






 , Yuntao Li 1,2,†, Yonggang Zhang 1, Wen Qin 3, Zhongzhou Su 1, Sheng Qiu 1,2,*
, Yuntao Li 1,2,†, Yonggang Zhang 1, Wen Qin 3, Zhongzhou Su 1, Sheng Qiu 1,2,* , Lifang Zheng 4,*
, Lifang Zheng 4,*1 Department of Neurosurgery, Fifth School of Clinical Medicine of Zhejiang Chinese Medical University (Huzhou Central Hospital), 313000 Huzhou, Zhejiang, China
2 Department of Neurosurgery, Huzhou Key Laboratory of Basic Research and Clinical Translation for Neuromodulation, 313000 Huzhou, Zhejiang, China
3 College of Pharmacy, Shenzhen Technology University, 518118 Shenzhen, Guangdong, China
4 Department of Neurology, Southern University of Sciences and Technology Yantian Hospital, 518081 Shenzhen, Guangdong, China
†These authors contributed equally.
Abstract
Oxidative stress and neuroinflammation are important secondary injury mechanisms in intracranial hemorrhage (ICH). V-set and immunoglobulin domain-containing 4 (VSIG4) has an inhibitory effect on oxidative stress and the inflammatory response. This study aimed to explore the possible role of VSIG4 in ICH-related neuropathology.
In this study, VSIG4 levels were investigated in an ICH mouse model and lipopolysaccharide (LPS)-stimulated RAW264.7 cells. Moreover, we examined oxidative stress levels, pro-inflammatory cytokine production, neuronal damage, inflammatory cell activation, brain water content, and neurological function. We performed these assays in ICH mice and macrophages with different VSIG4 levels. Additionally, the critical role of the nuclear factor erythroid 2 related factor 2/heme oxygenase-1 (NRF2/HO-1) signaling pathway in VSIG4 function was verified.
VSIG4 ameliorated neurological deficits in ICH mice (p < 0.01), alleviated cerebral edema (p < 0.05), and increased glutathione (p < 0.05) and decreased superoxide dismutase (SOD) levels (p < 0.01) in the perihematomal area and LPS-stimulated RAW264.7 cells. It also reduced Malondialdehyde (MDA) accumulation (p < 0.01), alleviated oxidative stress, and decreased interleukin-1β (IL-1β) (p < 0.01) and tumor necrosis factor-alpha (TNF-α) levels (p < 0.01), thereby attenuating the inflammatory response. Additionally, treatment of LPS-stimulated RAW264.7 cells with VSIG4 resulted in less damage to HT22 cells (p < 0.05). To further validate the involvement of the NRF2/HO-1 pathway in VSIG4-mediated neuroprotection, brusatol (an NRF2 inhibitor) was administered.
Our study demonstrates the neuroprotective effect and mechanism of action of VSIG4 in ICH.
Keywords
- intracranial hemorrhage
- NRF2/HO-1 signaling pathway
- VSIG4
- neuroinflammation
- oxidative stress
Intracranial hemorrhage (ICH) is a significant global health burden and a leading cause of disability and mortality [1]. The severity of ICH is attributed to its complex pathophysiological mechanism, which involves both direct and secondary injuries [2]. The former mainly results from the mechanical compression of the hematoma on brain tissue and subsequent tissue necrosis caused by ischemia and hypoxia. The latter includes cascading effects such as neuroinflammation, oxidative stress, calcium overload, ferroptosis, and blood-brain barrier disruption [2, 3]. Although direct damage caused by ICH can be alleviated with appropriate neurosurgical treatment, effective treatment options for secondary injury mechanisms remain limited [4]. However, secondary injuries are key factors affecting patient prognosis [5, 6]. This presents a significant challenge for ICH treatment strategies. Therefore, it is imperative to conduct comprehensive research on the potential mechanisms of secondary damage following ICH. This will facilitate the development of new treatments in the future, thereby enhancing patient recovery and quality of life.
V-set and immunoglobulin domain-containing 4 (VSIG4) represents a novel class of immunomodulatory proteins predominantly expressed on the cell membrane of tissue-resident macrophages. It can inhibit excessive immune responses by suppressing the complement pathway, regulating macrophages, and influencing T cells [7, 8, 9]. In previous studies, VSIG4 was found to alleviate the progression of hepatitis, inflammatory bowel disease, and myocardial infarction by inhibiting inflammatory responses and oxidative stress [10, 11, 12]. In central nervous system diseases, Parkinson’s disease and ischemic stroke can also benefit from high VSIG4 expression levels [13, 14]. Furthermore, it has been suggested that the protective effect of VSIG4 may be achieved by increasing nuclear factor erythroid 2 related factor 2 (NRF2) expression and promoting its translocation [15]. Neuroinflammation and oxidative stress are important forms of secondary injury after ICH and have a significant impact on patient prognosis [16, 17]. Based on these findings, VSIG4 likely mitigates ICH-induced secondary damage through dual anti-inflammatory and antioxidant mechanisms within the central nervous system. Nevertheless, the precise mechanistic role of VSIG4 in ICH pathogenesis is yet to be fully elucidated.
In this study, we investigated the function of VSIG4 in the regulation of ICH injury. Understanding this mechanism may provide new insights into potential therapeutic targets, ultimately enhancing patient recovery and improving the prognosis associated with secondary ICH injury.
Male C57BL/6 mice (8–10 weeks old, 22–25 g) were sourced from Shanghai Laboratory Animal Center and acclimatized for 7 days at Huzhou Central Hospital’s Animal Experiment Center before surgical procedures. The animal room is a barrier environment for specific pathogens (SPF) with a temperature controlled at 20–26 °C, relative humidity of 40%–70%, and a 12-hour light/dark cycle. The mice were housed in polycarbonate cages with 3–5 mice per cage, with a heat-sterilized bedding material at the bottom, which was replaced twice a week. The laboratory animals had free access to standard autoclaved laboratory mouse feed and sterile drinking water. All animal experiments complied with institutional ethical guidelines approved by the hospital’s Animal Care Committee (Approved number: 202411005).
RAW264.7 and HT22 cells were obtained from Central Laboratory of Huzhou Hospital (Huzhou, China). Cells were cultured in DMEM (Gibco, 11965, Waltham, MA, USA) supplemented with 10% heat-inactivated FBS (Gibco, A3161002) and 1% penicillin-streptomycin (Gibco, 15140122) to ensure optimal growth. The cells were incubated at 37 °C in a 5% CO2 atmosphere. All cell lines underwent short tandem repeat profiling for authentication and tested negative for Mycoplasma.
