










1 Department of Neurosurgery, Tianjin Medical University General Hospital, 300052 Tianjin, China
2 Department of Neurosurgery, Heji Hospital Affiliated to Changzhi Medical College, 046011 Changzhi, Shanxi, China
3 Department of Neurosurgery, Gaoping People’s Hospital, 048400 Gaoping, Shanxi, China
Abstract
Traumatic brain injury (TBI) is a disease caused by external forces that damage brain structure and function. After TBI, iron accumulation and reactive oxygen species (ROS) increase lipid peroxidation, promoting ferroptosis. Methyltransferase-like 3 (METTL3) inhibits ferroptosis by modulating related signaling pathways. This study investigates the effects of METTL3 on neuronal ferroptosis in TBI, offering new insights and potential therapies.
TBI mouse and neuron cell models were established and treated with METTL3 overexpression. The Morris Water Maze (MWM) test evaluated cognitive function. Histological staining of brain tissues was conducted to assess brain injury, nuclear pyknosis, and iron accumulation. The activation of neurons, microglia, and astrocytes were detected using immunofluorescence staining. Neuron cell proliferation was measured using the Cell Counting Kit 8 (CCK-8). Quantitative PCR (qPCR) and western blot detected the mRNA and protein expression. Ferroptosis was assessed by measuring the accumulation of iron, malondialdehyde (MDA), superoxide dismutase (SOD), and ROS. The quantification of the N6-methyladenosine (m6A) RNA methylation levels in cells was quantified using the m6A-ELISA assay. Methylated RNA immunoprecipitation (MeRIP) assays were conducted to analyze the m6A modification on GPX4 mRNA. The interaction between YTHDF2 and GPX4 mRNA was measured using RNA pulldown and RNA immunoprecipitation (RIP) assays.
METTL3 expression was downregulated in TBI-injured brain tissues. Overexpression of METTL3 improved cognitive function and brain recovery while simultaneously reducing ferroptosis and neuroinflammation. METTL3 overexpression upregulated GPX4 expression both in vitro and in vivo. Further studies indicated that m6A reader protein YTHDF2 binds to GPX4 mRNA, consequently mediating the METTL3-regulated m6A enrichment and RNA stability of GPX4. Knockdown of GPX4 and treatment with ferroptosis inducer abolished the protective effects of METTL3 on neurons.
METTL3 exhibits anti-ferroptosis properties and promotes brain injury recovery after TBI by regulating the m6A modification and RNA stability of GPX4.
Keywords
- traumatic brain injury
- ferroptosis
- m6a modification
- epigenetic regulation
- METTL3
Traumatic brain injury (TBI) is a significant global health issue that leads to substantial morbidity, mortality, and economic costs worldwide, with over 50 million new cases occurring annually [1]. Despite the high burden of severe TBI, no effective pharmacological intervention or treatment has been identified for TBI patients in randomized controlled trials, indicating that the key mechanisms leading to cell death and functional deficits after TBI remain elusive [2]. In the pathophysiological process of TBI, various forms of cell death occur, including apoptosis, necrosis, autophagy, pyroptosis, and ferroptosis [3, 4, 5]. However, the mechanisms of ferroptosis in the context of TBI have not been extensively explored.
Ferroptosis is a recently identified form of regulated cell death that is dependent on iron and lipid peroxidation products [6]. It is implicated in a variety of brain diseases and injuries, including ischemic stroke, intracerebral hemorrhage, and TBI [7, 8, 9]. The dysregulation of key components of the ferroptosis mechanism includes iron homeostasis, glutathione (GSH) depletion, and lipid peroxidation [10]. Research demonstrates that alterations in phosphatidylethanolamine oxidation, protein expression, and GSH levels following TBI are indicative of ferroptosis activation. Transmission electron microscopy further corroborates these findings by revealing characteristic features of ferroptosis, such as mitochondrial shrinkage. Moreover, studies have shown that inhibiting ferroptosis can potentially improve the long-term prognosis of TBI patients, underscoring the significant role of ferroptosis in the progression and outcome of TBI [11, 12, 13].
Traditionally, epigenetic regulation refers to the chemical modifications of DNA or histones, which can regulate gene expression independently of changes in the genomic sequence [14, 15]. Dysregulation of enzymes involved in epigenetic modifications has profound implications for human diseases and is frequently reported in various types of cancer [16, 17]. Similarly, RNA also carries hundreds of different sites for various post-transcriptional modifications. N6-methyladenosine (m6A) is the most prevalent mRNA modification in eukaryotic cells [18]. Methyltransferase-like 3 (METTL3) is the principal “writer” that introduces m6A methylation to RNA. YTHDF2, an m6A “reader”, recognizes and binds to these modifications [19]. METTL3 and YTHDF2 frequently collaborate to modulate the levels of m6A modifications in RNA. In glioma, YTHDF2 promotes UBXN1 degradation by recognizing METTL3-catalyzed m6A modifications, thereby activating the NF-kB pathway and accelerating glioma progression [20]. In liver cancer, METTL3 suppresses SOCS2 expression via a YTHDF2-dependent post-transcriptional mechanism, promoting hepatocarcinogenesis [21]. GPX4 is a critical enzyme that clears lipid peroxides. GPX4 dysfunction causes mitochondrial contraction, reactive oxygen species (ROS) accumulation, and increased lipid peroxide levels. Studies show that METTL3 inhibits ferroptosis by maintaining GPX4 expression at the translational level. METTL3 also regulates GPX4 to prevent ferroptotic signaling, which affects diseases such as glioblastoma, asthma, and aortic dissection [22, 23, 24].
A recent study has shown that METTL3 protein expression is increased in inflammatory microglia in human and mouse TBI models. Selective knockout of METTL3 has been shown to inhibit microglial pathogenic activity and enhance functional recovery post-TBI [25], yet the underlying regulatory roles and molecular mechanisms remain elusive. In this study, we aimed to explore the role of METTL3 in ferroptosis during TBI and to investigate the molecular mechanisms.
Animal experiments were conducted in accordance with the guidelines of the
Animal Ethic Committee of Medical Discovery Leader (MDL, Approval number:
MDL2023-08-26-01). Eight-week-old male C57BL/6J mice (wild-type,
METTL3+/+, and METTL3+/+GPX4-/-),
weighing between 20 and 25 grams, were purchased from Cyagen Biotechnology Co.,
Ltd. (Suzhou, China). A total of 60 mice were used in the experiment. The mice
were placed in a temperature-controlled room (23
Before surgery, the mice were assigned codes and evenly distributed into the following groups: sham, TBI model, model+NC, model+METTL3, model+METTL3+siGPX4, model+METTL3+erastin groups (10 mice in each group). Mouse TBI models were created through a controlled cortical impact (CCI) procedure [26]. The mice were positioned face-down in a stereotaxic frame. A surgical incision was made to expose the skull, and a 3 mm craniotomy was performed on the left side, positioned roughly halfway between the bregma and lambda, lateral to the midline. The skull flap was meticulously removed without damaging the underlying dura mater. The CCI was inflicted perpendicularly to the brain surface using a pneumatic cortical impactor (AmScien Instruments, Richmond, VA, USA). The settings for the impactor were as follows: an impact pressure of 10 KPa, a depth of 1.0 mm, and a duration of 70 milliseconds. The craniotomy site was immediately sutured shut using standard surgical materials after TBI. Mice in the sham group underwent all surgical steps except for the actual CCI. For the model+METTL3+erastin groups, the mice were intraperitoneally administrated with erastin (329600, Sigma-Aldrich, St. Louis, MO, USA) at a dose of 20 mg/kg body weight, 1 h after TBI. To ensure consistency and reduce variability, the surgical procedures, drug administrations, and CCI operations were conducted by the same experienced personnel. All surgical interventions were performed under deep anesthesia induced by intraperitoneal injection of sodium pentobarbital (1% w/v in sterile saline, 50 mg/kg body weight). Anesthesia depth was confirmed by the absence of corneal reflex and negative response to the toe pinch test.
