






 , Juan Sauca Pérez 2, Rodrigo S. Hernández 1, Rocío Sarasa 1, Moisés Garcés 1, M José Luesma 2, Juan J Badiola 1
, Juan Sauca Pérez 2, Rodrigo S. Hernández 1, Rocío Sarasa 1, Moisés Garcés 1, M José Luesma 2, Juan J Badiola 11 Centre for Encephalopathies and Transmissible Emerging Diseases, Institute for Health Research Aragón (IIS), WOAH Reference Laboratory for BSE and Scrapie, University of Zaragoza, 50013 Zaragoza, Spain
2 Department of Human Anatomy and Histology, Faculty of Medicine, University of Zaragoza, 50009 Zaragoza, Spain
Abstract
In recent years, neuroglia has become a therapeutic target for neurodegenerative diseases. Despite the recognition of a variety of microglial morphologies associated with the neuroinflammatory process that involve diverse functionalities for this glial type, it is still unknown its beneficial or harmful role to the surrounding tissue.
The study presented here proposes a novel approach to the neurodegenerative progression based on the reliability of its results due to the use of a natural model. Morphological alterations in microglia were assessed in cerebellar samples from prion-affected individuals at different stages of the natural disease (pre-clinical, clinical and terminal).
Immunohistochemical profiles confirmed that the abundance and morphology of the cells were found irrespective of the stage of the disease. Only an evident association of dystrophic pattern with advanced stages of the neurodegenerative process of scrapie was consistently demonstrated.
Overall, we conclude that the observations described here support a potential failure of microglial cells that could perhaps lead to their inability to perform some of their physiological functions, maybe due to a senescent state. Gaining insight into the multifaceted roles of neuroglia in central nervous system (CNS) diseases is of critical importance in knowledge and understanding of CNS disease pathogenesis, but also in generating novel therapeutic strategies.
Keywords
- microglia
- progression of neurodegeneration
- astroglia
- prion diseases
- cerebellum
Although there is no consensus on the key mechanisms underlying neurodegeneration, today, neuroinflammation is considered its main possible cause [1]. As a consequence, it is considered essential to know how the main components of the neuroprotection system presented by the host interact with the neuronal system in this deterioration process.
Neuroinflammation as a central mechanism in the pathogenesis of neurodegenerative diseases consists of a complex network of interrelated processes in which astrocytes and microglial cells play essential roles. It is thought that disruption in the crosstalk between microglia, astrocytes and neurons causes neurotoxicity and cognitive impairment [2]. Recent studies describe the presence of reactive states of these cell lines in neurodegenerative pathologies, relating these changes to the loss of their protective and repair function. The failure in the phagocytic work of damaged neurons and incorrectly folded proteins that microglia inherently have, together with the deterioration of the homeostatic function of astrocytes, would aggravate the initiated inflammatory process, exacerbating neurodegeneration [3].
The existence of heterogeneity of astrocytes [4, 5] and microglia [6, 7, 8] in response to these and other pathological states supports that different phenotypes are associated with diverse functionalities. However, the activation mechanisms of one or the other phenotype continue to be discussed, as well as the confirmation of whether they involve neurotoxic or neuroprotective properties [9]. This study aims to understand how these non-neuronal cells, specifically microglia, behave in the neurodegenerative progression.
Microglia are the resident immune cells of the central nervous system (CNS),
playing a critical role in maintaining brain homeostasis or in neurogenesis [10],
synaptic density and synaptic plasticity. This glial population is responsible
for phagocytosis and clearance of pathological protein aggregates, dead cells,
and other pathological particles [11]. Microglial cells are very sensitive to
changes in their microenvironment, presenting an extremely plastic and
chameleon-like phenotype [12]. In response to internal or external stimuli, these
glial cells are activated and release numerous inflammatory factors, thus leading
to neuroinflammation. In neurodegenerative conditions, they undergo metabolic
reprogramming in response to pathological stimuli, including plaques, tau tangles
and protein aggregates such as
On the other hand, it is well-known that prion diseases comprise a group of
neurodegenerative diseases caused by conformational alterations in the structure
of host-encoded glycoprotein protein (PrPc), forming the prion protein (PrPsc
[13]). Moreover, prion-like disorders share mechanisms of propagation with prion
diseases and consequently, histopathological and molecular aspects. Misfolded
endogenous proteins, such as
We assessed the morphological alterations of these glial cells in affected individuals at different stages of the natural progression of the disease in order to approach their functional relationship with the disease progression. Phenotypical changes have been widely demonstrated to be correlated with functional alterations in Biology and Neurobiology in particular. The final goal of this study is to contribute to the knowledge of the global neuroinflammatory process that occurs in the process of neurodegeneration. A better understanding of the evolution of these pathologies will make it possible to establish possible therapeutic strategies that are currently nonexistent.