An ICH model was established in 10 mice, with cohorts euthanized (See section 2.11 for euthanasia methods) at 1-day (n = 6) and 3-day (n = 6) post-operation for behavioral assessment (neurological deficit scoring) and brain tissue collection. In vitro, RAW264.7 macrophages were stimulated with lipopolysaccharide (LPS) (1 µg/mL) for 6, 12, or 24 hours to model ICH-associated inflammation, with untreated cells as controls.
45 ICH model mice were randomized into three treatment groups (n = 15/group): Vehicle (PBS, intraventricular), Isotype IgG (10 mg/kg, intraventricular), Recombinant VSIG4 (5 mg/kg, intraventricular). For in vitro validation, RAW264.7-HT22 co-cultures received: IgG (1 µg/mL), VSIG4 (100 µg/mL), LPS + IgG, LPS + VSIG4.
60 Mice were allocated to four cohorts (n = 15/group): ICH, ICH + Brusatol (1 mg/kg, intraventricular), ICH + VSIG4, ICH + Brusatol + VSIG4. Parallel in vitro experiments subjected LPS-stimulated RAW264.7 cells to: LPS, LPS + Brusatol (10 µg/mL), LPS + VSIG4, LPS + Brusatol + VSIG4.
The mice were anesthetized using a 2% isoflurane/air mixture in a general-purpose small animal anesthesia machine (RWD R500, Shenzhen, China) before being placed on a stereotaxic apparatus. After drilling a hole in the skull, a 32-gauge needle was inserted into the right striatum using coordinates of 2.0 mm from the median line, 1.0 mm from the bregma, and a depth of insertion of 4.0 mm below the brain’s surface. Autologous whole blood (obtained from the caudal vein) was injected into each mouse over 10 min using a microsyringe. After injection, the needle was left in place for 5 minutes to prevent blood reflux before being gently withdrawn. The wound was then sutured. The overall generation rate of mice within 72 h post-ICH was approximately 85%, and mice with a modified Neurological Severity Score (mNSS) greater than 2 were considered successfully modeled. The mice in the sham-operated group underwent the same procedure, except that no blood was injected.
Cell viability was assessed via the Cell Counting Kit-8 (CCK8) assay (Sigma-Aldrich, 96992, St. Louis, MO, USA) in accordance with the supplier’s standardized protocol.
We used Beyotime assay kits to measure various biochemical activities: a Superoxide Dismutase Assay Kit (Beyotime, S0101S, Shanghai, China) for SOD activity, Lipid Peroxidation MDA Assay Kit (Beyotime, S0131S, Shanghai, China) for MDA levels, and Micro Reduced Glutathione Assay Kit (Beyotime, S0053, Shanghai, China) for GSH measurement. All experimental steps were executed as specified in the manufacturer’s guidelines.
After treatment, tissues and cells were lysed using a radioimmunoprecipitation assay-based solution containing protease inhibitors. Extracted proteins were resolved by SDS-polyacrylamide gel electrophoresis, transmitted to a polyvinylidene difluoride membrane, blocked with FBS, and cleared with PBS. Subsequently, the membrane was incubated overnight at 4 °C with primary antibodies, namely rabbit anti-mouse NRF2 (1:2000; CST, 12721, Danvers, MA, USA), heme oxygenase-1 (HO-1) (1:2000; CST, 70081, USA), B-cell lymphoma 2 (Bcl-2) (1:2000; CST, 3498, USA), Bax (1:2000; CST, 2772, USA), VSIG4 (1:2000; Abcam, ab252933, Cambridge, UK), and GAPDH (1:2000; CST, 2118, USA). After three 5-minute wash cycles with PBS, the substrate was coupled to a goat anti-rabbit IgG horseradish peroxidase secondary antibody (1:3000; Affinity, S0001, USA) for 1 hour at room temperature. Protein signals were visualized using an enhanced chemiluminescence system and quantified via scanning densitometry and computer-aided image analysis. Protein levels were reported as the ratio of each detected band to that of GAPDH.
RNA isolation from tissue and cell samples was carried out with TRIzol reagent
per the manufacturer’s protocol, followed by reverse transcription of RNA into
cDNA using commercial kits. RT-qPCR was performed using a specific PCR system
according to the manufacturer’s instructions. The PCR cycling conditions
consisted of the following steps: The initial step was a pre-denaturation phase,
which was performed at 95 °C for 10 min. After that, 40 cycles were
carried out. Each cycle involved denaturation at 95 °C for 15 s and then
an annealing and extension phase at 60 °C for 35 s. PCR was performed in
triplicate for each sample. Each experiment was repeated at least three times.
Data were analyzed using the comparative CT (
The interleukin-1
Three days post-ICH, the mice were evaluated using the mNSS as described [18, 19]. The scoring scale ranged from 0 (no visible neurological impairment) to 18 (the most severe neurological impairment). The mNSS assesses motor function, sensory responses, balance, and reflexes. Motor deficits, sensory impairments, balance dysfunction, and reflex abnormalities were scored on a scale of 0–6, 0–2, 0–6, and 0–4, respectively. All evaluations were conducted by trained investigators who were blinded to the experimental groups.
72 h after ICH, the mice were sacrificed by cervical dislocation. Immediately
thereafter, the brain specimens were divided into four parts: the ipsilateral
cortex (Ipsi-CX), basal ganglia on the ipsilateral side (Ipsi-BG), contralateral
cortex (Cont-CX), and basal ganglia on the contralateral side (Cont-BG). First,
the specimens were weighed to determine their wet weight. They were then placed
in an oven at 105 °C and dried for 72 h. After drying, the specimens
were weighed again to determine their dry weight. As described [20, 21], the
percentage of brain water content was derived by applying the following formula:
(wet weight–dry weight)/wet weight
On day 3 after ICH, samples were obtained, and paraffin-embedded slices were prepared according to the previously described method [17]. Brain slices were incubated with 5% BSA to block unspecific binding. After overnight incubation at 4 °C with primary antibodies—anti-Iba-1 (1:500, CST 17198, USA), anti-NeuN (1:500, Abcam, ab104224, UK) and anti-GFAP (1:500, CST, 3670)—procedures proceeded to subsequent steps. The samples were then incubated with fluorescence-conjugated secondary antibodies (Abcam, ab150115 and ab150077, UK) at room temperature for 2 h. After incubation with secondary antibodies, the cells were washed three times with PBS. Next, the nuclei were stained with DAPI (Abcam, ab228549, UK). Finally, the ImageJ software (1.54f, National Institutes of Health, Bethesda, MD, USA) was used to calculate and quantify the average number of positively stained cells.