To assess the efficacy of the interventional treatments, the Morris Water Maze (MWM) test was performed 7 days post-TBI. Prior to the TBI, mice underwent a 3-day training period to acclimate to locating a submerged platform in the maze, with three attempts per day, each limited to 90 seconds. Mice unable to find the platform within this period were guided to it and remained there for 15 seconds to familiarize themselves with the location. A probe trial, conducted 24 hours after training, tracked the latency to first reach the platform and the frequency of traversing its former location. These metrics were recorded and analyzed using a computer-assisted video tracking system (EthoVision XT 16.0, Noldus Information Technology, Wageningen, Netherlands), providing precise records of the swimming paths and times.
Neurological severity score (NSS) was used to evaluate motor, balance, and reflex functions in mice post-TBI, offering a comprehensive assessment of neurological status [27]. Trained professionals conducted NSS assessments, ensuring accuracy and consistency through specialized training. The NSS ranges from 0 to 10, with increasing scores signifying more pronounced neurological deficits. The grading system is as follows: 0–3 points indicate nearly normal neurological function or mild deficits; 4–6 points suggest moderate deficits; and 7–10 points represent severe impairments.
After the completion of the experimental treatments, the mice were anesthetized using an intraperitoneal injection of sodium pentobarbital (1.0% w/v in normal saline, 50 mg/kg body weight) to ensure minimal distress. The anesthetized mice were then euthanized by cervical dislocation. Subsequently, the brain tissues were carefully excised and immediately placed in a chilled phosphate-buffered saline (PBS) solution to prevent degradation. Brain tissues were fixed in 4% paraformaldehyde for 24 hours at 4 °C, dehydrated through a 90% ethanol solution, and then cleared in xylene. The tissues were sectioned into 5-µm-thick slices using a microtome.
Brain tissue sections were stained with 0.1% crystal violet solution (C0121, Beyotime, Shanghai, China) for 30 minutes, then quickly rinsed with PBS to remove excess stain. Subsequent dehydration with 95% and 100% ethanol for 5 minutes each, followed by clearing with xylene for 5 minutes (repeated twice), preceded mounting with a neutral medium. The stained sections were examined under a Nikon TE300 microscope (Nikon, Tokyo, Japan).
Brain tissue sections were immersed in Nissl staining solution (C0117, Beyotime, Shanghai, China) for 5 minutes at room temperature. After staining, the sections were rinsed with distilled water for 5 minutes. The stained sections were examined under a light microscope (Nikon, Tokyo, Japan) to visualize the morphology of neuron cells.
To identify degenerating neurons, FJB staining was performed on the brain sections according to the manufacturer’s protocol (AG310, Millipore, Darmstadt, Germany). The sections were first incubated in a 0.06% potassium permanganate solution for 10 minutes, washed with distilled water for 2 minutes, and stained with a 0.0004% solution of Fluoro-Jade B for 30 minutes. The stained sections were examined using a Nikon TE300 microscope to visualize degenerative neurons. The stained sections were examined under a Nikon TE300 fluorescence microscope equipped with filters to visualize degenerating neurons.
To identify iron deposition within cellular structures, Perls’ Prussian blue staining was performed using the Prussian Blue Iron Stain Kit (G1422, Solarbio, Beijing, China). Brain sections were immersed in the Perls Stain Solution for 30 minutes and rinsed with distilled water for 5 minutes. Subsequently, the sections were dehydrated with anhydrous ethanol for three times, each for 5 minutes, followed by clearing in dimethylbenzene for 5 minutes. The sections were then removed, and neutral resin was applied dropwise for mounting. Iron deposits were visualized as blue particles within cellular structures using a light microscope. These deposits were quantified with ImageJ software (version 1.53; National Institutes of Health, Bethesda, MD, USA).
Brain slices were rehydrated with PBS for 10 minutes and then blocked with a buffer consisting of 0.3% Triton X-100 and 10% BSA in PBS for 1 hour to reduce non-specific binding. The sections were incubated overnight at 4 °C with primary antibodies targeting GPX4 (ab125066, 1:250, Abcam, Cambridge, UK), SLC7A11 (ab307601, 1:500, Abcam), METTL3 (ab195352, Abcam), NeuN (ab104224, 1:3000, Abcam), GFAP (ab7260, 1:5000, Abcam), and Iba-1 (ab178846, 1:2000, Abcam). After washing with PBS, the sections were incubated with secondary antibodies for 1 hour at room temperature: Anti-rabbit IgG H&L (ab205718, 1:1000, Abcam) for GPX4, SLC7A11, METTL3, and GFAP; Anti-mouse IgG H&L (ab205719, 1:1000, Abcam) for NeuN; and Anti-goat IgG H&L (ab205720, 1:1000, Abcam) for Iba-1. For immunofluorescence staining, sections were mounted with a solution containing 4′,6-diamidino-2-phenylindole (DAPI, 62248, Thermo Scientific, Rockford, IL, USA) to stain cell nuclei. The slides were examined under a fluorescence microscope (Leica, Wetzlar, Germany) to visualize cell density and specific protein expression within the brain tissue.
The levels of total iron and ferrous irons in the ipsilateral cortex were assessed using an iron assay kit (ab83366, Abcam, Cambridge, UK). Malondialdehyde (MDA), superoxide dismutase (SOD) and ROS levels in cortex tissues were measured using the lipid peroxidation MDA assay kit (S0131S, Beyotime, Shanghai, China), Total SOD detection kit (S0101M, Beyotime, Shanghai, China) and Tissue ROS test kit (BB-460512, Bestbio, Shanghai, China), respectively.
Mouse primary cortical neurons were extracted from E15.5 mouse embryos as previously described [28], and the mouse hippocampal neuronal cell line HT-22 was from the Wanwu Biotechnology Co., Ltd. (Hefei, China). Primary cortical neurons exhibited a clear cell body with multiple long, slender processes extending from the soma, while HT-22 cells displayed typical neuronal-like morphology with elongated cell bodies and processes. Both primary neurons and HT-22 cells were cultured in DMEM (Gibco, New York, NY, USA) supplemented with 10% fetal bovine serum (Gibco, USA), 1% streptomycin, and penicillin mix at 37 °C with 5% CO2. To ensure cell purity and authenticity, mycoplasma detection and short tandem repeat (STR) profiling were conducted. No mycoplasma contamination was detected in any of the cell cultures.
To overexpress METTL3 and YTHDF2, mouse METTL3 (NM_019721.2) and YTHDF2 (NM_145393.4) were synthesized and cloned into the pLVX plasmid vector (General Biosystems, Anhui, China) and the complete plasmid sequence have been provided in Supplementary Fig. 1. The siRNA targeting METTL3 (5′-UCUAUCUCCAGAUCAACAUCG-3′; 5′-GCUACAAUCACAUCGCAGUCC-3′; 5′-GAUAUCACAACAGAUCCACUG-3′), siRNA targeting YTHDF2 (5′-UAGUAACUGGGUAAGUAGGAG-3′; 5′-UGGUUUAUUCUCGUUGUUCUC-3′; 5′-UUUGAAAUCAAAUUAAUCCUG-3′), siRNA targeting GPX4 (5′-AGUUUACGUCAGUUUUGCCUC-3′) and nonsense siRNA (5′-UUCUCCGAACGUGUCACGU-3′) were constructed by RiboBio (Guangzhou, China). Cells were transfected for 24 hours, after which a mechanical scratch injury was created using a 200 µL pipette tip to establish the cell injury model. Transfections were performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA).