All samples were provided by the Centre for Encephalopathies and Transmissible Emerging Diseases Tissue Bank (Zaragoza, Spain). The region selected for the study was the cerebellum, due to the fact that it is one area of the brain most affected in prion diseases (in both classical and atypical agents) and properly reflects the neuroinflammatory profile. The cerebella came from sheep affected by natural scrapie, which originated from different flocks from the region of Aragón (age: 1–9 years; sex: female). They were classified as preclinical, clinical or terminal cases. The classification was based on the presentation of the clinical signs associated with the diseases (a terminal classification was made when the animal was exhaustively debilitated and prostrated) and by using prion protein diagnosis (or by lymph reticular biopsy for preclinical cases). There were 6, 8 and 8 animals at the pre-clinical, clinical and terminal stages, respectively. Different PrP (prion protein) genotypes (ARQ/ARQ, ARQ/VRQ and VRQ/VRQ) were selected in order to assess the possible effect of this variable on microglial behavior.
All of the applicable international, national and/or institutional guidelines for the care and use of animals were followed. All of the procedures performed in studies involving animals were in accordance with the ethical standards of the Institution or practice at which the studies were conducted (Ethical Committee for Animal Welfare from the University of Zaragoza, reference number: PI 21/13).
A total of 6 control animals were also included in the study. They all came from flocks where cases of scrapie had never been reported.
The method of euthanasia was intravenous pentobarbital injection (Dolethal, 200 mg/mL; 40 mg/kg live weight), followed by exsanguination to ensure death. After removal of the whole brain, 3 cm thick sections of cerebellum were selected. These pieces were immersed in 4% paraformaldehyde for 48 hours for fixation. Five mm transversal sections, including granular, Purkinje cell and molecular layers, and white matter, were selected and immersed in formaldehyde for 24 additional hours to complete fixation.
Three µm slides were then sectioned and immunohistochemically processed.
A pan-microglial and specific activated microglial cells immunolabelling were both applied in the present study.
For the detection of microglia, heat treatment for 20 min was applied before the 5-minute endogenous peroxidase blocking (DAKO, Glostrup, Denmark). Afterward, sections were incubated with a polyclonal antibody against ionized calcium binding adaptor molecule 1 (IBA-1, at 1/1000 overnight, 4 °C; 019-19741, FUJIFILM WAKO, Richmond, VA, USA [15]).
For specific immunolabelling of the reactive microglial cells, a pre-treatment consisting of immersion in formic acid (15 min), digestion with proteinase K (15 min, 37 °C) and heating was necessary to be included prior to endogenous peroxidase blockade and incubation with three primary monoclonal mouse antibodies was performed for 72 h at 4 °C: MHC II (1/200; LS-C57747, LSBio, Newark, CA, USA), CD68 (1/500; M087601-2CN, DAKO, Denmark) and IB4 (1/500; I21411, Invitrogen, Eugene, OR, USA). The combination of these three antibodies was essential to mark active microglia cells in the ovine species [16, 17]. EnVisionTM polymer (rabbit or mouse, DAKO) and DAB PLUS (10 min) were used as a visualization system and chromogen, respectively. Hematoxylin counterstaining was finally performed before mounting.
Samples from cerebella originating from animals affected by meningoencephalitis were used as positive controls for this technique. Negative controls with no primary antibody were also included.
Marker immunostaining was scored from negative (–) to maximum (++++), by evaluating the density and the extent of the labeled deposits in each layer from each cerebellum. The immunostaining morphologies of each marker were also assessed for reactive microglia.
