







 , Xingjiang Chen 3,*
, Xingjiang Chen 3,*1 School of Biological & Chemical Engineering, Zhejiang University of Science & Technology, 310023 Hangzhou, Zhejiang, China
2 Guizhou Provincial Tobacco Company Zunyi Company Yuqing County Branch, 564400 Zunyi, Guizhou, China
3 Guizhou Academy of Tobacco Science, 550001 Guiyang, Guizhou, China
Abstract
The Ralstonia solanacearum species complex (RSSC) is a group of destructive plant-pathogenic bacteria that targets a wide range of economically important crops across the globe, including tomato, pepper, and tobacco. Extensive research on this plant pathogen is essential due to the severe losses it inflicts on agricultural production.
We isolated strain MLY158 from diseased tobacco, identifying it as Ralstonia pseudosolanacearum. The strain was characterized genomically by biochemical profiling, genome sequencing, compositional and functional annotation, and comparative genomics.
MLY158 was capable of utilizing D-glucose (dGLU), sucrose (SAC), and D-trehalose dihydrate (dTRE). The genome had a total size of 5.88 MB and consisted of a circular chromosome and a circular megaplasmid. It contained 5485 coding genes and had a GC content of 67.50%. Comparative genomic results revealed that MLY158 is closely related to R. pseudosolanacearum strain GMI1000 (average nucleotide identity (ANI) value of 99.03%). MLY158 has 527 special genes and 13 homologous genes of species-specific gene families. The primary differences between MLY158 and genomes from other strains are located in the phage protein region and show characteristics of high genomic uniqueness.
Complete genome sequence analysis of MLY158 has contributed important information regarding the genome of the bacterial wilt disease pathogen R. pseudosolanacearum. This work provides useful references for future research into molecular disease control strategies and disease-resistant breeding.
Keywords
- Ralstonia solanacearum
- genomics
- plant disease
- tobacco
The United Nations (UN) Food and Agriculture Organization and the Organization for Economic Cooperation and Development have reported that rapid growth of the agricultural sector is now facing a major challenge from crop diseases, which are impeding further development [1]. Ralstonia solanacearum species complex (RSSC) is a phytopathogenic bacterium and a member of the Proteobacteria [2]. RSSC ranks among the most devastating phytopathogenic bacteria globally [3], with a wide distribution in tropical, subtropical and temperate regions [4]. RSSC infects over 450 plant species across 54 families, including economically important crops like tomato, peanut and tobacco [5, 6]. RSSC infection of tobacco causes rapid wilting of the plant, resulting in a 10–30% loss in production worldwide [7].
RSSC is a soil-borne pathogen. Upon infecting a plant, it enters the vascular tissue and multiplies, ultimately leading to plant death [8]. RSSC can return to the soil through withered plants and survive under nutrient-poor conditions for several years in preparation for the next infestation [9]. The classification of RSSC is based on its geographic distribution, with four phylotypes identified to date: phylotype I (Asia), phylotype II (America), phylotype III (Africa), and phylotype IV (Indonesia) [10]. RSSC was also classified into five biochemical variants based on the utilization of carbohydrates (lactose, maltose, fibrous dextran, and mannitol, etc.) [11]. The RSSC classification system was revised in 2014, with division into three distinct species: R. solanacearum, R. pseudosolanacearum, and R. syzygii. These species are distinguished by their distinct phylotypes, with R. solanacearum predominantly comprising phylotype II, R. pseudosolanacearum primarily consisting of phylotypes I and III, and R. syzygii comprising phylotype IV [12, 13, 14]. This classification system is currently employed by the majority of researchers in the field.
Significant biochemical differences exist among different bacterial strains. The VITEK 2 Compact fully automated microbial identification system offers highly efficient and rapid biochemical identification and characterization of strains. This system enables rapid biochemical characterization by utilizing gram-negative identification (GN) cards designed specifically for Gram-negative bacteria, with the cards containing multiple, independent biochemical reactions [15]. It has been widely and successfully applied in comparative studies of clinical, foodborne, and environmental pathogens [16]. The continuous development of microbial genome sequencing technology provides an important means to identify bacterial pathogenicity at the genetic level. Salanoubat et al. [17] isolated R. pseudosolanacearum strain GMI1000 from tomato plants and performed whole genome sequencing, thereby laying the foundation to study its pathogenesis. Subsequently, an increasing number of strains have undergone complete genome sequencing. The RSSC genome is approximately 5.9 MB in size and is primarily composed of a circular chromosome and a circular megaplasmid [18]. The pathogenicity of R. pseudosolanacearum is closely related to its virulence factors (VFs). R. pseudosolanacearum causes plant death by colonizing the vascular tissues and inducing the synthesis of large quantities of extracellular polysaccharides that impede water transport [19]. In addition, VFs including effector proteins, flagella, and cell surface appendages allow R. pseudosolanacearum to attach to plant roots during the early stages of infection [20]. The Type III secretion system (T3SS) has been studied extensively [21]. R. pseudosolanacearum utilizes T3SS to inject effector proteins into plant cells, thereby suppressing the host’s defense mechanisms and in some cases triggering immune responses [22, 23].