Cell viability was assessed by the Cell Counting Kit-8 (C0038, Beyotime,
Shanghai, China). Briefly, cells were harvested and resuspended in complete
culture medium to prepare a cell suspension with a density of 2
The samples were homogenized in lysis buffer (P0023, Beyotime, Shanghai, China)
and sonicated. The homogenate was then centrifuged at 4 °C for 20
minutes at 12,000 g to separate soluble proteins. Protein concentrations were
measured using a BCA kit (P0009, Beyotime, Shanghai, China). Proteins were
resolved on SDS-PAGE gels ranging from 7.5% to 12.5% (Bio-Rad, Hercules, CA,
USA) and transferred onto polyvinylidene fluoride membranes (Millipore,
Darmstadt, Germany), and incubated overnight at 4 °C with primary
antibodies against SLC7A11 (ab307601, 1:1000, Abcam, Cambridge, UK),
IL-6 (ab290735, 1:1000, Abcam),
TNF-
Total RNA was extracted from the samples using TRIzol reagent (15596018CN,
Invitrogen, CA, USA), and quantified using a NanoDrop 2000C spectrophotometer
(Thermo, USA). Reverse transcription was performed using
PrimeScript™ RT Master Mix (RR036A, Takara, Japan) to generate
cDNA. qPCR reactions were conducted using a SYBR Green master mix (04913850001,
Roche, Basel, Switzerland) to ensure accuracy. mRNA levels were normalized to the
housekeeping gene
GPX4-F: 5′-TGGTTTACGAATCCTGGCCT-3′, GPX4-R: 5′-GGCATCGTCCCCATTTACAC-3′;
SLC7A11-F: 5′-GTCTGCCTGTGGAGTACTGT-3′, SLC7A11-R: 5′-ATTACGAGCAGTTCCACCCA-3′;
METTL3-F: 5′-TGGCCTCTTCAGCATCAGAA-3′, METTL3-R: 5′-ACTGACCTTCTTGCTCTGCT-3′;
YTHDF2-F: 5′-TGCTTGGTCTACTGGAGGTG-3′, YTHDF2-R: 5′-AATGGAGTGCTACCTAGGGC-3′;
IL-6-F: 5′-ACTTCCATCCAGTTGCC-3′, IL-6-R: 5′-ATGTGTAATTAAGCCTCCGAC-3′;
TNF-
IL-1
The production of inflammatory factors IL-6 (ab222503, Abcam, Cambridge, UK),
TNF-
The level of m6A RNA methylation in cells was quantified using an m6A RNA Methylation Quantification Kit (ab185912, Abcam, Cambridge, UK). In brief, mRNA was extracted and bound to a strip well for 90 minutes. After washing, captured, detection, and enhancer antibodies were added sequentially. The color-developing solution was then added, and the absorbance was measured at 450 nm using a microplate reader.
The MeRIP assay was performed using the Magna MeRIP m6A Kit (17-10499, Millipore, Darmstadt, Germany) to analyze the m6A modification in RNA samples. The enrichment of GPX4 mRNA was measured by qPCR.
The interaction between YTHDF2 and GPX4 mRNA was measured by Pierce™ Magnetic RNA-Protein Pull-Down Kit (PI20164, Thermo, USA), which contains streptavidin-coated magnetic beads (1 µm diameter, silica core, binding capacity: 1 µg biotinylated probe per 1 mg beads). In brief, the biotin-labeled GPX4-specific probe was reacted with magnetic beads, which were then mixed with protein lysate samples. After washing and elution, YTHDF pulled down by GPX4 mRNA was detected by western blot.
The RIP assay was performed using antibodies against YTHDF2 or a negative IgG control with Magna RIP™ RNA-Binding Protein Immunoprecipitation Kit (17-700, Millipore, Darmstadt, Germany). Protein A/G-coated magnetic beads (1 µm diameter, agarose matrix, provided in the kit) were pre-washed with RIP wash buffer and incubated with 5 µg of anti-YTHDF2 antibody or negative control IgG (I9145, Sigma-Aldrich, St. Louis, USA) for 1 hour at 4 °C. Cells were lysed in RIP lysis buffer, and the lysates were incubated with RIP immunoprecipitation buffer containing immunoprecipitated with A/G magnetic beads. The magnetic frame was used to fix bead-bound complexes and wash away unbound material. RNA was extracted from the bead-bound complexes, and GPX4 mRNA levels were analyzed by qPCR.
Cells transfected with siMETTL3, siYTHDF2, or siNC were treated with actinomycin D for 0, 3, and 6 h. RNA was extracted, and GPX4 mRNA levels were measured by qPCR.
Statistical analyses were conducted using GraphPad Prism 9 (San Diego, CA, USA).
Comparisons between two groups were analyzed using Student’s t-test,
while multiple groups were analyzed by
one-way ANOVA followed by Tukey’s post hoc test. Statistical significance was
defined as p
We established a mouse model of TBI to
investigate the ferroptosis. MWM test was employed to assess cognitive function
in mice. Compared to the sham group, TBI resulted in increased path lengths (Fig. 1A) and a decreased number of dentate gyrus neurons (Fig. 1B). Brain tissue
images and the NSS confirmed the successful creation of the TBI model (Fig. 1C,D). Histological analyses, including HE, Nissl, Perls, FJB staining, and
immunofluorescence, revealed significant brain injury (Fig. 1E), cytoplasmic
shrinkage or nuclear pyknosis (Fig. 1F), iron-positive cells (Fig. 1G,
Supplementary Fig. 2), and degeneration of cortical neurons (Fig. 1H, Supplementary Fig. 2) in TBI tissues, along with
reduced expression of the neuronal marker NeuN (Fig. 1I, Supplementary
Fig. 2). Additionally, TBI tissues exhibited significantly elevated levels of
MDA, Fe2+, total iron, and lipid ROS, along with significantly decreased SOD
levels (Fig. 1J, p
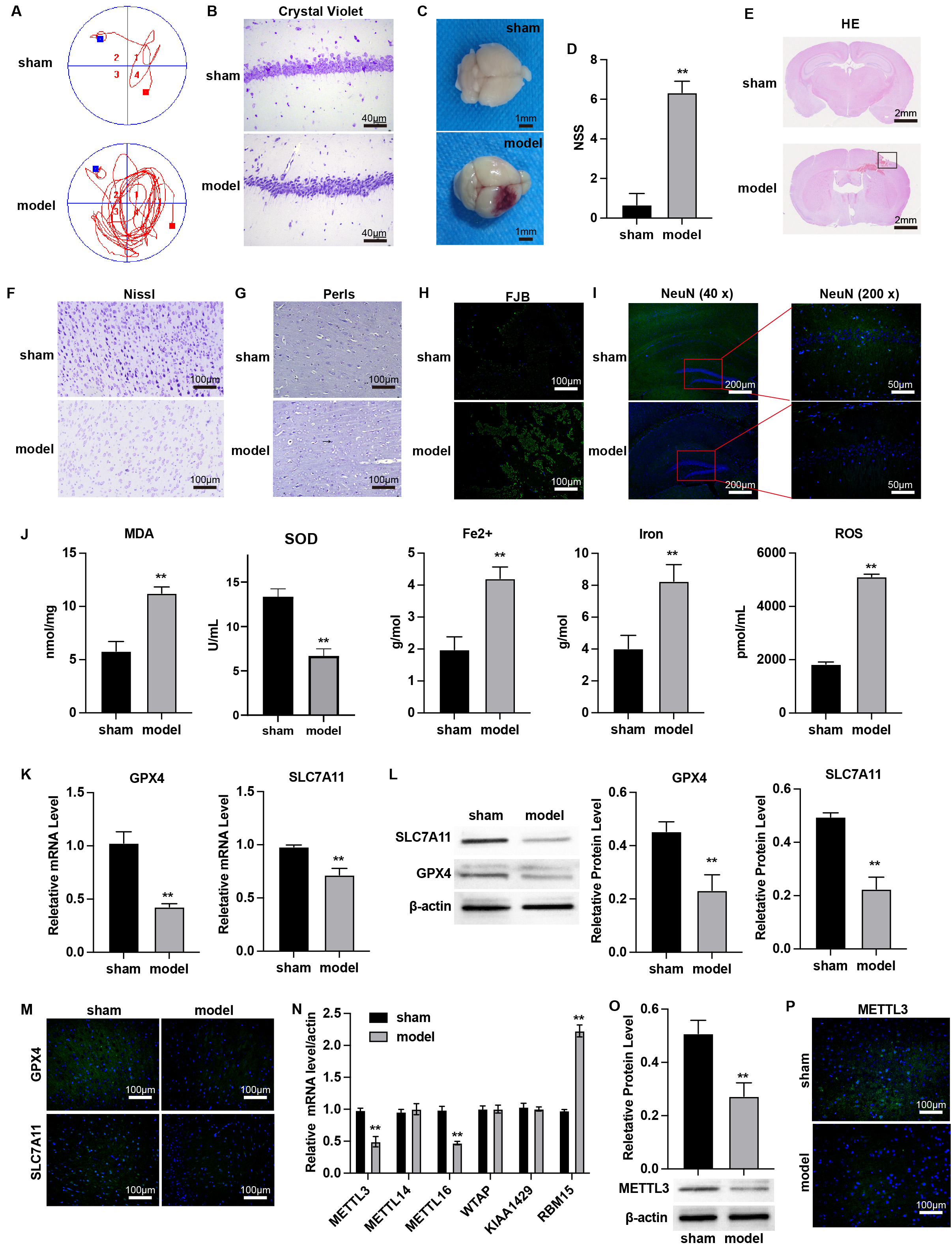 Fig. 1.
