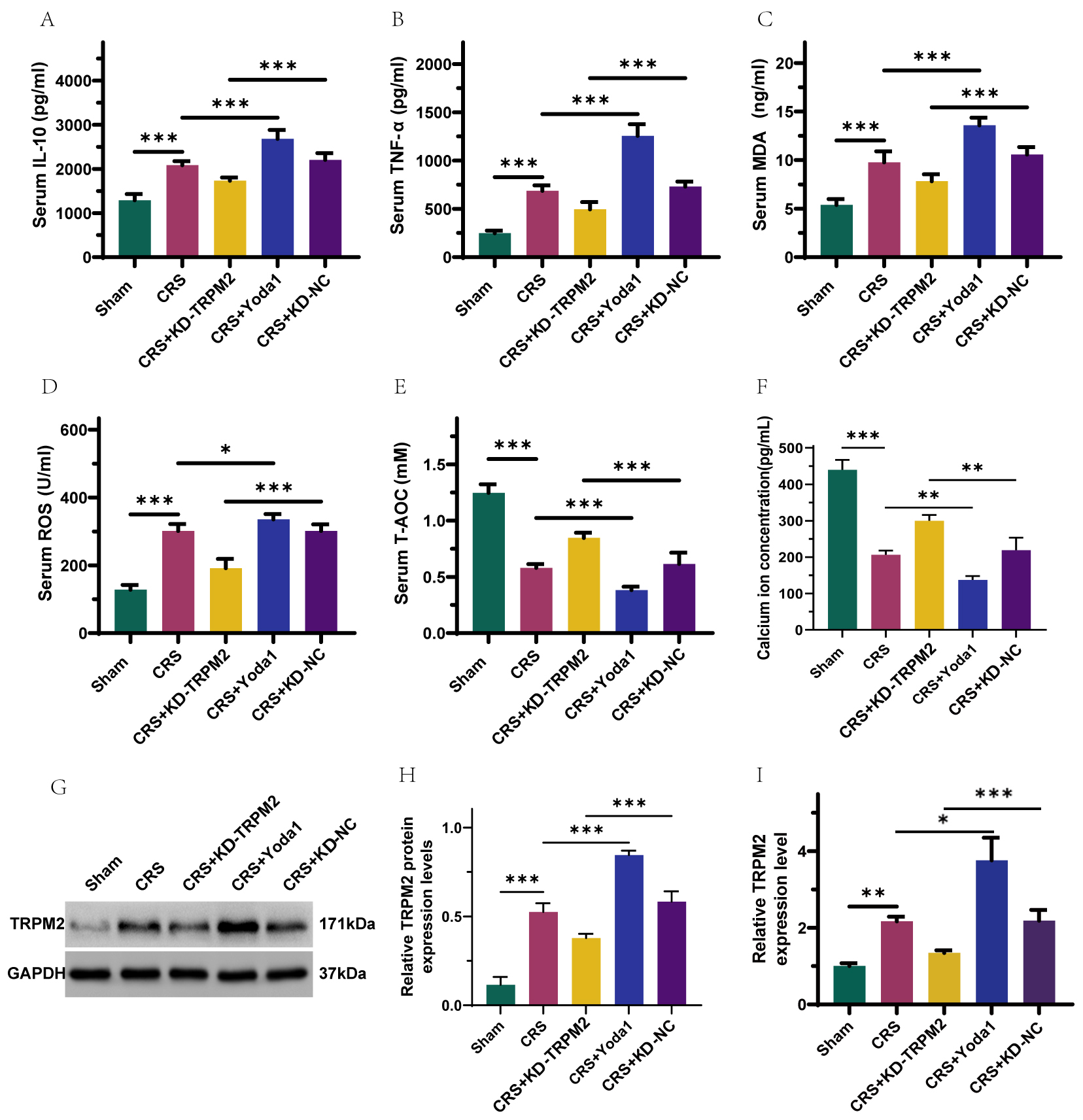 Fig. 3.
Fig. 3.