Although a growing number of strains have been sequenced, additional genomic data are required for a comprehensive analysis of this species. Furthermore, substantial variation in virulence is observed among strains from different geographical origins and host plants. Albuquerque et al. [24] reported two R. pseudosolanacearum strains that exhibit differing compatibility with the tomato cultivar Hawaii 7996, indicating the importance of variations in effector genes and pathogenic interactions among strains. Ding et al. [12] recently compared four strains derived from sunflowers. Despite originating from the same host, these strains exhibited significant differences in pathogenicity and genetic composition [12].
In the present study, we sequenced the complete genome of R. pseudosolanacearum strain MLY158. Compared to the reference genome, MLY158 carries special phage-associated genes and plasmid fragments. These findings not only suggest potential genomic plasticity and differences in virulence, but also provide valuable clues for deepening our understanding of gene transfer and evolution at the genetic level in R. pseudosolanacearum. Moreover, the comprehensive genome sequencing and annotation of strain MLY158 enriches the genomic database of R. pseudosolanacearum and provides a crucial reference for further research into molecular disease control strategies and disease-resistant breeding.
R. pseudosolanacearum strain MLY158 was isolated from a diseased tobacco stem leachate from Dejiang County, Tongren, Guizhou Province. After incubation in nutrient agar (NA) medium at 30 °C for 48 h, single colonies were selected for further inoculation into conical flasks containing nutritious broth (NB) liquid medium. This medium was shaken at 180 rpm/min at 30 °C overnight, and the cells were then collected by centrifugation at 4000 rpm/min for 5 min. The prepared strains were used for subsequent experiments. The OD600 was adjusted to 0.1 using sterile water, and the suspension injected into the leaf veins of tobacco seedlings using a sterile syringe. After inoculation, the seedlings were placed in a light incubator (Shanghai Boxun Industrial Co., Ltd. Medical Equipment Factory, Shanghai, China) at 30 °C with a 16-h light cycle and an 8-h dark cycle. The pathogen was then re-isolated and identified from the infected tobacco plants.
The prepared strains were suspended in sterile saline (0.45% to 0.50% NaCl solution, pH 4.5 to 7.0) and the McFarland turbidity was adjusted to 0.50–0.63. The bacterial suspension tubes and GN cards were placed in a card rack for online analysis of the biochemical reaction patterns. The 16S ribosomal RNA gene was amplified using the universal primers Eubac27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and Eubac1492R (5′-GGTTACCTTGTTACGACTT-3′). The resulting DNA sequences were measured and subjected to sequence comparisons on GenBank (https://www.ncbi.nlm.nih.gov/).
DNA from MLY158 was extracted using the MGIEasy Microbiome DNA Extraction Kit (1000027955, MGI Tech, Shenzhen, Guangdong, China) according to the manufacturer’s instructions. The genome for this strain was sequenced using Pacbio and DNBSEQ platforms.
Gene prediction of the assembly results was performed using Glimmer v3.02 (The Center for Computational Biology at Johns Hopkins University, Baltimore, MD, USA) [25]. Non-coding RNA prediction was performed using RNAmmer v1.2 (The Center for Biological Sequence Analysis at the Technical University of Denmark, Kongens Lyngby, Denmark) [26], tRNA prediction by tRNAscan v1.3.1 (Lowe Lab at the University of California, Santa Cruz, CA, USA) [27], and sRNA prediction by Rfam v9.1 (https://rfam.org) [28]. Tandem repeats were predicted by Tandem Repeats Finder (TRF) v4.04 (Benson Lab at Boston University, Boston, MA, USA) [29]. Clustered Regularly Interspaced Short Palindromic Repeats (CRISPRs) were identified using CRISPRCasFinder v4.2.20 (https://crisprcas.i2bc.paris-saclay.fr/).