Tissue sections were deparaffinized in xylene, rehydrated through a graded ethanol series, and rinsed with water. Antigen retrieval was performed by microwave heating in sodium citrate buffer (pH 6.0). Endogenous peroxidase activity was quenched with 3% H₂O₂ for 10 minutes. After washing with TBST, sections were blocked with 5% BSA for 30 minutes at room temperature. The sections were incubated with primary antibodies against VSIG4 (1:500, Abcam, ab252933, UK), TMEM119 (1:500, CST 90840, USA), and IBA1 (1:500, CST 17198, USA) for 1 hour at room temperature, followed by incubation with HRP-conjugated goat anti-rabbit secondary antibody (absin, abs50012, Shanghai, China) for 10 minutes. Fluorescence signal amplification was achieved using tyramide conjugates (absin, abs50012, Shanghai, China) for 10 minutes. Nuclei were counterstained with DAPI (Abcam, ab228549, UK), and sections were mounted with anti-fade medium. Images were acquired using a fluorescence microscope (Olympus DP74, Olympus Corporation, Tokyo, Japan).
Brain tissues from each group of mice were collected and fixed in 4% paraformaldehyde at 4 °C for one week. Following paraffin embedding and sectioning, HE staining was performed according to the manufacturer’s instructions (Solarbio, G1120, Beijing, China). Tissue sections were dehydrated using graded ethanol, and staining results were observed under an inverted fluorescence microscope (Olympus DP74, Olympus Corporation, Japan).
Quantitative data are presented as mean
Microglia and macrophages have similar functions and play essential roles in
neuroinflammation after ICH [22]. It has been reported that VSIG4 can be
expressed in peripheral macrophages [23]. To investigate VSIG4 expression in
microglia/macrophages in brain tissue after ICH, we constructed an ICH mouse
model and performed immunostaining (Fig. 1A). Immunostaining revealed VSIG4
expression exclusively in IBA1+ TMEM119– cells (peripheral
macrophages) post-ICH, with no detection in IBA1+ TMEM119+ microglia
(Fig. 1B). In addition, compared to 1 day after ICH, the fluorescence intensity
of VSIG4 in IBA1+ cells decreased 3 days after ICH (p
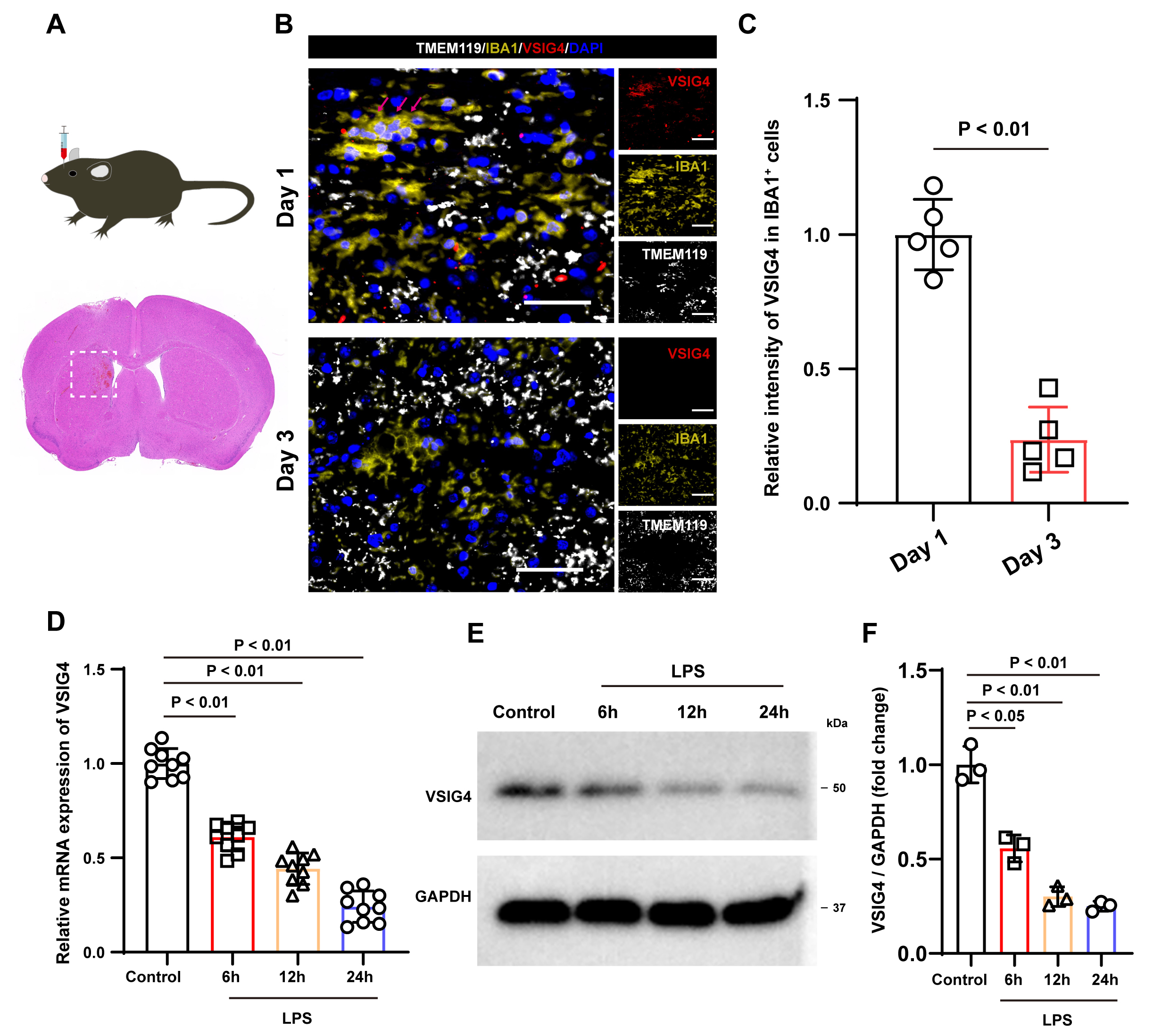 Fig. 1.
Fig. 1.