Fig. 1.
Downregulation of METTL3 in TBI-induced brain injury.
(A) The Morris Water Maze (MWM) test was performed on day 7 post-TBI. (B) Crystal
violet staining of brain tissues. Scale bar = 40 µm. (C) Brain
representative images. Scale bar = 1 mm. (D) Neurological severity score (NSS)
scores indicating brain tissue damage. (E–I) Representative images of (E) HE,
scale bar = 2 mm, (F) Nissl, scale bar = 100 µm, (G) Perls (arrows indicate
iron-positive cells), Scale bar = 100 µm, (H) Fluoro-Jade B (FJB), scale
bar = 100 µm, and (I) immunofluorescence staining of markers of neuron
(NeuN) in brain sections, scale bar = 200 µm or 50 µm; cortical contusion areas are
represented by rectangles. (J) The cortical levels of MDA, SOD, Fe2+, total
iron, and lipid ROS were measured. (K–M) The mRNA and protein levels of
GPX4 and SLC7A11 in cortex tissues of TBI mice were measured by
(K) qPCR, (L) western blot, and (M) immunofluorescence staining, scale bar = 100
µm. (N) The mRNA levels of m6A modification regulators in cortex tissues
were measured by qPCR assay. (O,P) The protein level of METTL3 in cortex tissues
was detected by (O) western blot and (P) immunofluorescence staining. Scale bar =
100 µm. **p
To assess the impact of METTL3 on TBI progression, we administered METTL3 overexpression in the TBI model.
qPCR and western blot analyses confirmed that this treatment successfully
restored the mRNA and protein levels of METTL3 in TBI-damaged brain
tissues (Fig. 2A–C, Supplementary Fig. 4, p
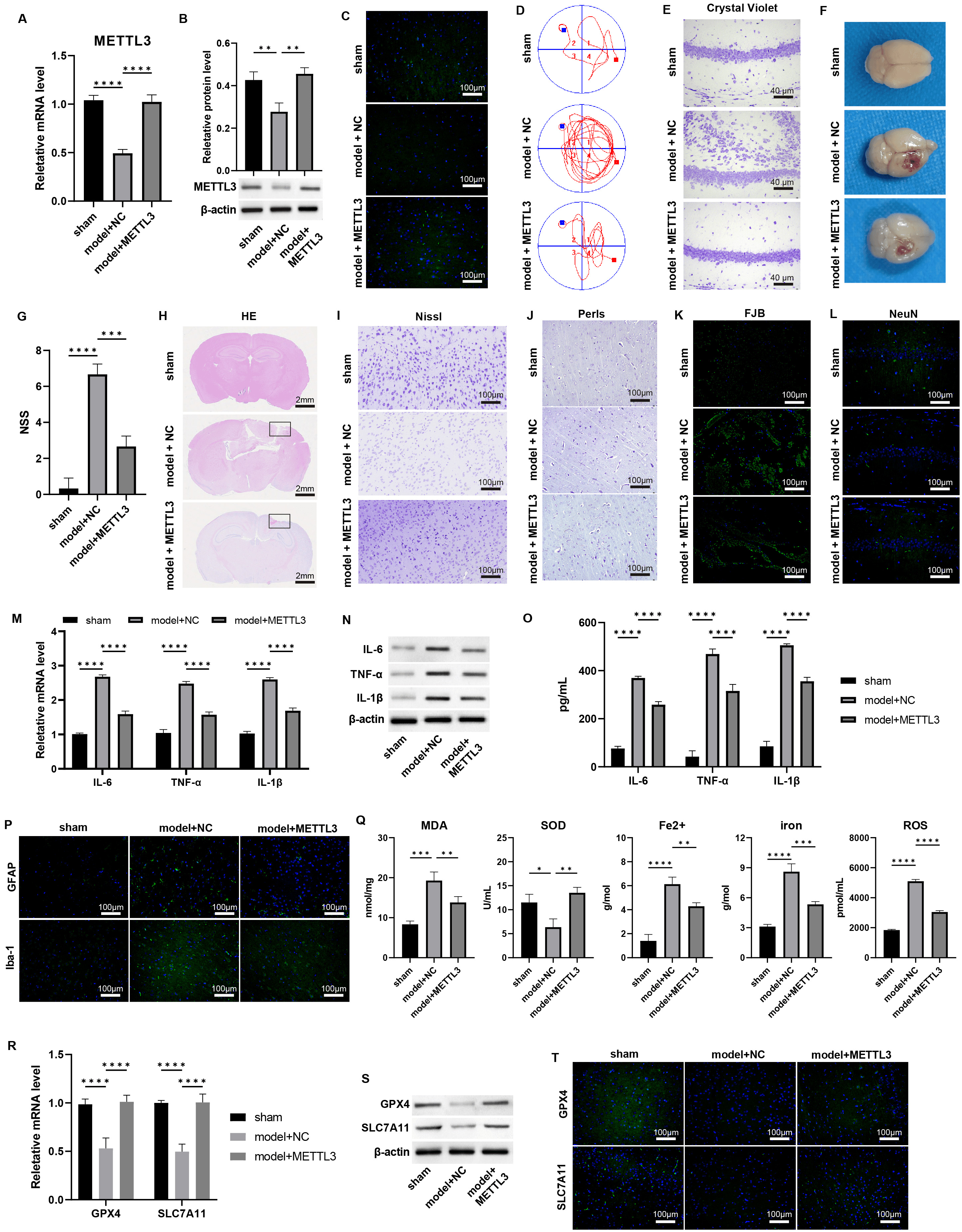 Fig. 2.
Fig. 2.
Overexpression of METTL3 alleviates the TBI damages and
ferroptosis. (A–C) The mRNA and protein levels of METTL3 in mouse
brain tissues were measured by (A) qPCR, (B) western blot, and (C)
immunofluorescence staining. Scale bar = 100 µm. (D) The Morris Water Maze
(MWM) test was performed on day 7 post-TBI. (E) Crystal violet staining of brain
tissues. Scale bar = 40 µm. (F) Representative images of the brain. (G) NSS
scoring of brain tissue damage. (H–L) Representative images of (H) HE, scale bar
= 2 mm, (I) Nissl, scale bar = 100 µm. (J) Perls, scale bar = 100 µm.