Gene function annotation was performed based on Non-Redundant Protein Database (NR) v2023-4-20 (https://www.ncbi.nlm.nih.gov/refseq/about/nonredundantproteins/), Cluster of Orthologous Groups of proteins (COG) v202011-25 (https://www.ncbi.nlm.nih.gov/research/cog/) [30], Gene Ontology (GO) v2019-07-01 (https://www.geneontology.org/) [31], Kyoto Encyclopedia of Genes and Genomes (KEGG) v106.0 (https://www.genome.jp/kegg/) [32], and Swiss-Prot v2022_03 databases (https://www.expasy.org/resources/uniprotkb-swiss-prot) [33]. The type III effector protein of strain MLY158 was predicted by the T3SS v1.0 database [21]. Additional annotations were performed using IPR (https://www.ebi.ac.uk/interpro/), Antibiotic Resistance Genes Database (ARDB) v1.1 [34], virulence factor database (VFDB) v2023-4-28 (https://www.mgc.ac.cn/VFs/) [35], Carbohydrate-Active enZYmes Database (CAZy) v2022-9-15 (https://www.cazy.org/) [36], and The Comprehensive Antibiotic Resistance Database (CARD) v3.0.9 (https://card.mcmaster.ca/) [37]. Default parameters were used for all analyses.
Genomic similarity was analyzed based on the average nucleotide identity (ANI) using fastANI v1.32 software (The Center for Computational Biology at the University of California, San Diego, CA, USA) [38]. In addition, MUMmer v3.22 (The Center for Computational Biology at Johns Hopkins University, Baltimore, MD, USA) was used to align five R. solanacearum strains (RS-UW251, CFBP2957, RS-CIAT078, Po82, RS-Rs5), two R. pseudosolanacearum strains (GMI1000, CMR15), and three R. syzygii strains (PSI07, RS-T95, RS-SL2064). These were compared with the reference genome of MLY158 to determine the percentage of co-lined genes [39]. We also analyzed the core and pan genomes of these 11 strains, mainly using the software CD-HIT v4.6.6 (The Li Lab at the University of Texas Health Science Center at Houston, Houston, TX, USA) [40]. Phylogenetic trees were constructed using the PHYML (maximum likelihood method) algorithm for the single nucleotide polymorphism (SNP) matrix of the population of the 11 strains. Default parameters were used for all analyses. Additionally, comparative genomic analysis of the MLY158 genome sequence was performed against 10 highly similar R. pseudosolanacearum strains. Plasmid-specific regions and mutation hotspots were identified by comparing the comparative genomic circles. We also performed comparative analysis of the 10 similar genome sequences against the large plasmid in the MLY158 genome and constructed comparative genomic circles, primarily using BRIG v0.9.5 software (The Australian Centre for Ecogenomics at the University of Queensland, Brisbane, QLD, Australia).
Following inoculation with MLY158, tobacco seedlings exhibited wilting symptoms, with blackened stems and extensive leaf yellowing and necrosis (Supplementary Fig. 1a). The strain was re-isolated from the blackened stems (Supplementary Fig. 1b), and 16S rRNA sequencing demonstrated 100% similarity to strain MLY158. Based on Koch’s postulates, MLY158 was identified as R. pseudosolanacearum, the pathogen causing tobacco wilt.
Table 1 shows the biochemical characteristics of strain MLY158, as determined by GN card. Carbon sources for MLY158 include D-glucose (dGLU), sucrose (SAC) and D-trehalose (dTRE), but not D-cellobiose (dCEL), Fermentation of glucose (OFF), D-maltose monohydrate (dMAL), D-mannose (dMNE), Paleo sugar (PLE) and D-tagatose (dTAG). With regard to chemical sensitivity, MLY158 exhibited a positive reaction only for succinate alkalization (SUCT) among the tested enzymatic reactions. This indicates it has the ability to metabolize succinate and induce environmental alkalization. Additionally, the MLY158 strain demonstrated resistance to the antibiotic O129R.