The protocol applied for the co-location studies was based on previously developed studies [18]. Briefly, 50 µm sections from fixed samples were collected in PBS (floating). Endogenous peroxidase blockade (H2O2 33%, 30 min) was performed before incubation with Triton X 100 for 3 h RT. Afterward, incubation with microglia-specific antibodies (MHC II, CD68 and IB4 - 1/200 and 1/500, respectively) and rabbit anti-cow GFAP (1/250; Z0334, DAKO, Denmark) for 72 h with agitation at 4 °C was performed. The secondary conjugated antibodies Alexa 488 (1/200; P11047, Invitrogen, Eugene, OR, USA) and Alexa 594 (1/200, A30678, Invitrogen, Eugene, OR, USA) were added in darkness 1 hour before prior assessment using a confocal microscope (Zeiss LSM 510, Zeiss, Germany).
The fluorescence emission was the result of excitation with 488 nm and 594 nm lasers and was detected with a two-channel multi-track configuration, using band-pass 505–530 nm and long-pass 615 nm filters, respectively. Z-stacks of digital images were captured using Zen 2008 software (Carl Zeiss Microimaging, Jena, Germany). They were collapsed into a single picture using the maximum intensity projection provided by the software described above.
All of the positive scrapie cases were diagnosed according to World Organisation for Animal Health (WOAH) standard protocols and demonstrated prion protein accumulation. And all of the negative controls were discarded to present this misfolded protein.
Concerning microglial detection, several morphological patterns were found regardless of the stage of disease (Fig. 1). Amoeboid and ramified morphology were mainly present in all cerebellar layers from all individuals studied, including healthy control samples. Dystrophic pattern was consistently associated with molecular and granular layers corresponding to those from terminal stages and also white matter from clinical cases.
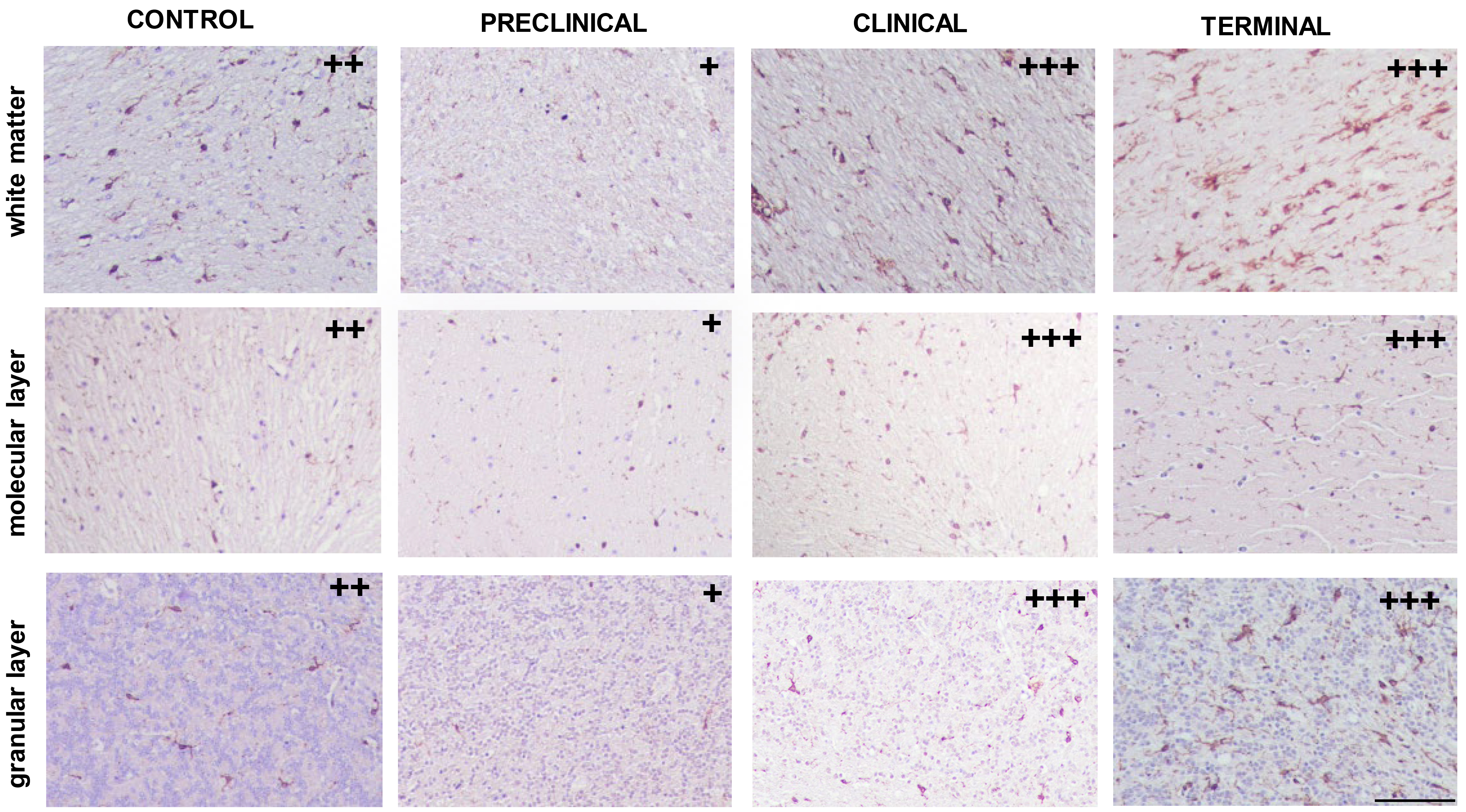 Fig. 1.