(K) Fluoro-Jade B (FJB), scale bar = 100 µm, and (L) immunofluorescence
staining of markers of neuron (NeuN) in brain sections, scale bar = 100 µm;
Cortical contusion areas are represented by rectangles. (M–O) The mRNA and
protein levels of IL-6, TNF-
To investigate the protective role of METTL3 on neurons in
vitro, we first isolated mouse primary cortical neurons, which expressed the
neuronal marker NeuN (Supplementary Fig. 9) and subsequently established
a cellular model to simulate mechanical injury akin to that experienced during
TBI. We treated these cells with METTL3 overexpression. The
downregulation of METTL3 observed in damaged neurons was effectively
reversed by its overexpression (Fig. 3A,B, p
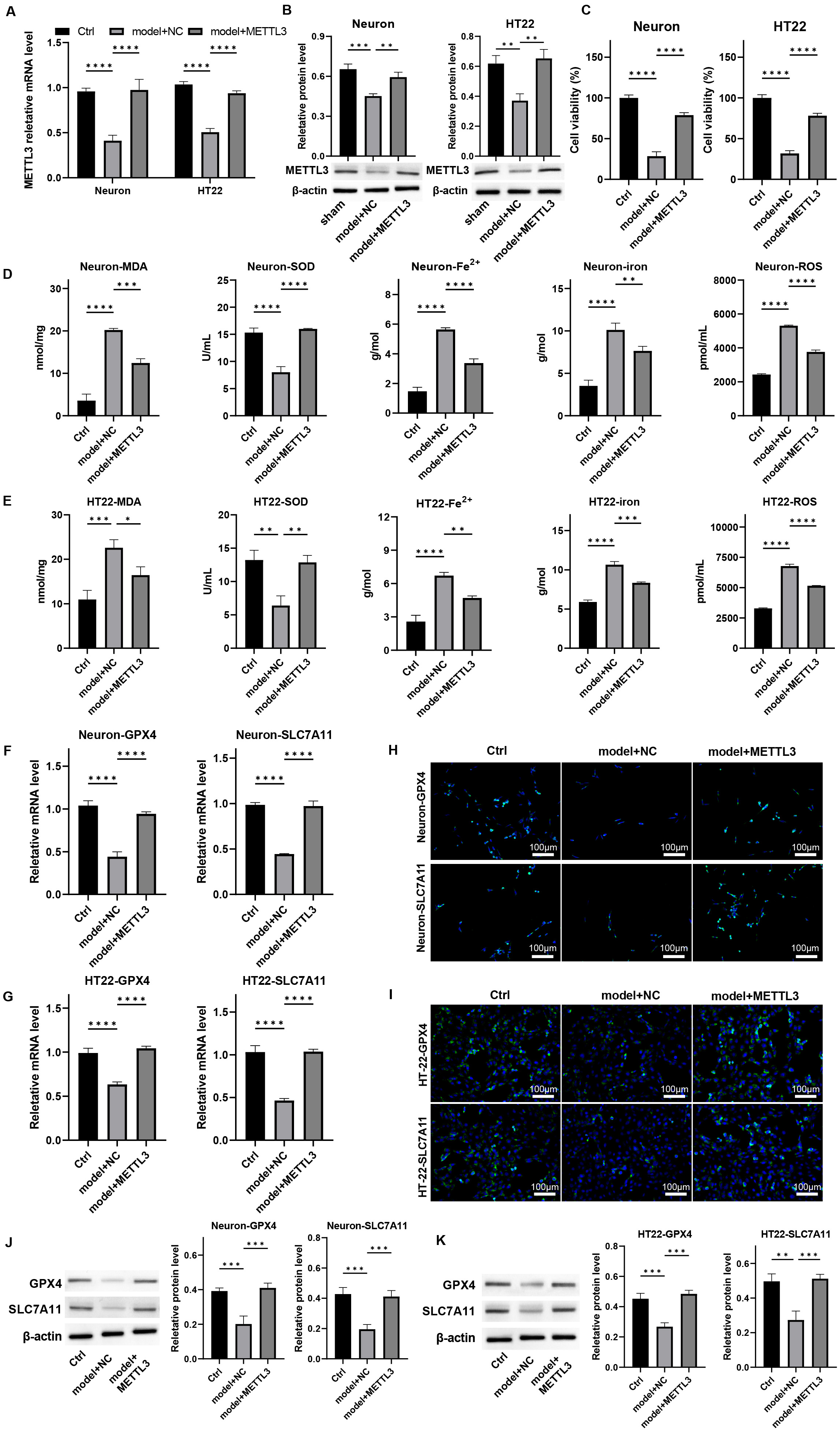 Fig. 3.
Fig. 3.
Overexpression of METTL3 protects neuron viability and
alleviates ferroptosis in vitro. Primary neurons and HT-22 cells were
processed by mechanical injury and treated with METTL3 overexpression.
The mRNA and protein levels of METTL3 were measured by (A) qPCR and (B)
western blot. (C) Cell viability was detected by CCK-8 assay. (D,E) The levels of
MDA, SOD, Fe2+, total iron, and lipid ROS were measured. (F,G) The mRNA
levels of GPX-4 and SLC7A11 in cells were detected by qPCR. (H–K) The protein
expression of GPX4 and SLC7A11 in cells was determined by (H,I)
immunofluorescence staining, scale bar = 100 µm, and (J,K) western blot
assay. *p
To explore the underlying mechanisms of METTL3-mediated neuronal
functions, we conducted a knockdown of METTL3 in primary neurons and
HT-22 cells, selecting siMETTL3-1 for further analysis (Fig. 4A,B,
p
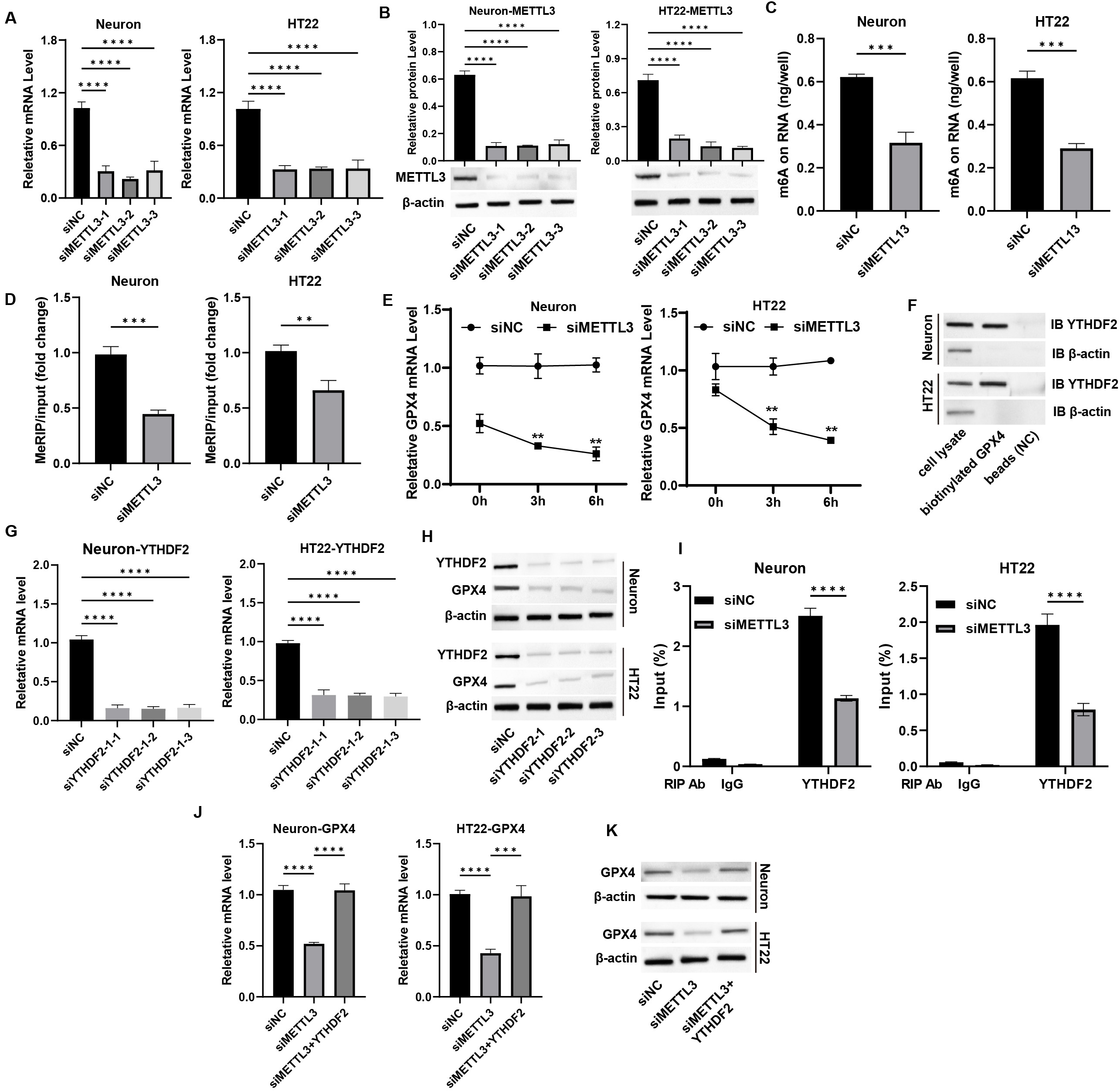 Fig. 4.