| Well no. | Tests | Results | Well no. | Tests | Results | Well no. | Tests | Results |
| 2 | APPA | – | 21 | BXYL | – | 42 | SUCT | + |
| 3 | ADO | – | 22 | BAIap | – | 43 | NAGA | – |
| 4 | PyrA | – | 23 | ProA | + | 44 | AGAL | – |
| 5 | IARL | – | 26 | LIP | – | 45 | PHOS | – |
| 7 | dCEL | – | 27 | PLE | – | 46 | GlyA | – |
| 9 | BGAL | – | 29 | TyrA | – | 47 | ODC | – |
| 10 | H2S | – | 31 | URE | + | 48 | LDC | – |
| 11 | BNAG | – | 32 | dSOR | – | 53 | IHISa | – |
| 12 | AGLTp | – | 33 | SAC | + | 56 | CMT | – |
| 13 | dGLU | + | 34 | dTAG | – | 57 | BGUR | – |
| 14 | GGT | – | 35 | dTRE | + | 58 | O129R | – |
| 15 | OFF | – | 36 | CIT | – | 59 | GGAA | – |
| 17 | BGLU | – | 37 | MNT | – | 61 | IMLTa | – |
| 18 | dMAL | – | 39 | 5KG | – | 62 | ELLM | – |
| 19 | dMAN | – | 40 | ILATk | – | 64 | ILATa | – |
| 20 | dMNE | – | 41 | AGLU | – | |||
| GN Card | Incubation time: 6.00 h | |||||||
“+” indicates that the bacterium is capable of producing a certain reaction or
possessing a certain characteristic under certain conditions; “–” indicates
that the bacterium is unable to produce a reaction or does not possess a
characteristic under certain conditions. APPA, alanine-phenylalanine-proline
arylamidase; ADO, adonitol; PyrA, L-pyrrolidonyl arylamidase; IARL, L-arabitol;
dCEL, D-cellobiose; BGAL,
The genome of R. pseudosolanacearum strain MLY158 was sequenced using
both the DNBSEQ and PacBio platforms to achieve a high-quality assembly. The 1270
MB (216
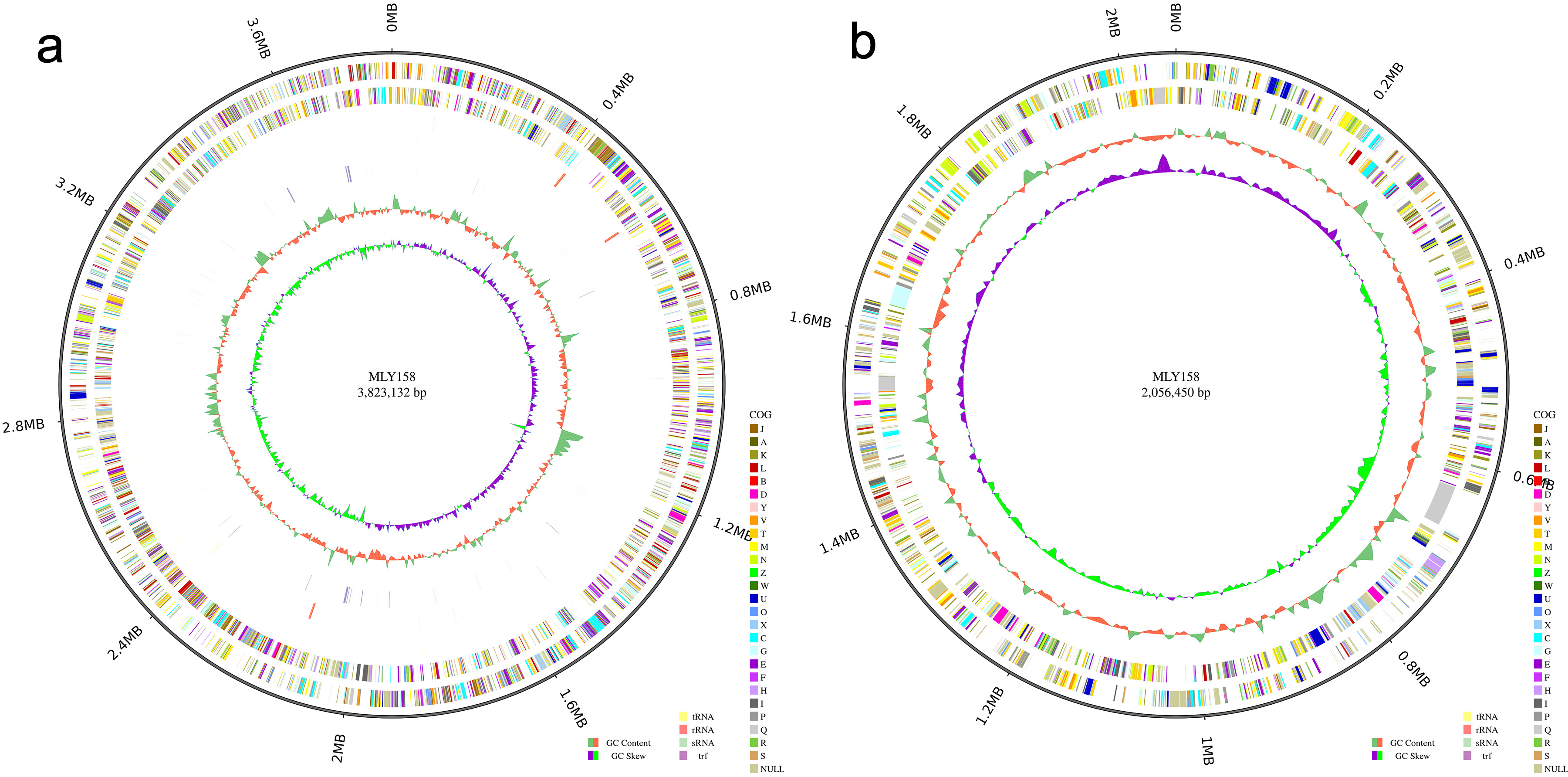 Fig. 1.