Fig. 1.
Immunolabelling patterns found in white and grey matters from cerebella corresponding to control and pre-clinical, clinical or terminal scrapie-affected animals after application of the immunohistochemical protocol for detection of microglia (ionized calcium binding adaptor molecule 1, IBA-1; scale bar: 100 µm). Immunointensity is graded according to the score established here (from negative, – to maximum, ++++), as shown on micrographs.
With respect to variations in intensity degree, it was only observed in preclinical cases, which evidenced a decreased immunostaining compared to the rest of the cases. Another noteworthy finding referring to quantitative changes consisted of some clinical individuals showing, in contrast to others at the same stage of the disease, scarcely scattered immunolabeling and nearly null in the granular layer (Fig. 2).
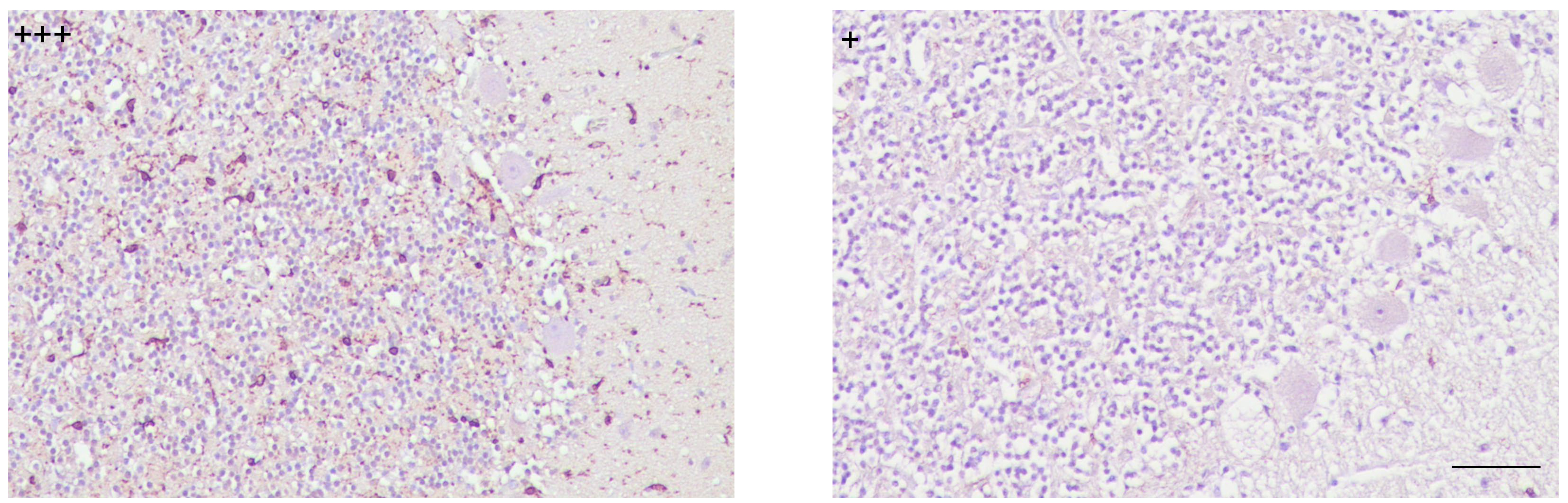 Fig. 2.
Fig. 2.
Patterns of immunostaining for microglial cells showed differing intensity degrees demonstrated in cerebella from scrapie clinically affected individuals (scale bar: 100 µm). Immunointensity is graded according to the score established here (from negative, – to maximum, ++++), as shown on micrographs.