Illustrates the expression characteristics of VSIG4 following ICH and the expression of VSIG4 in macrophages following LPS stimulation. (A) A schematic diagram outlining the experimental protocol carried out in mice. (B) Representative immunofluorescence images of VSIG4-positive cells, IBA1-positive cell and TMEM119-positive cells in the tissue surrounding the hematoma on days 1 and 3 after ICH, n = 5, scale 50 µm. The red arrow shows VSIG4+IBA+TMEM119– cells. (C) The relative fluorescence intensity of VSIG4-positive and IBA1-positive cells in the tissue surrounding the hematoma on day 1 and day 3 after ICH, with n = 5. (D) The RT-qPCR results of VSIG4 mRNA in RAW264.7 cells after LPS stimulation. (E,F) Western blotting analysis of VSIG4 in RAW264.7 cells after LPS stimulation. VSIG4, V-set and immunoglobulin domain-containing 4; ICH, intracranial hemorrhage; LPS, lipopolysaccharide.
Inflammatory stimulation is an important factor that causes changes in
macrophages. To examine VSIG4 expression in macrophages under inflammatory
stimulation, RAW264.7 cells were treated with LPS (1 µg/mL) in
vitro. RT-qPCR findings revealed that, compared to the control group, LPS
treatment resulted in a time-dependent decrease in VSIG4 mRNA levels (p
To explore the effects of VSIG4 on macrophages, we introduced recombinant VSIG4
(100 µg/mL) into RAW264.7 cells. Western blot analysis showed that VSIG4
treatment significantly upregulated the expression of NRF2 and HO-1 in RAW264.7
cells (p
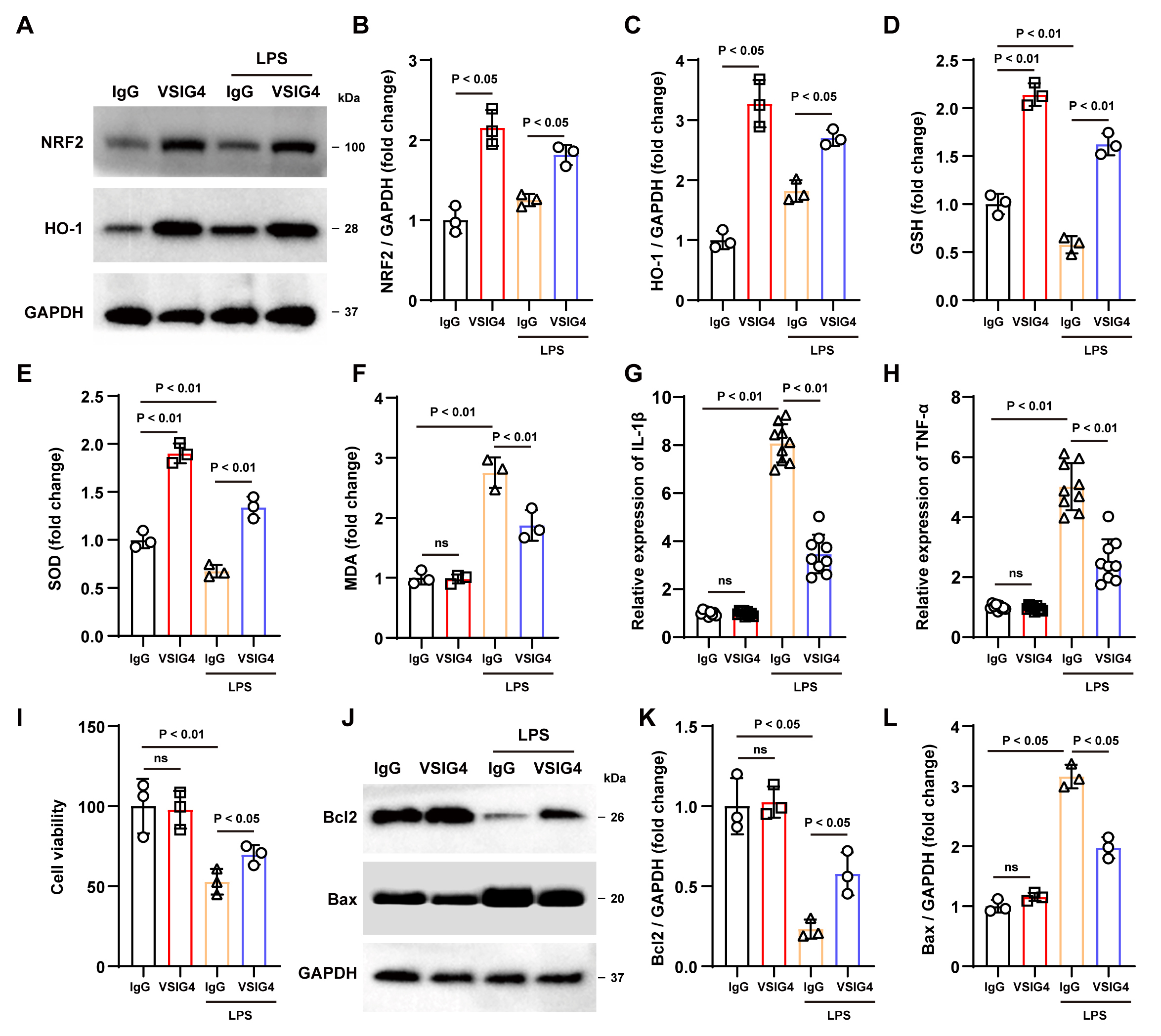 Fig. 2.
Fig. 2.
Recombinant VSIG4 activates the NRF2 signaling pathway to
inhibit oxidative stress and inflammatory responses in macrophages. (A–C)
Western blotting analysis of NRF2 and HO-1 in RAW264.7 cells after the addition
of recombinant VSIG4, n = 3. (D–F) Levels of GSH, SOD, and MDA in RAW264.7 cells
after the addition of recombinant VSIG4, n = 3. (G,H) Levels of IL-1
Oxidative stress has also been shown to activate inflammatory cells and release
pro-inflammatory cytokines. As expected, ELISA findings demonstrated a notable
increase in TNF-
Subsequently, we conducted a co-culture experiment with HT22 and RAW264.7 cells
treated with various treatments to investigate the potential protective effect of
VSIG4-induced macrophages in vitro. According to the CCK-8 assay
results, no significant difference was observed in cell viability between the two
groups without LPS exposure. However, under LPS stimulation, VSIG4-treated
RAW264.7 cells displayed improved viability compared to the IgG group (p
Next, we evaluated the potential protective effects of VSIG4 in ICH mice. To
this end, VSIG4 or IgG was injected into the mouse brain ventricle after ICH
induction, and neurological function scoring was performed on day 3 after ICH.