Fig. 1.
Circle diagram. (a) The chromosome circle diagram of MLY158. (b) The giant plasmid circle diagram of MLY158.
| Feature | Genome |
| Genome size | 5.88 MB |
| GC content (%) | 67.50 |
| tRNA | 60 |
| 5s_rRNA | 4 |
| 16s_rRNA | 4 |
| 23s_rRNA | 4 |
| sRNA | 22 |
| CRISPRs | 2 (questional) |
| TRF | 307 |
| Minisatellite DNA | 209 |
| Microsatellite DNA | 34 |
GC, G+C; CRISPRs, Clustered Regularly Interspaced Short Palindromic Repeats; TRF, Tandem Repeats Finder.
In total, 5404 genes were annotated. The gene annotation rates are shown in Supplementary Table 1. Gene function annotation successfully categorized 3940 genes into 24 COGs (Fig. 2a). Among the assigned genes, most were involved in transcription (n = 399), general function (n = 336), amino acid transport and metabolism (n = 402), signal transduction mechanisms (n = 295), and cell wall/membrane/envelope biogenesis (n = 293). In addition, 3060 and 3097 annotated genes were categorized by GO and KEGG, respectively (Fig. 2b,c). Among these were murI (glutamate racemase), tatC (component of the Tat secretion system), and dsbA (catalyzes disulfide bond formation in periplasmic proteins), which are related to xylem sap fitness factors. Comparison with the VF database revealed that MLY158 contained 