Immunolabelling using markers for specifically detecting activated microglia was very scarce (+), but was present in all samples analyzed here. No specific layer showed a higher concentration of immunostaining in comparison to the rest. The reactive microglial morphology found in this study was almost exclusively ramified; amoeboid morphology was only very rarely observed. These immunohistochemical profiles, confirming the very low number and morphology of the cells, were found irrespective of the stage of the disease and PrPsc deposition. They also did not differ from those observed in control cerebella (Fig. 3).
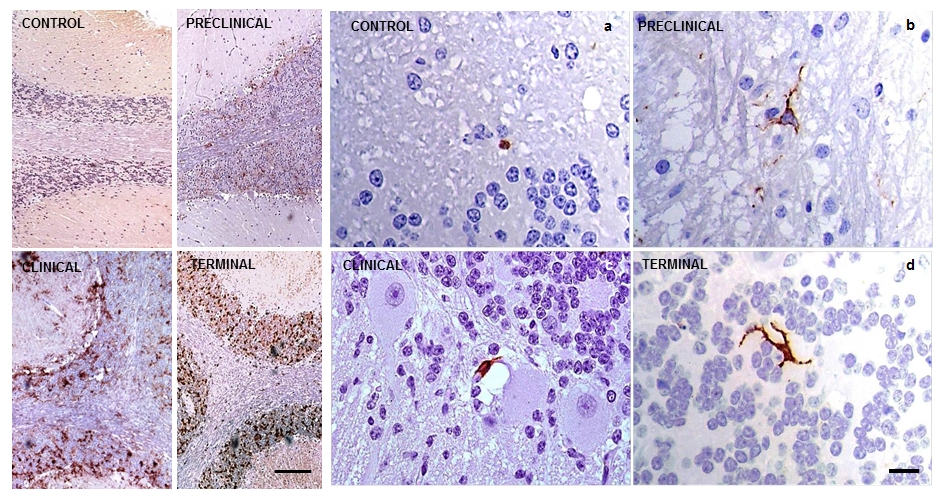 Fig. 3.
Fig. 3.
Immunolabelling patterns after application of the immunohistochemical protocol for detection of reactive microglia (MHC II+CD68+IB4; brown) in sheep cerebella. They resulted in extremely discrete (+, minimum) in all cases, as shown in CONTROL (a) and scrapie-affected animals at PRE-CLINICAL (b), CLINICAL (c) and TERMINAL (d) stages of the disease (scale bar: 20 µm). PrPsc accumulation representative profiles of each stage of the disease are illustrated with comparative aims on the left (scale bar: 100 µm). Immunointensity graded according to the score established here (from negative, – to maximum, ++++). PrPsc, prion protein; MHC II, major histocompatibility complex II; IB4, ionized calcium binding adaptor molecule 1.
However, an increased intensity (+++) of immunostaining was found in four individuals: two clinical and two terminal sheep. We determined that the common variable causing this quantitative difference was that all four of these sheep shared genotypes that were different from those found in the rest of the animals (ARQ/VRQ or VRQ/VRQ vs. ARQ/ARQ; Fig. 4).
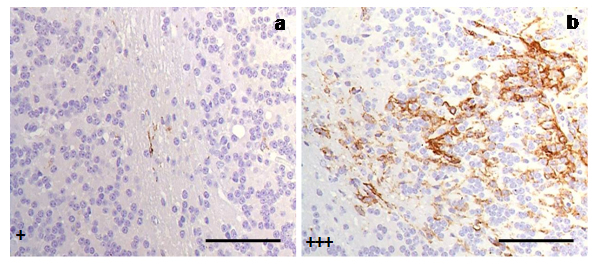 Fig. 4.
Fig. 4.
Immunostaining of reactive microglial cells in cerebella from scrapie-affected sheep demonstrated quantitative differences of reactive microglia (MHC II+CD68+IB4) in relation to PrP genotypes presented in (a) ARQ/VRQ, (b) VRQ/VRQ. Both cerebella corresponded to animals at terminal stages of the disease (scale bar: 100 µm). Immunointensity is graded according to the score established here (from negative, – to maximum, ++++), as shown on micrographs.
Confocal studies were carried out in order to illustrate a possible relationship between microglia and astrocytes. Although it was not a constant finding in all of the samples studied, colocalization between both glial cells was demonstrated on several occasions (Fig. 5). This interaction was not dependent on the clinical stage of the disease.