Fig. 4.
METTL3 epigenetically regulates GPX4 expression in
neurons. (A,B) Neurons were transfected with siMETTL3, and the (A) mRNA
and (B) protein levels of METTL3 were measured by qPCR and western blot
assay. (C) The total m6A level was measured by m6A-ELISA assay. (D) The m6A
enrichment on GPX4 mRNA was detected by MeRIP assay. (E) Neurons were
treated with actinomycin D, and the mRNA level of GPX4 was detected by
qPCR assay. (F) The interaction between YTHDF2 and GPX4 mRNA
was measured by RNA pulldown assay. (G,H) Neurons were transfected with
siYTHDF2, and the (G) mRNA and (H) protein levels of YTHDF2were measured by qPCR and western blot assay. (I) RIP-qPCR assay was performed
to measure the binding of YTHDF with GPX4 mRNA. (J) Neurons
were transfected with siMETTL3 and YTHDF2 overexpression
vectors, and the mRNA level of GPX4 was measured by qPCR assay. (K) The
protein level of GPX4 was measured by western blot assay. **p
To determine if METTL3 influences ferroptosis in TBI-induced neuronal
damage through GPX4 regulation, we conducted further analyses. qPCR and
western blot assays revealed that GPX4 knockdown or erastin treatment
did not affect METTL3 expression in neurons (Fig. 5A,B,
Supplementary Fig. 14). Notably, the neuronal proliferation enhanced by
METTL3 in the damage model was counteracted by GPX4 knockdown
and erastin treatment (Fig. 5C, p
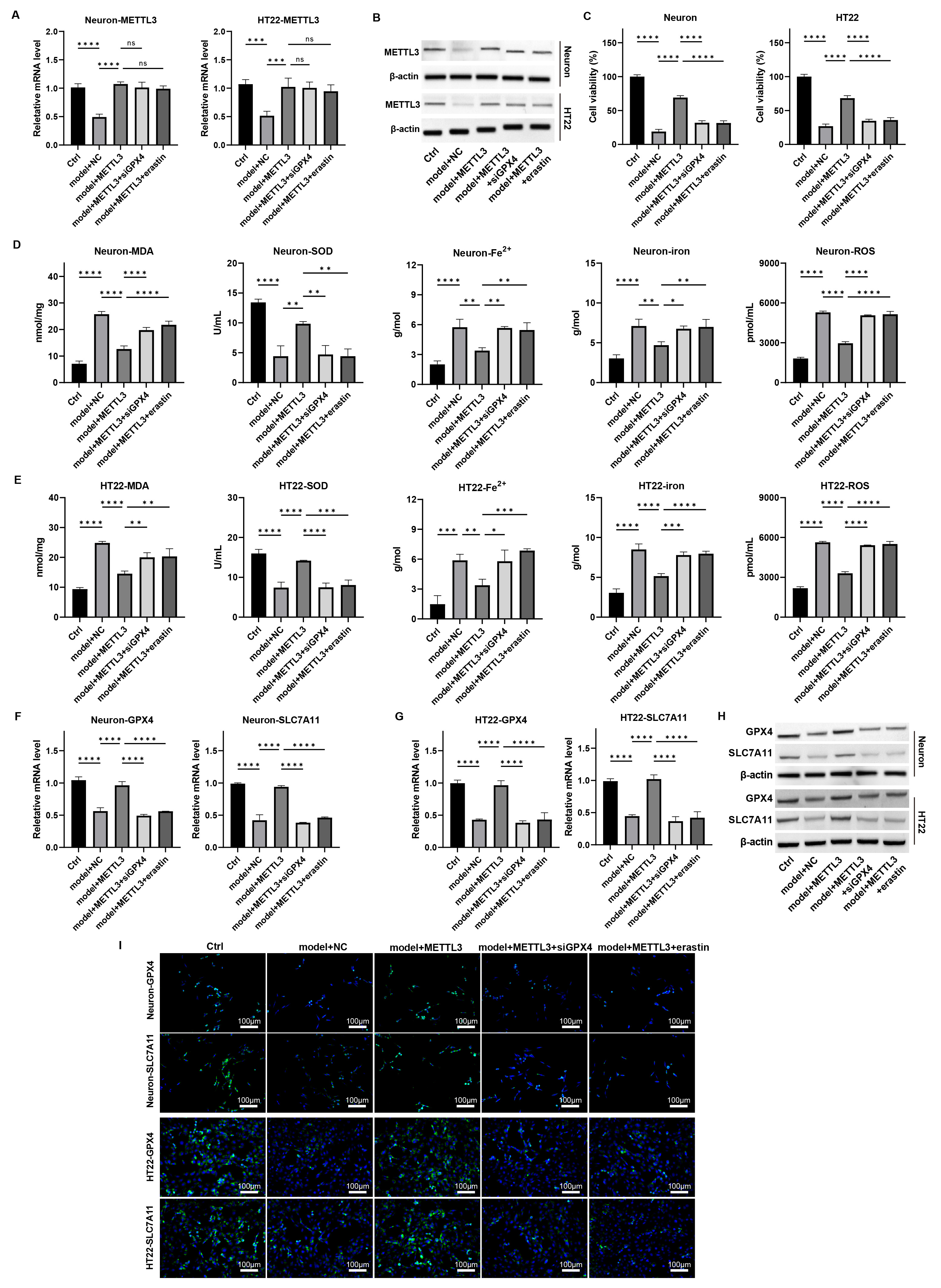 Fig. 5.
Fig. 5.
METTL3 modulates ferroptosis in
neurons via regulating GPX4 in vitro. Primary neurons
and HT22 cells were induced with mechanical damage and treated with
METTL3 overexpression, siGPX4, and erastin. (A,B) The mRNA and