478 homologous genes, including Cya, Hsp, and flagella. We also annotated VF-related genes, including those involved in stress tolerance (bcp, acrA, acrB, and dps), motility (pilA and filC), cell wall degradation (cbhA), and exopolysaccharide (EPS) biosynthesis (epsABCDEFP). The antibiotic resistance genes identified include varG (from Vibrio cholerae) and adeF. These were mainly involved in antibiotic inactivation and efflux.
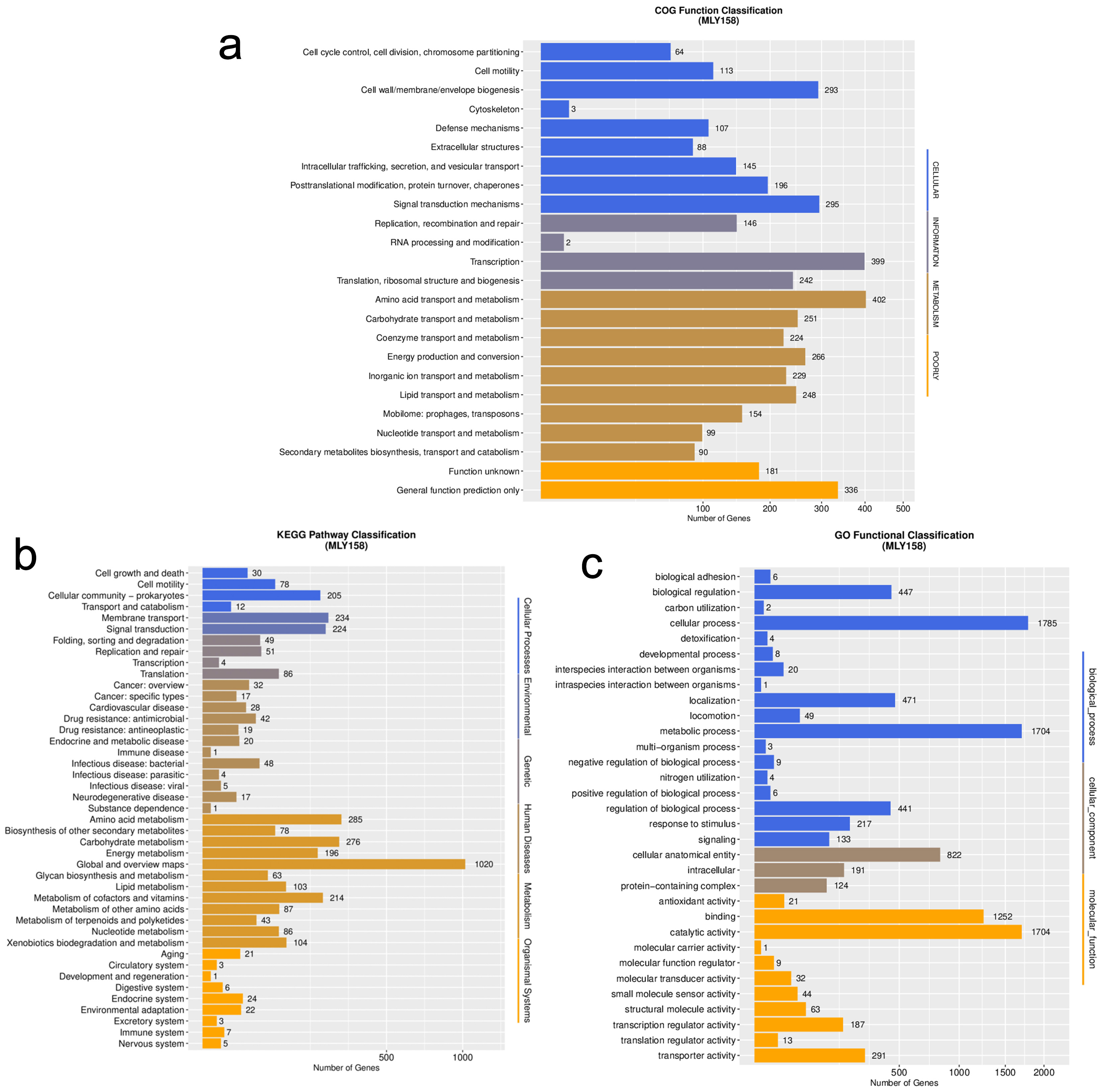 Fig. 2.
Fig. 2.
Functional classification map of genes in MLY158. (a) The function of COG annotation. (b) The function of KEGG annotation. (c) The function of GO annotation. COG, Cluster of Orthologous Groups; KEGG, Kyoto Encyclopedia of Genes and Genomes; GO, Gene Ontology.
Next, we determined the ANI (parameter: -c 1024) between the genome of strain
MLY158 and the genome of the reference strains (CFBP2957, CMR15, GMI1000, PSI07,
Po82, RS-CIAT078, RS-Rs5, RS-SL2064, RS-T95, RS-UW251). The results showed that
MLY158 was most closely related to strain GMI1000 (ANI 99.03%), followed by
strain CMR15 (ANI 96.13%) (Fig. 3). The ANI values between MLY158 and the
remaining reference strains ranged from 91.32% to 92.82%. The ANI values
between PSI07 and strains RS-T95 and RS-SL2064 were 98.32% and 98.30%,