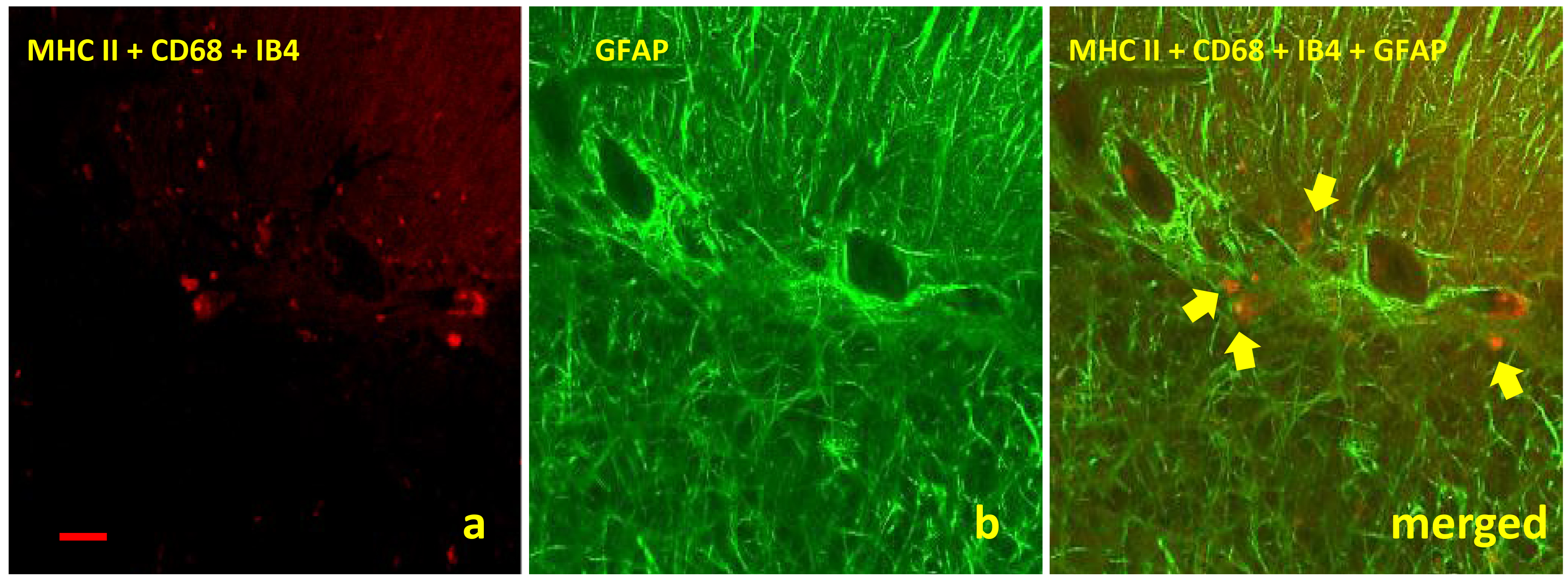 Fig. 5.
Fig. 5.
Images obtained by confocal microscopy for detection of both
glial cells assessed in this study showed an interaction between microglia (a) and
astroglia (b) regardless clinical stage of disease. The micrograph shows this glial
interaction in cerebella from clinical scrapie sheep by using specific markers
MHC II+CD68+IB4 (red immunolabelling, arrows) and GFAP (green immunolabelling),
respectively (scale bar: 100 µm; objective: 40
This is the first study where microglial alterations were assessed based on the clinical stage of the disease in a natural model of prion disorders using samples from naturally occurring scrapie-affected sheep.
The microglial response to these diseases has increasingly received attention in recent years, but in vivo and natural models have been less widely used to investigate this. The progressive changes have never been assessed in accordance with the natural evolution of the disease. Investigations so far have essentially been developed in experimental models based on organotypic slices, transgenic animals [19, 20] or in the end-stage of the human diseases [21], and support the growing recognition of the relevance of microglia in the development of prion disease. To confirm or otherwise that similar trends occur in the brain samples of naturally occurring scrapie-affected sheep would add to this body of knowledge, which is the rationale for the objective proposed in this study. The main aim of our study was to provide reliable results that reflect the actual behavior of this cellular type in natural prion disease, to finally establish this neuroprotector component as a turning point in the therapeutic strategy of these neurodegenerative diseases.