protein levels of METTL3 were measured by (A) qPCR and (B) western blot. (C) Cell
viability was detected by CCK-8 assay. (D,E) The levels of MDA, SOD, Fe2+,
total iron, and lipid ROS were measured. (F,G) The mRNA levels of GPX4
and SLC7A11 in cells were detected by qPCR. (H,I) The protein
expression of GPX4 and SLC7A11 in cells was determined by (H) western blot and
(I) immunofluorescence staining assay. Scale bar = 100 µm, ns: p
Additionally, both mRNA and protein levels of GPX4 and SLC7A11 were elevated with METTL3 overexpression, but this increase was
attenuated by GPX4 knockdown and erastin treatment
(Fig. 5F–I, Supplementary Figs. 14,15, p
We investigated the role of METTL3 in regulating GPX4 during
TBI using a mouse model with METTL3 overexpression and treatments with
siGPX4 or erastin. MWM test results showed that METTL3 enhanced
cognitive function in TBI mice (Fig. 6A), reduced NSS scores (Fig. 6B, p
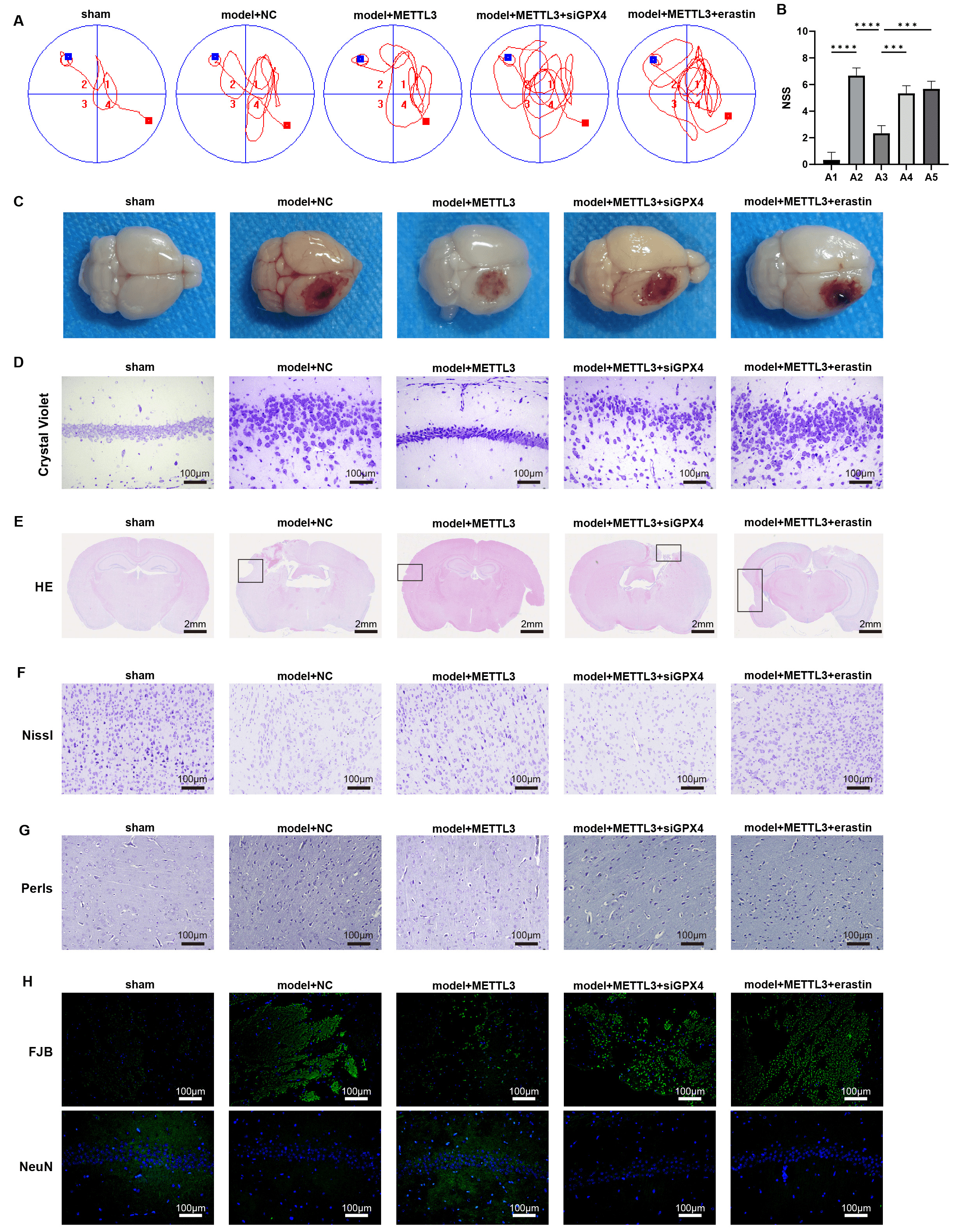 Fig. 6.
Fig. 6.
METTL3 alleviates brain damage during TBI via
regulating GPX4 in vivo. (A) The Morris Water Maze (MWM) test
was performed on day 7 post-TBI. (B) NSS score of brain damage. (C)
Representative images of the brain. (D) Crystal violet staining of brain tissues.
Scale bar = 100 µm. (E–H) Representative images of (E) HE, scale bar = 2
mm, (F) Nissl,scale bar = 100 µm, (G) Perls,scale bar = 100 µm, (H)
Fluoro-Jade B (FJB) and immunofluorescence staining of markers of neuron (NeuN)
in brain sections, scale bar = 100 µm; Cortical contusion areas are
represented by rectangles. ***p
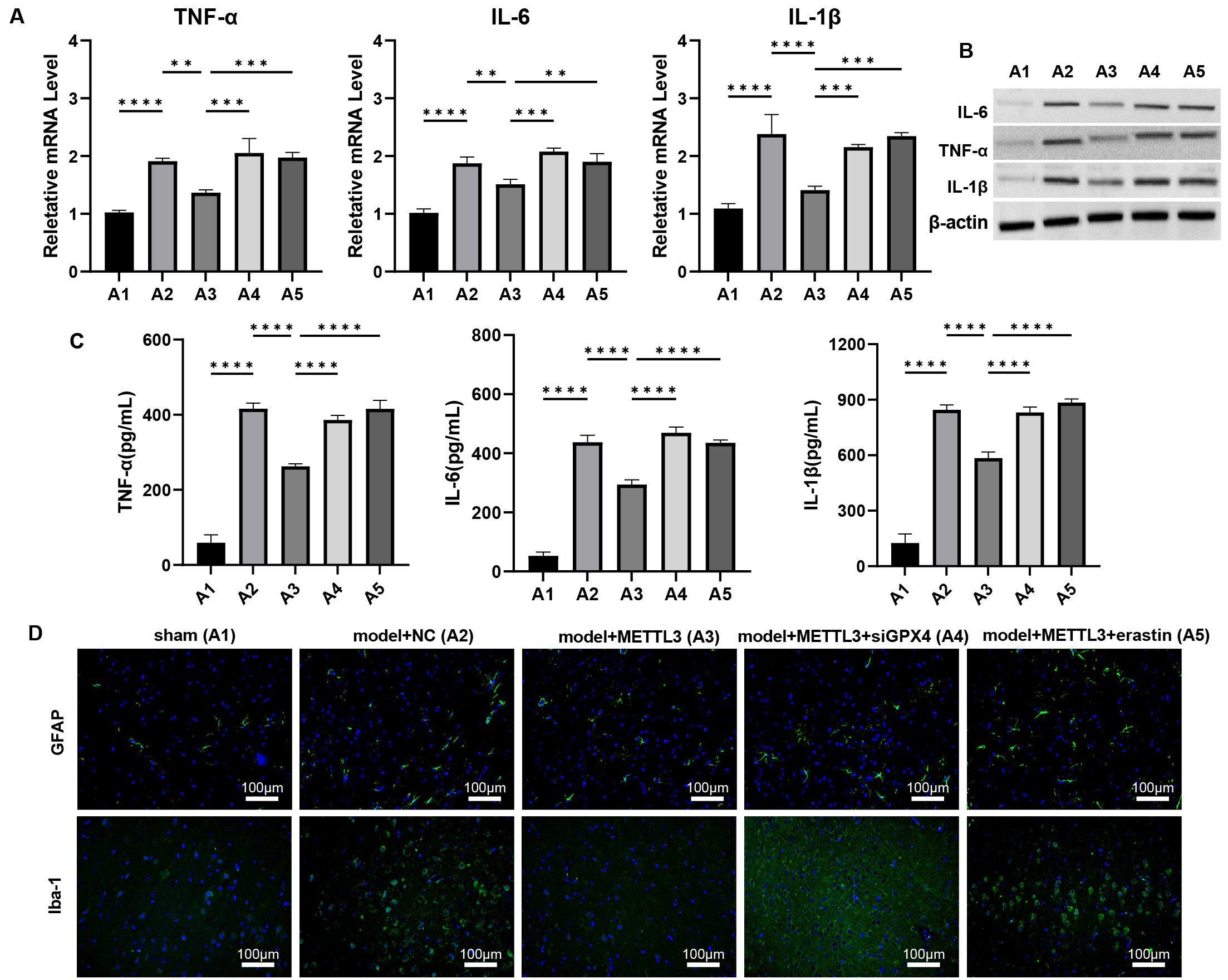 Fig. 7.
Fig. 7.