Parameters including cerebral edema severity, antioxidant/apoptotic protein
expression, and inflammatory marker dynamics were further analyzed. Neurological
function scores in the VSIG4 group showed significant improvement relative to the
IgG controls (p
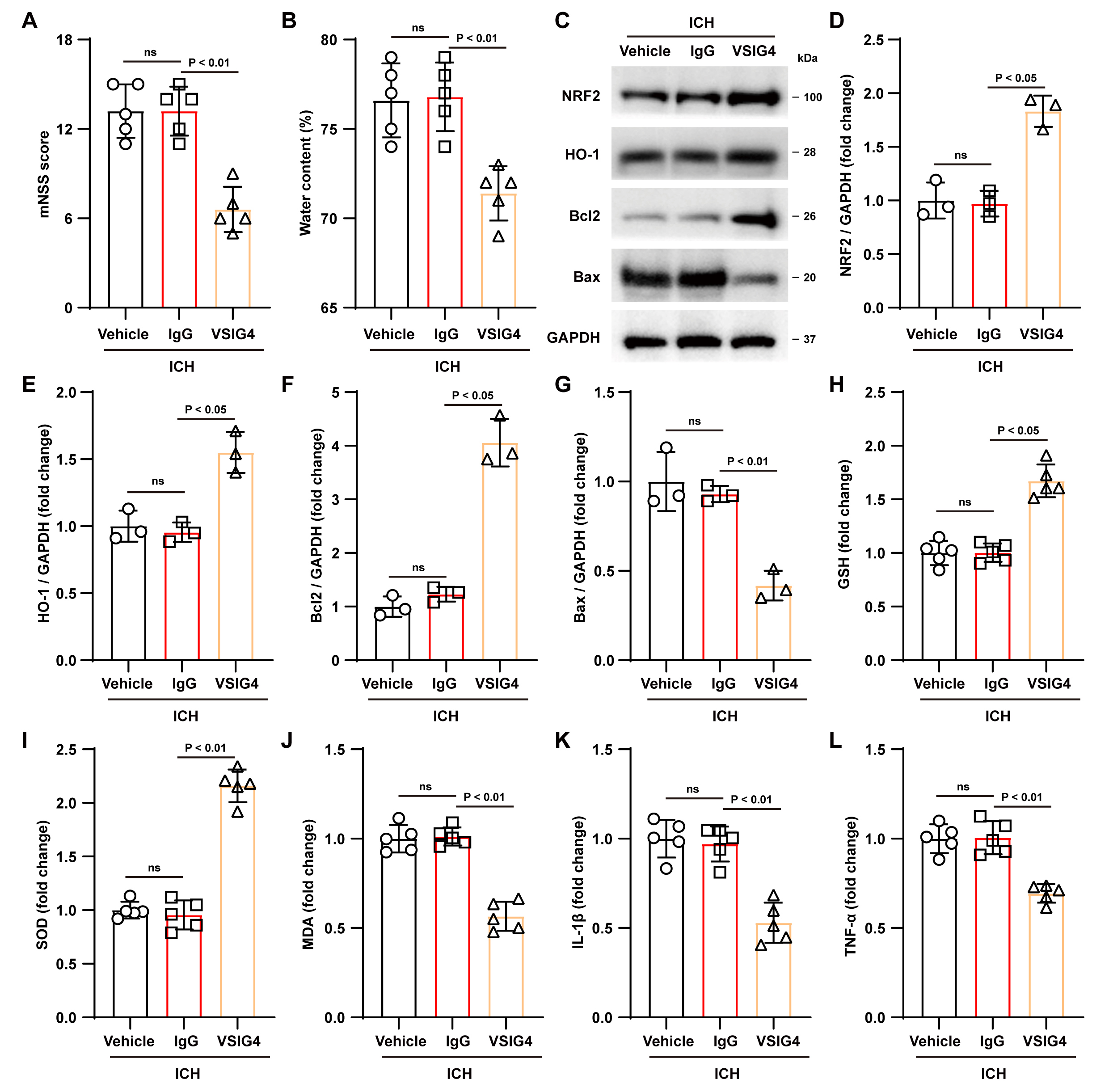 Fig. 3.
Fig. 3.
Effect of VSIG4 on ICH mouse outcomes. (A)
Neurological scores, n = 5. (B) Brain water content, n = 5. (C–G) Western blot
assays were conducted in ICH mice to examine NRF2, HO-1, Bcl-2, and Bax, n = 3.