TRPM2 is highly expressed in cardiorenal syndrome (CRS) rats,
and silencing TRPM2 suppresses inflammatory responses and oxidative stress
reactions in CRS rats, which can be reversed by Yoda1. (A–F) ELISA detection of
IL-10, TNF-
To investigate the effects of intervening TRPM2 expression on renal tissue
damage and cell apoptosis, this study focused on type 2 CRS rats. H&E staining
revealed focal interstitial fibrosis, inflammatory cell infiltration, irregular
tubular structure, minimal tubular dilation, decreased glomeruli with unclear
structure in type 2 CRS rat kidney tissues. Conversely, silencing TRPM2
reduced focal interstitial fibrosis, decreased inflammatory cell infiltration,
restored tubular structure with increased tubular dilation, and clearer
glomerular structure. After injection of Yoda1 into type 2 CRS rats, compared
with CRS model rats, Yoda1 further inhibited renal tubular atrophy, renal tubular
wall thickening with lumen expansion and irregular arrangement, which was
different from the results after silencing TRPM2 (Fig. 4A). TUNEL
staining showed that TRPM2 silencing reversed cell apoptosis in type 2
CRS rat kidney tissues, which was promoted upon Ca2+ channel activation
(p
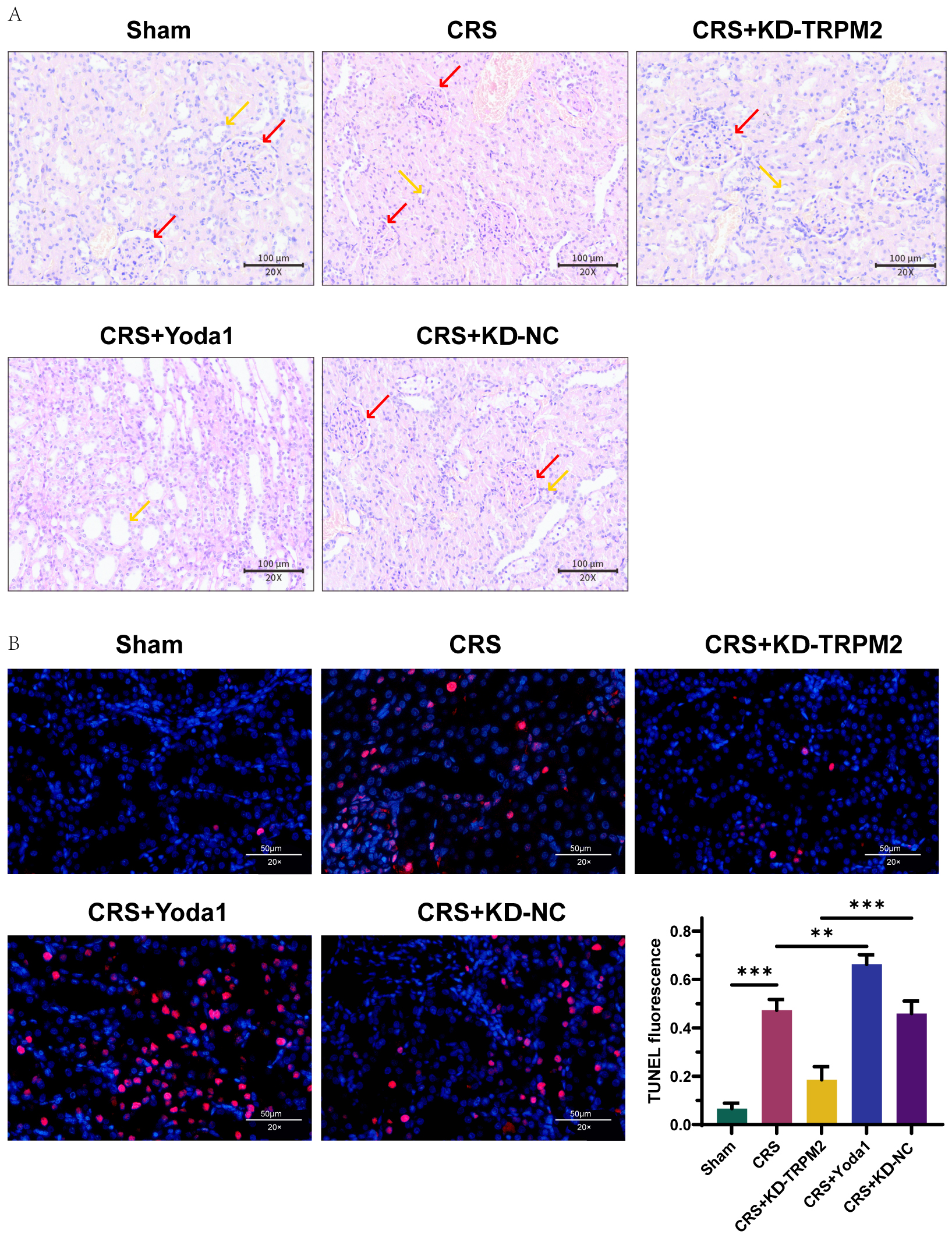 Fig. 4.
Fig. 4.