METTL3 alleviates brain inflammation response during TBI via
regulating GPX4 in vivo. (A–C) The mRNA and protein levels of
IL-6, TNF-
Assessments of ferroptosis biomarkers revealed that METTL3 mitigated
TBI-induced ferroptosis, as evidenced by decreased levels of MDA, Fe2+,
total iron, and lipid ROS, coupled with increased SOD levels (Fig. 8A, p
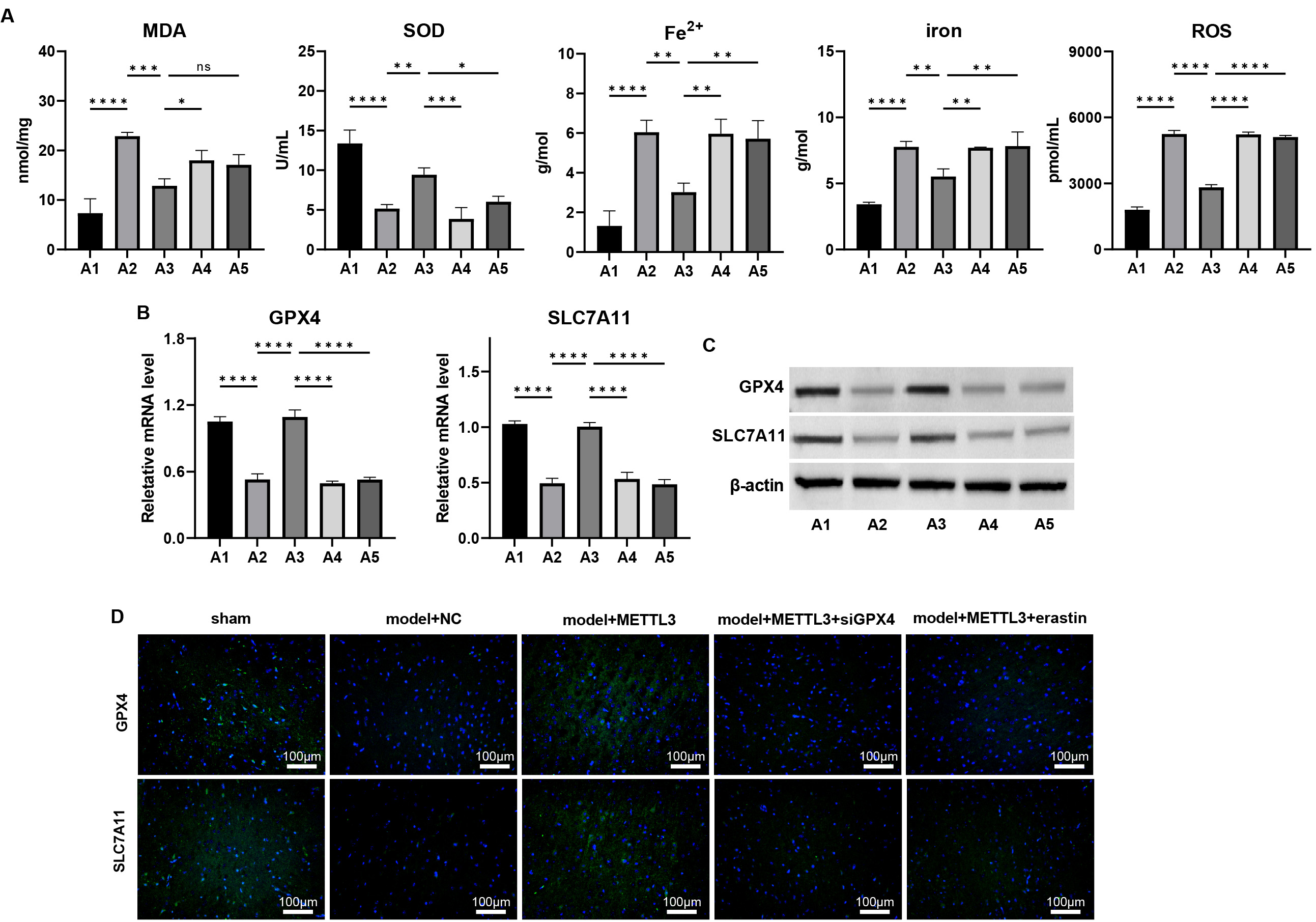 Fig. 8.
Fig. 8.
METTL3 alleviates ferroptosis during TBI via regulating
GPX4 in vivo. (A) The cortical levels of MDA, SOD, Fe2+, total
iron, and lipid ROS were measured. (B–D) The mRNA and protein levels of
GPX4 and SLC7A11 in cortex tissues of TBI mice were measured by
(B) qPCR, (C) western blot, and (D) immunofluorescence staining. Scale bar = 100
µm, ns: p
Conversely, siGPX4 silencing and erastin treatment negated these
protective effects. Additionally, siGPX4 and erastin treatment
downregulated the METTL3-induced upregulation of GPX4 and
SLC7A11 at mRNA and protein levels (Fig. 8B–D, Supplementary
Figs. 18,19, p
TBI is recognized as a significant risk factor for neurodegenerative conditions, including Alzheimer’s disease and chronic traumatic encephalopathy, and is associated with persistent cognitive deficits [29]. Our study revealed a decrease in METTL3 levels within the brain tissues of TBI-afflicted mice. The enhancement of METTL3 expression significantly mitigated cognitive impairments triggered by TBI, reduced brain lesion severity, diminished neuronal damage, and regulated iron accumulation in brain tissue. These observations suggest that METTL3 may play a neuroprotective role against TBI-induced injuries, fostering the repair and recovery of brain tissue as well as cognitive functions.
Neuroinflammation is a pivotal secondary injury mechanism following TBI, with
the activation of microglia and astrocytes playing a crucial role [30, 31]. As
the primary immune cells of the central nervous system, microglia are swiftly
activated upon TBI, modulating inflammatory responses through morphological
changes and cytokine release [32]. These activated microglia can manifest either
pro-inflammatory or anti-inflammatory phenotypes. Specifically, the M1 subtype,
characterized by classical activation, secretes pro-inflammatory cytokines like
TNF-
Lipid peroxidation, catalyzed by iron, is a hallmark of ferroptosis [34]. Our study suggests that METTL3 may offer neuroprotection by mitigating the buildup of lipid ROS and iron (Fe2+) within neurons in both cellular and animal models of TBI. Furthermore, METTL3 modulates the expression of critical genes GPX4 and SLC7A11, hinting at a mechanism of action that likely involves the inhibition of ferroptosis. Ferroptosis is characterized by iron accumulation, extensive lipid peroxidation, weakened antioxidant defenses, and increased ROS production, all of which are intimately linked to oxidative stress and the pathogenesis of neurological disorders and brain injuries [12, 13]. Consistent with previous research [12, 35], our study verified that TBI causes iron homeostasis disruptions and elevated lipid ROS levels, both indicative of ferroptosis. These findings support the notion that ferroptosis substantially contributes to TBI pathophysiology. METTL3 seems to counter these effects, suggesting its potential as a protective agent against the ferroptosis phenotype linked to TBI.
The N6-methyladenosine (m6A) modification is the prevalent mRNA modification in eukaryotes [36]. This mRNA modification is dynamic and reversible, regulated by proteins categorized as “writers”—including METTL3—“erasers” such as FTO and ALKBH5, and “readers” comprising YTH domain-containing proteins like YTHDF1-3 and YTHDC1-2 [37, 38]. These reader proteins identify m6A sites on RNA, influencing RNA processing aspects such as stability, degradation, splicing, and translation. METTL3, a key to the m6A methyltransferase complex, regulates gene expression via this modification. Notably, METTL3’s irregular expression has been linked to various cancers [39], implying a connection between m6A modification and carcinogenesis. However, the mechanisms underlying METTL3 dysregulation in TBI remain obscure. Our study reveals that YTHDF2 directly interacts with GPX4 mRNA, serving as a reader protein that enhances m6A modification and RNA stability on GPX4 mRNA. Modulating METTL3 levels or activity could potentially alleviate cognitive deficits, curb neuroinflammation, and shield neurons from ferroptosis-induced damage. Further research is essential to clarify the neuroprotective mechanisms of METTL3 and to assess the therapeutic potential of targeting the m6A pathway in TBI and other neurological disorders.
Our findings demonstrate that METTL3 exerts a protective influence against TBI-induced neurological deficits, lesion volume, and neurodegeneration by suppressing ferroptosis. METTL3’s anti-ferroptosis properties likely facilitate functional recovery post-TBI through the regulation of m6A modification and RNA stability of GPX4. This study advances our understanding of how METTL3-mediated epigenetic modifications relate to TBI-induced brain damage, indicating METTL3’s potential as a therapeutic target for TBI. Addressing the limitations and challenges identified will be crucial for the translation of METTL3-based therapies into clinical practice.
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
FQ, FL and XY designed the research study. FQ, WZ and FL performed the research. SZ and JS analyzed the data. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
All animal experiments were conducted in strict accordance with the ethical guidelines and regulations established by the Animal Ethics Committee of Medical Discovery Leader (MDL). The study protocol was reviewed and approved by the committee under approval number MDL2023-08-26-01.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL31304.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.