Microglia, being the major immune cell in the brain, are characterized as a very multi-functional cell type with a spectrum of heterogeneous phenotypes and functions being observed spatially and temporally in various disease states. The versatility of their phenotypes beyond the classical M1/M2 classification, as well as their double-edged actions in neurodegeneration, indicates the need for further exploration of these cell dynamics and their contribution to neurodegenerative processes. This glial population is involved in the neurological repair processes, such as neurovascular unit restoration and synaptic plasticity, and also manages the extent of the brain damage due to their phenotype switching. They are the main functional cells that mediate neuroinflammation in the CNS. When activated, they release inflammatory factors to promote neuroinflammation. In fact, chronic inflammation mediated by them has been found to cause damage to healthy neural tissue in neurodegenerative diseases [22]. It has also been postulated that microglia iron accumulation and inflammation interact to jointly drive the development of neuroinflammation, aggravating CNS damage [23].
However, the findings presented here support a potential failure of microglial cells instead of a role in enhancing pathology. Defects in microglia could perhaps lead to their inability to perform some of their physiological functions, such as debris clearance, or enhance other functions, such as the secretion of neurotoxic factors. A lack of normal microglial function could possibly contribute to the establishment of the disease [24]. It has been demonstrated that microglial activation occurs early in the course of prion disease [25], as a response to abnormal PrP deposition rather than to neuronal loss [26]. They have been hypothesized to play a major role in initiating the pathological changes seen in this group of diseases [27]. Nevertheless, all the findings described in the present study are as follows: no correlation between morphological alterations and the progress of neurodegeneration; dystrophic pattern in advanced stages of the disease; and a decrease in preclinical or even null microglial presence and clinical stages, which would contribute to this support.
Some of our previous works had suggested a possible astroglial paralysis in the clinical stage of scrapie [15, 28], proposing a possible impairment of communication between microglia and astroglia at this stage of the disease. This clearly emphasizes the relevance of the complex network of neuromodulator peptides in these diseases.
It is known that pro-inflammatory cytokines released by microglia trigger and modulate astrogliosis [29]. Microglia and astrocytes are known not to act independently of each other; there is an important molecular crosstalk between them. The different types of molecular signals secreted by microglia determine the neuroprotective or neurotoxic phenotypes of astrocytes and vice versa [30]. In fact, microglia activated by prion proteins have been shown to release cytokines that stimulate astroglial proliferation. And astrogliosis has been indicated to take on special significance in the evolution of the disease according to our previously published study [31]. As a consequence, the most consistent hypothesis to justify all of these findings could be that the exacerbation of toxic effects in the brain is related to both microglial and astroglial cells. Results provided by confocal microscopy studies would support this close interaction.
Our observation of an increase in reactive microglia, matching up with individuals presenting more susceptible genotypes, reinforces the relevance of glia in the progression of the disease. The VRQ haplotype is generally classified as being at high risk for scrapie infection [32], and samples coming from scrapie-affected animals with this haplotype showed a higher microglial response. This observation might reflect the essential role of microglial cells in the neurodegenerative process, even if it is not a direct effect. Considering that these VRQ individuals have been confirmed to have a higher susceptibility to developing prion disease, it is reasonable to think that microglial cells may enhance the propagation of the disease in these cases.
Neurons, glial cells and blood vessels are organized in neurovascular units,
which together constitute the blood-brain barrier. The function of these units
has been demonstrated to be altered in some diseases, such as Alzheimer’s
disease.
The most relevant contribution of this study is the reliability of its results. It has become a common practice in Neurobiology, and research in general, to extrapolate natural disorders from experimental models. However, the existence of great differences has been repeatedly confirmed in recent years [36, 37]. Meanwhile, the use of a natural model, as in this study, has consistently provided reliable results reflecting the real process of the disease. Furthermore, the possibility of assessing different stages of the disease is usually unavailable, conferring an added value to the results presented here. Moreover, taking into account that all neurodegenerative prion and prion-like disorders, on the basis of sharing aberrant protein accumulation as a molecular basis, share molecular pathogenesis pathways [14]. One of the main conclusions drawn here was the validity of the prototype of natural prion disease as a useful study tool to approach the pathogenic process of neurodegeneration in all the remaining proteinopathies.