TRPM2 mitigates renal tissue damage and alleviates cell
apoptosis in type 2 CRS rats by inhibiting calcium ion channel activation. (A)
H&E staining to observe histopathological changes in kidney tissue. Red,
glomerulus; Yellow, renal tubule. Scale bar = 100 μm. (B) TUNEL staining to observe cell apoptosis in
kidney tissue. Scale bar = 50 µm, n = 3, **p
CRS involves a complex interaction between the heart and kidneys, where primary
damage in one organ, whether it occurs suddenly or persists over time, leads to
secondary abnormalities in the other organ [23, 24]. In recent years, with a
deeper understanding of the pathogenesis of CRS, researchers have gradually
identified many related factors such as ischemia-reperfusion injury, inflammatory
response and OS, and their interactions can initiate a multiorgan malfunction and
trigger the pathological cascade of CRS [25, 26, 27, 28]. Our study found that TRPM2 was
highly expressed in LPS-induced HK-2 cells. After silencing TRPM2, the
viability of LPS-induced injured HK-2 cells increased and the apoptosis rate
decreased. However, after adding the calcium channel activator Yoda1 to the
cells, as the expression of TRPM2 further increased, cell viability decreased,
and apoptosis was promoted. Further studies found that low expression of TRPM2
inhibited inflammatory factors TNF-
ROS refers to a group of substances within cells that possess oxidative
capabilities. And as a ROS-sensitive calcium channel, TRPM2 plays a regulatory
role in intracellular calcium homeostasis [14, 29, 30]. Studies have shown that
when ROS accumulate within the cell to a certain extent, they can interact with
TRPM2 channels, activating the channels and mediating Ca2+ influx into the
cell. This disrupts intracellular Ca2+ homeostasis and triggers a series of
physiological or pathological reactions [30, 31]. Therefore, the activation of
the ROS-TRPM2 signaling axis can regulate Ca2+ homeostasis and affect the
normal function of cells. In some diseases, abnormal activation of the ROS-TRPM2
signaling axis can lead to an imbalance in Ca2+ homeostasis, resulting in
cell damage or death [13, 14, 32]. Consequently, our study indicates that
silencing TRPM2 affects the activation of calcium channel proteins in injured
HK-2 cells and influences ROS expression. This suppression leads to decreased
levels of inflammatory markers such as TNF-
Further, in order to confirm that TRPM2 does have an impact on CRS. We silenced the expression of TRPM2. Similar to the expected results, after TRPM2 low expression, the levels of inflammatory factors, ROS and MDA decreased, the level of T-AOC increased, and the level of calcium ion decreased. Yoda1 increased the fluorescence intensity of Ca2+. In the study of Cao et al. [33], primary neutrophils from TRPM2-/- mice showed reduced ROS production and calcium influx, and in TRPM2-/- mice, inhibition of inflammatory cell infiltration was shown. Tian et al. [34] showed that in lupus nephritis, silent information regulator 1 (SIRT1) regulates the progression of lupus nephritis by affecting the ROS/TRPM2/Ca2+ channel. This indicates that the ROS-TRPM2 signaling pathway plays an important role in the progression of type 2 CRS. It has been reported that polymerase (PARP)-1 interacts with TRPM2, and its activation leads to the production of adenosine diphosphate ribose (ADPR), which in turn activates the TRPM2 channel [35, 36]. In addition, TRPM2 interacts with other calcium channels (such as SUPPRESSOR OF OVEREXPRESSION OF CO (SOC)1/2) and NADPH oxidase (such as NOX1), which may be important in the context of type 2 CRS [37]. Because these interactions are essential for the transmission of calcium signals and the regulation of ROS levels. However, whether these molecules also play a role in the ROS-TRPM signaling pathway of type 2 CRS needs to be further explored in future studies.
Ca2+ influx elicited by TRPM2 brings about drug-evoked hepatotoxicity, ischemia-reperfusion damage, and the development of non-alcoholic fatty liver disease to cirrhosis, fibrosis, and hepatocellular carcer [38]. This study is the first to demonstrate that TRPM2 is abnormally overexpressed in CRS and is induced by ROS activation. It is worth mentioning that the role of the ROS-TRPM2 signaling axis in CRS have not been taken seriously and elaborated fully, and our findings broaden the current understanding of the molecular mechanisms regulating TRPM2 activation, as well as its involvement in inflammation and oxidative stress reactions during cell damage of CRS. Furthermore, because of the double-edged effect of TRPM2 [34], and it plays protective and detrimental parts in cellular damage, future research is necessary for us to elucidate the intricate biological networks of the ROS-TRPM2 signaling axis in CRS detailedly.
This research systematically uncovered the position of the ROS-TRPM2 signaling axis in the pathogenesis of CRS. Experimental evidence demonstrated that ROS accumulation activates TRPM2 channels, leading to elevated intracellular calcium levels, thereby promoting pathological changes in the heart and kidneys. Silencing the activity of TRPM2 channels effectively attenuated pathological damage in an animal model of CRS. This study elucidates the critical role of the ROS-TRPM2 signaling axis in the progression of CRS and underscores the potential of targeting this pathway as a novel therapeutic strategy for CRS treatment.
All data generated or analyzed in this study are included in the present manuscript.
YL: data collection and analysis, drafting the manuscript, investigation. FW: study design, data analysis, drafting the manuscript and revision of the manuscript. Both authors read and approved the final version of the manuscript. Both authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
All animal experiments were carried out with approval by the Institutional Laboratory Animals Care and Use Committee of Liaoning University of Traditional Chinese Medicine (Approval No. 21000042024002) and adhered to the 3R principles.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.