respectively. The ANI values between RS-Rs5 and RS-UW251 (97.31%), RS-CIAT078
(96.07%), Po82 (96.09%), and CFBP2957 (97.30%) strains were all
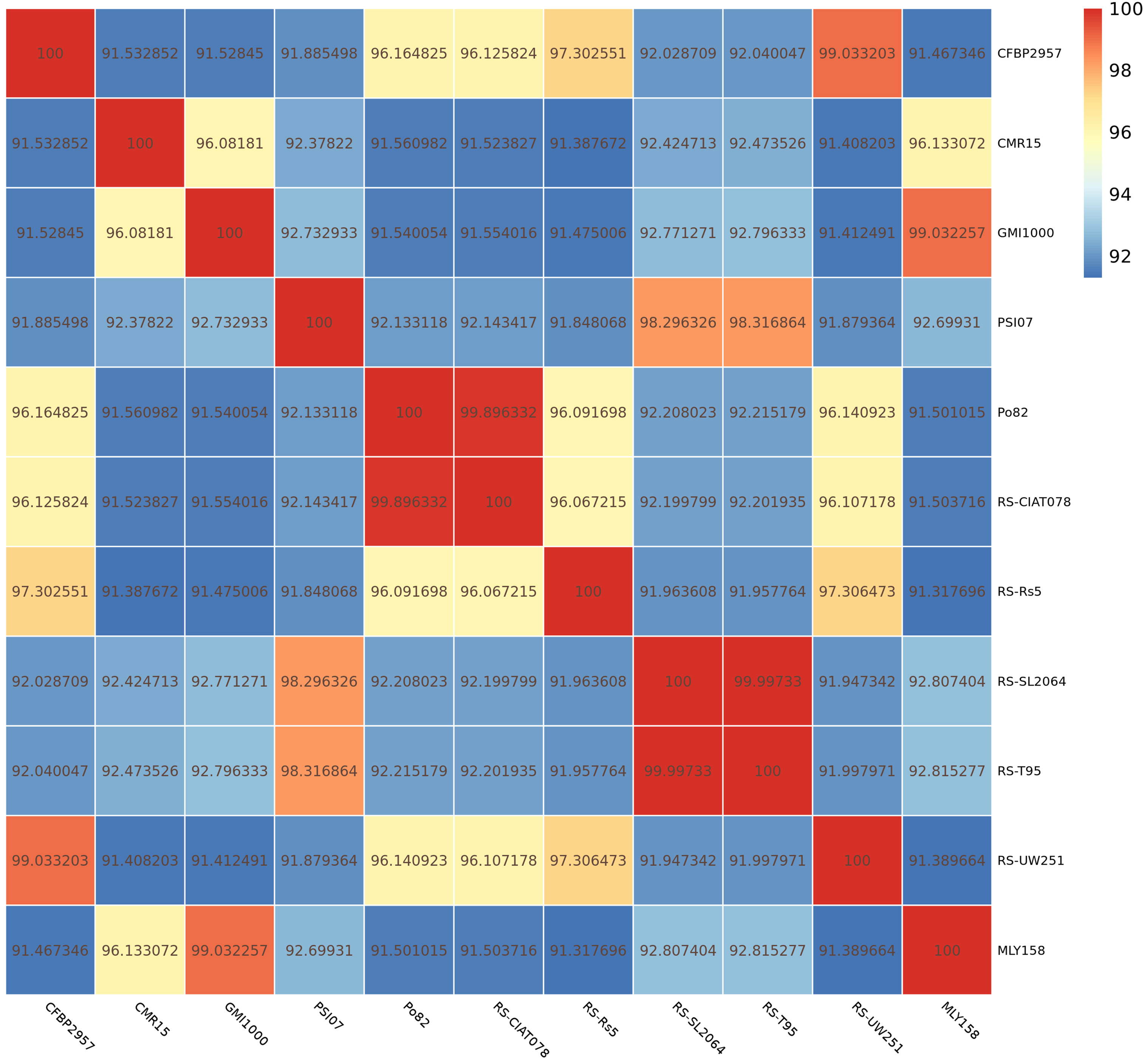 Fig. 3.
Fig. 3.
ANI heat map of MLY158 and reference strains.
Pan-genome analysis of strain MLY158 alongside the 10 reference strains identified 5505 homologous gene groups. Among these, 2998 core genes were shared across all 11 genomes (Fig. 4a). MLY158 contained 527 unique genes, the second-highest count among the different strains. MLY158-specific genes were annotated using COG (Supplementary Fig. 3), with 4 genes related to cellular, 17 genes related to information, 8 genes related to metabolism, and 8 genes related to poorly. According to gene family analysis (Table 3), the MLY158 and GMI1000 strains had the highest number of special gene families (n = 13), with MLY158 containing the most unclustered genes (n = 530). A heatmap of dispensable genes was also drawn based on their distribution in different samples (Fig. 4b). MLY158 and GMI1000 clustered together, forming a group with CMR15 that exhibited high similarity. RS-T95, RS-SL2064, and PSI07 clustered together. RS-UW251, CFBP2957, and RS-Rs5 also clustered together, whereas RS-CIAT078 and Po82 clustered separately.