The results provided in this work contribute to the elaboration of the global neuroinflammatory pattern that occurs in the progression of the neurodegenerative pathology characteristic of scrapie as a prototype of prion disease. But in addition, this model could be extrapolated to the neurodegenerative process that occurs in the rest of prion and prion-like diseases, since, as previously stated, all of them present the deposit of aberrant protein as a common pathological stimulus. In relation to Alzheimer’s disease (AD) as a neurodegenerative disease with a higher prevalence in the population, the key role of astroglia has been previously demonstrated [38, 39, 40]. Those results support the hypothesis raised in our previous works on prion disease, which have already been discussed about a possible failure of astroglial neuroprotective functions against the stimulus that the accumulation of aberrant proteins supposes [15, 18, 28, 31]. It would be of great interest to confirm whether the proteins associated with myelin also play a role in these pathologies, as other authors have postulated [2, 41].
And regarding microglial cells, the toll-like receptor (TLR) family had already been found to be involved in its function regulation in the pathogenesis and progression of AD or Parkinson’s disease (PD), making it a double-edged sword in these diseases. Altered function of peripheral innate immune cells was found in both neurodegenerative disorders and thus contributed to their development and progression. In fact, alteration of different subsets of T cells was found in the peripheral blood and CNS from AD and PD patients. The CNS-infiltrating T cells can exert both neuroprotective and neurotoxic effects in their pathogenesis and progression. Therefore, evidence for the role of these peculiar immune cells as microglia has also been described in the pathogenesis and progression of both AD and PD. A role of senescent microglia in brain aging-related neurodegenerative diseases has recently emerged [42]. It is hypothesized that the senescence of microglia, which is notably vulnerable to aging, could potentially serve as a central catalyst in the progression of neurodegenerative diseases. That is why, further confirming as here, a normally functioning interaction between the astroglial and microglial populations points out anti-inflammatory therapies as having a certain therapeutic potential, especially in early stages of the disease.
Additionally, under physiological conditions, neuroinflammation plays a
neuroprotective role against damaging factors. However, chemokine overexpression
breaks the integrity of the blood–brain barrier (BBB), facilitating the
infiltration of inflammatory cells into the brain. The prolonged inflammation
loses its protective role, which, together with the increase in the aggregation
of aberrant proteins, contributes to neuronal deterioration. Thus, the modulation
of these chemokines could also represent therapeutic strategies against
neurodegeneration [43]. Based on all these assumptions, interventions that
address inflammation mediated by glial cell interaction or these cells themselves
may be a suitable strategy against neurodegenerative diseases [22]. Wood has recently
pointed to microglial senescence as a potential target in AD [44]. Functional
assays to confirm this hypothesis regarding this and all the neurodegenerative
diseases are essential. Nevertheless, results provided by Fancy et al.
[45] identifying prematurely senescent microglia in the vicinity of
amyloid-
One of the main conclusions drawn here was that the findings described support a potential failure of microglial cells instead of a role for enhancing neurodegenerative pathology. As some of our previous works had suggested a possible astroglial paralysis in the clinical stage of scrapie, all findings together would point to a possible impairment of microglia or communication between microglia and astroglia at this stage of the disease, which would finally justify the exacerbation of toxic effects found in the encephalic region studied in prion-affected individuals. Based on all this, senescent microglia would emerge as a promising target for mitigating neurodegeneration. Conceivably, anti-inflammatory therapies addressing inflammatory processes mediated by glial cells themselves or through the communication between them modulated by chemokines may constitute a potential therapeutic tool, especially in early stages of the neurodegenerative process.
The prototype of natural prion disease is reconfirmed here as a useful study tool to approach the pathogenic process of neurodegeneration, not only in the rest of prionopathies, but also in all the proteinopathies.
Gaining insight into the multifaceted roles of neuroglia in CNS diseases is of critical importance in the knowledge and understanding of CNS disease pathogenesis, but also in generating novel therapeutic strategies.
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Conceptualization, MM; methodology, JSP and RSH; validation, RS and MG; writing-original draft preparation, MM; writing-review and editing, MJL and MM; visualization, JSP, RSH; supervision, MM and JJB; search references, MJL and JJB; funding acquisition, JJB. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The research protocol followed national and international animal 3R guidelines established and was approved by the Ethics Committee for Animal Welfare of the University of Zaragoza (PI 21/13).
We gratefully acknowledge the excellent technical support from Centre for Encephalopathies and Transmissible Emerging Diseases technical staff.
This research was funded by University of Zaragoza, grant number UZ 2014 - BIO4.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.