(H–L) Levels of GSH, SOD, MDA, and the pro-inflammatory factors IL-1
Correspondingly, analysis of antioxidant indicators showed that VSIG4 treatment
significantly increased GSH and SOD levels in brain tissue (GSH, p
Additionally, we observed that VSIG4 therapy reduced Iba-1+ and GFAP+
cell counts while increasing the NeuN+ cell population in mouse brain tissue
compared to the IgG group (p
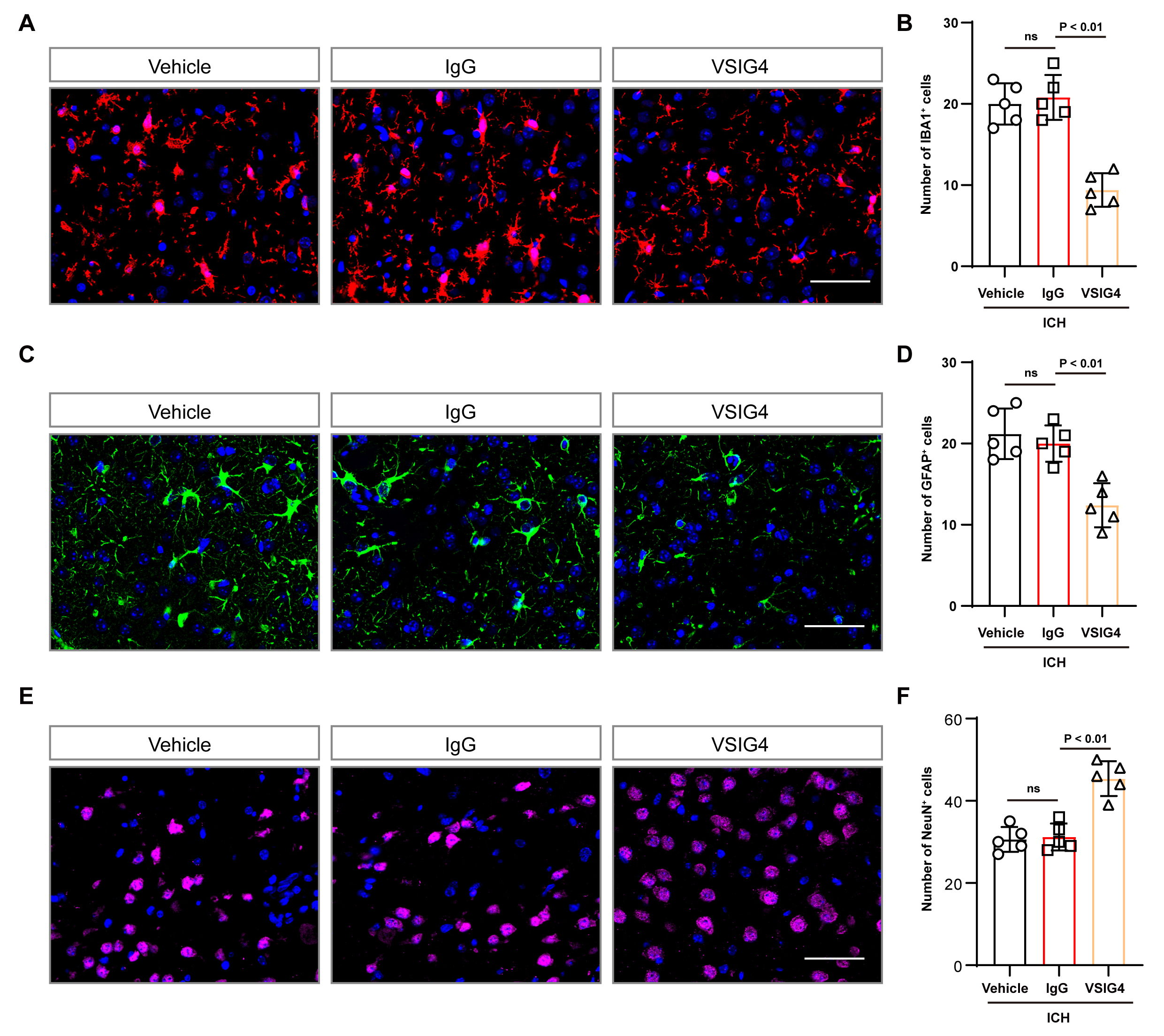 Fig. 4.
Fig. 4.
Effect of VSIG4 on IBA1-positive cells and GFAP-positive cells in ICH mice. (A,B) Representative immunofluorescence images and cell numbers of IBA1-positive cells in the tissue surrounding the hematoma after ICH, n = 5, scale 50 µm. (C,D) Illustrative immunofluorescence images and corresponding cell counts of GFAP-positive cells in the tissue surrounding the hematoma after ICH, n = 5, scale 50 µm. (E,F) Illustrative immunofluorescence images and corresponding cell counts of NeuN-positive cells in the tissue surrounding the hematoma after ICH, n = 5, scale 50 µm. “ns” means no significant difference.
To verify whether VSIG4 alleviates oxidative stress and inflammatory responses
in macrophages in vitro through the NRF2/HO-1 pathway, we treated
RAW264.7 cells with the NRF2 inhibitor brusatol [27]. Western blot results showed
that brusatol reduced NRF2 and HO-1 expression and inhibited VSIG4-induced
upregulation of NRF2 and HO-1 expression (p
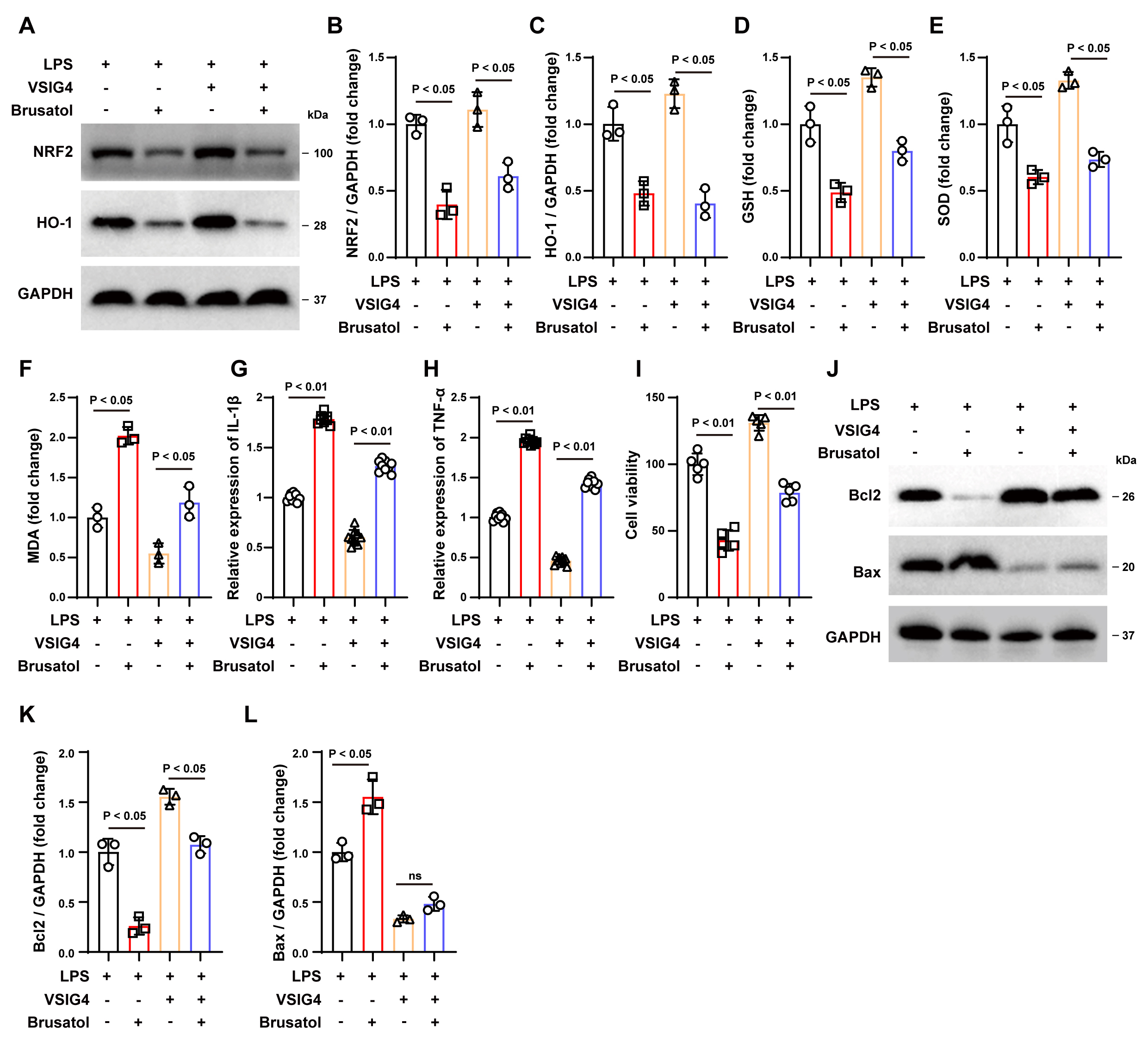 Fig. 5.
Fig. 5.
The NRF2 inhibitor Brusatol inhibits the protective
effect of VSIG4 in vitro. (A–C) Western blotting analysis of NRF2 and
HO-1 in RAW264.7 cells exposed to Brusatol, n = 3. (D–F) Levels of GSH, SOD and
MDA levels in Brusatol-exposed RAW264.7 cells, n = 3. (G,H) Concentrations of
IL-1
Subsequently, we co-cultured brusatol-treated RAW264.7 cells with HT22 cells to
determine whether VSIG4 confers a protective effect on neurons through the
NRF2/HO-1 signaling pathway. As shown in Fig. 5I, brusatol treatment reduced the
survival rate of LPS-treated HT22 cells (p
To further elucidate the protective effect of VSIG4 in the ICH model and its relationship with the NRF2/HO-1 pathway, we conducted a comparative analysis of neurological function, histopathological changes, and molecular-level changes between the VSIG4-only treatment group and the brusatol intervention group following ICH.
The results demonstrated that brusatol administration, which inhibits NRF2
signaling, worsened the neurological deficits in ICH mice, increased brain tissue
water content (p
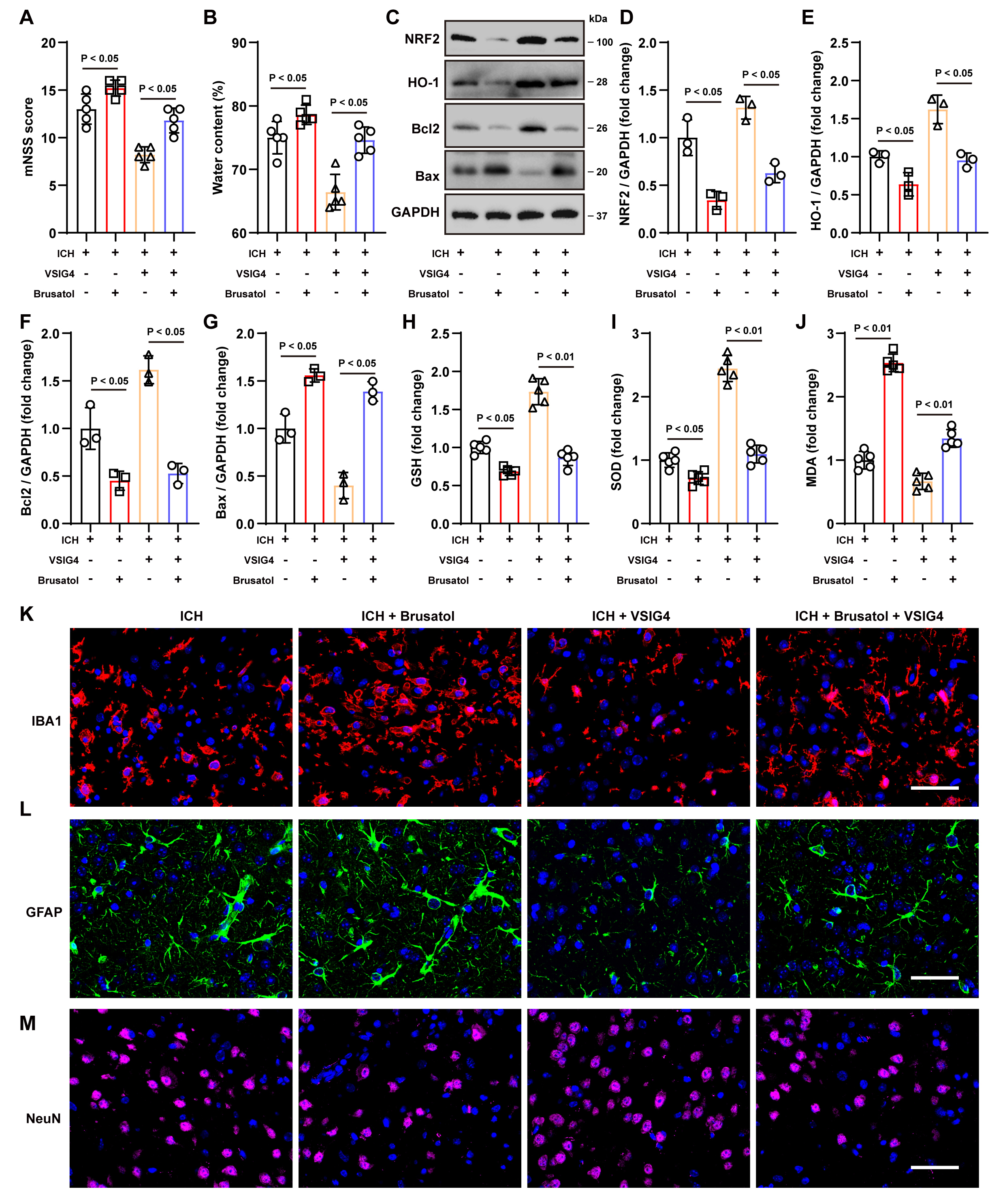 Fig. 6.
Fig. 6.
The NRF2 inhibitor Brusatol inhibits the protective effect of VSIG4 in vivo. (A) Neurological scores, n = 5. (B) Brain water content, n = 5. (C–G) Western blot analysis was conducted to measure the protein levels of NRF2, HO-1, Bcl-2, and Bax in brain tissues from ICH mice treated with Brusatol, n = 3. (H–J) Levels of GSH, SOD and MDA levels in ICH mice exposed to Brusatol, n = 5. (K–M) Representative immunofluorescence images and quantification of cells positive for IBA1, GFAP, and NeuN in the perihematomal region following Brusatol treatment, n = 5, scale 50 µm. “ns” means no significant difference.
Consistent with the in vitro findings, brusatol administration resulted
in a significant reduction in the increased expression of NRF2 (p
Tissue immunofluorescence staining and cell counting demonstrated that VSIG4
markedly diminished the aberrant proliferation of IBA1+ and GFAP+
cells, thereby attenuating the pro-inflammatory response and increasing
NeuN+ cells survival. Conversely, brusatol increased the number of
inflammatory cells (p
In conclusion, VSIG4 exerts notable neuroprotective effects in vivo by upregulating the NRF2/HO-1 pathway. These effects include improving neurological function, reducing cerebral edema, inhibiting apoptosis and oxidative stress, and suppressing excessive inflammatory responses. However, adding the NRF2 inhibitor brusatol significantly diminished the protective effects of VSIG4.
Intracranial hemorrhage is linked to high rates of fatality and disability and represents a serious threat to human health. Its pathophysiological mechanisms include both direct and secondary damage. To date, intracerebral hematoma removal remains the central clinical intervention for patients with ICH and can help reduce mortality and prevent further neurological deterioration. However, studies investigating the long-term functional recovery benefits of surgical intervention have yielded inconsistent results and currently lack robust, high-quality evidence [28]. These inconsistencies may stem from the difficulty of hematoma removal, surgeon expertise, and treatment time window. Therefore, targeting secondary damage after ICH, such as oxidative stress and neuroinflammation, is a potential treatment strategy for functional recovery.
This study comprehensively investigated the alterations and therapeutic effects
associated with VSIG4 in ICH and LPS stimulation models and explored the
underlying mechanisms in detail. Our findings indicate that VSIG4 has a
neuroprotective effect. Microglia are generally considered prominent participants
in the immune reaction of the central nervous system [29]. Unlike previous study
[18], we found that VSIG4 was mainly expressed in macrophages rather than
microglia. In addition, our data showed that VSIG4 expression decreased as ICH
duration increased, suggesting a negative correlation between VSIG4 expression
levels and ICH severity, providing insight into the function of VSIG4. After
exogenous VSIG4 intervention, we found that VSIG4 upregulated the expression of a
series of anti-oxidative enzymes, such as HO-1 and SOD, decreased levels of
substances, such as GSH, and reduced the production of oxidative damage markers,
such as MDA [30]. These changes effectively relieved oxidative stress and
inflammatory damage in brain tissue and cells subjected to ICH and LPS
stimulation, thereby increasing HT22 cell survival rate, improving neurological
function, and reducing cerebral edema. Furthermore, VSIG4 upregulated Bcl-2 and
reduced Bax expression, which supports its positive role in apoptosis regulation
[31]. Bcl-2 functions as an anti-apoptotic protein, preventing the formation of
holes in the outer mitochondrial membrane. However, Bcl-2 activity is inhibited
by binding to Bax, which can damage cell integrity, release apoptotic factors,
and initiate an apoptotic cascade [32, 33]. A high Bcl-2/Bax ratio inhibits the
mitochondrial apoptotic pathway, prevents cytochrome C escape from mitochondria,
enables caspase-9 and caspase-3 activation, and promotes cell survival.
Additionally, VSIG4’s inhibitory effect on microglial and astrocyte activation in
an inflammatory environment further underscores its importance in maintaining the
balance of the neural microenvironment. By decreasing the secretion of
pro-inflammatory mediators, including IL-1
Second, our study clarified that VSIG4 exerts its protective effects by
regulating the NRF2/HO-1 signaling pathway. NRF2 is considered a key
transcription factor in cells responding to oxidative stress and inflammatory
responses. Under physiological conditions, NRF2 is bound to Keap1, retained in
the cytoplasm, and steadily degraded via the ubiquitin-proteasome pathway [38].
However, under stress conditions, NRF2 is released and transferred to the
nucleus, promoting the expression of antioxidant genes, including HO-1 [25]. HO-1
can achieve antioxidant effects by promoting heme degradation and inhibiting
signaling pathways such as NF-
Moreover, the present study has certain limitations. For instance, the study was exclusively verified in a mouse model, and there are significant differences in pathophysiology between humans and mice, including differences in hematoma size, inflammation duration, and immune cell infiltration pattern. Furthermore, human patients with ICH exhibit larger hematoma volumes, which consequently extend the time window for the inflammatory response. Additionally, VSIG4 and NRF2 signaling pathway activation may undergo distinct dynamic changes. There are also differences in the response patterns of immune cells, such as macrophages and microglia, between humans and mice. Although the protective role of VSIG4 has been confirmed in animal experiments, further exploration is necessary to assess its feasibility in clinical settings, determine the optimal administration route, and evaluate potential safety concerns.
The current study provides experimental evidence for the neuroprotective mechanism of VSIG4 in ICH. VSIG4 improves neurological deficits and mitigates tissue damage after ICH by activating the NRF2/HO-1 pathway, enhancing antioxidant capacity, and inhibiting excessive inflammatory responses. Future studies should focus on elucidating the molecular mechanism underlying the interaction between VSIG4 and NRF2 and evaluating its potential value in clinical applications.
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
SQ and LZ were responsible for experimental design and project management. HL and YL conducted experimental procedures and data analysis. HL drafted the initial manuscript and participated in revisions, while YL prepared the figures/charts and contributed to manuscript editing. YZ and WQ assisted in figure/chart preparation and manuscript revisions. ZS participated in experimental design and manuscript polishing. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
For mice, all experimental protocols followed the ARRIVE guidelines and were approved by the Laboratory Animal Management and Ethics Committee of Huzhou Central Hospital (Approval number: 202411005).
We would like to express our gratitude to all those who helped us during the writing of this manuscript.
This work was supported by the Shenzhen Natural Science Fund (No. JCYJ20210324134800001 and JCYJ20190808103401655 to Lifang Zheng), the Huzhou City Public Welfare Applied Research Project (No. 2022GZ63 to Sheng Qiu) and the Zhejiang Chinese Medical University Affiliated Hospital Scientific Research Project (No. 2023FSYYZZ16 to Sheng Qiu).
The authors declare no conflict of interest.
During the preparation of this work the authors used Grammarly in order to check spell and grammar. After using this tool, the authors reviewed and edited the content as needed and takes full responsibility for the content of the publication.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL37810.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.