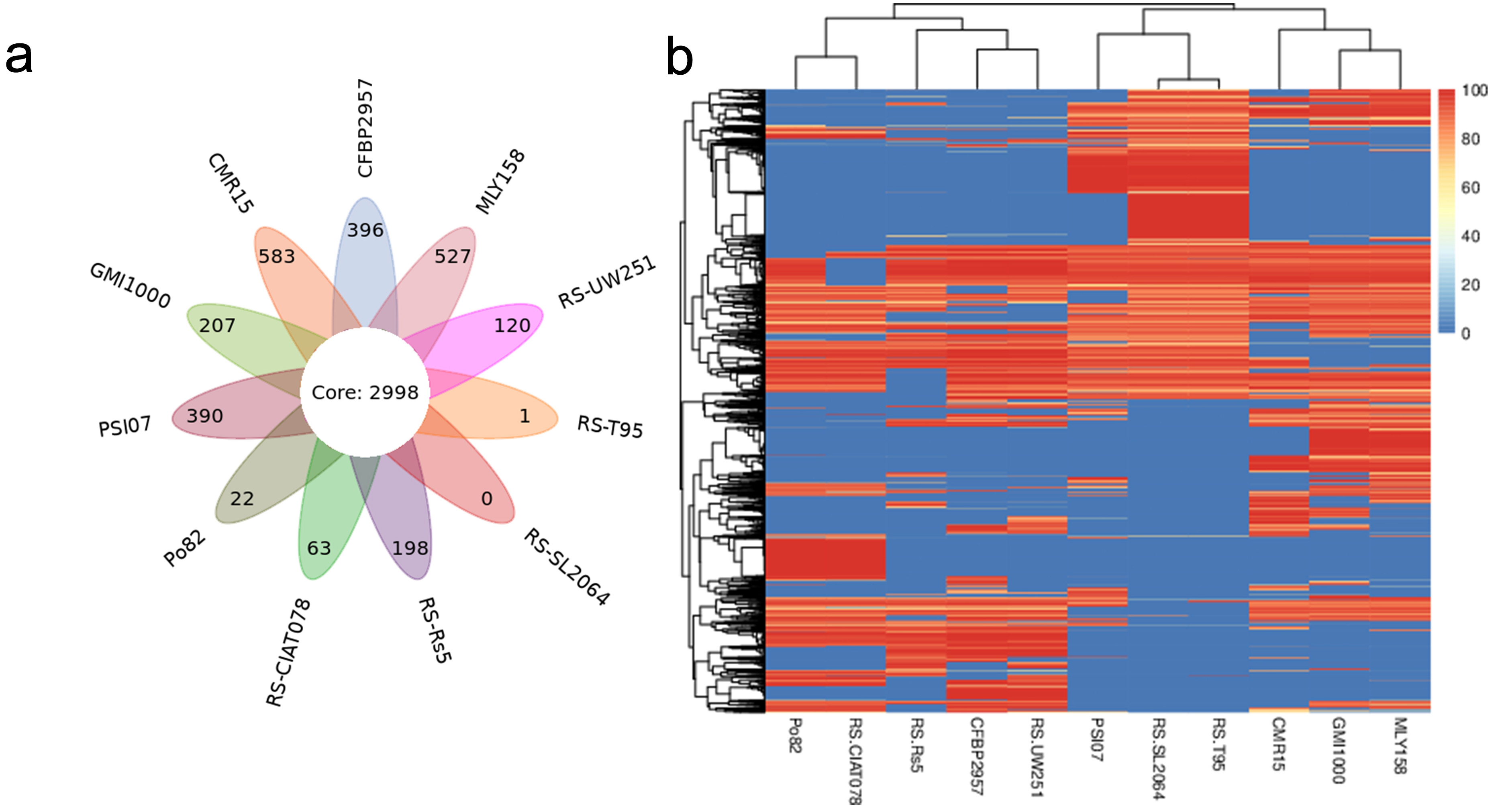 Fig. 4.
Fig. 4.
Comparison of genes between MLY158 and the reference strains. (a) Venn diagram of Pan gene set for MLY158 and the reference strains. (b) Heatmap of dispensable genes for MLY158 and the reference strains.
| Sample ID | Gene number | Clustered gene | UnClustered gene | Family num | Unique family |
| CFBP2957 | 5065 | 4669 | 393 | 3097 | 3 |
| CMR15 | 5123 | 4609 | 505 | 3056 | 9 |
| GMI1000 | 5109 | 4928 | 181 | 3137 | 13 |
| PSI07 | 4967 | 4611 | 356 | 2982 | 1 |
| Po82 | 4649 | 4622 | 27 | 3089 | 0 |
| RS-CIAT078 | 4548 | 4453 | 95 | 3031 | 2 |
| RS-Rs5 | 4560 | 4405 | 155 | 2977 | 9 |
| RS-SL2064 | 4719 | 4713 | 6 | 3174 | 0 |
| RS-T95 | 4729 | 4727 | 2 | 3176 | 0 |
| RS-UW251 | 4758 | 4660 | 98 | 3083 | 2 |
| MLY158 | 5485 | 4955 | 530 | 3128 | 13 |
A phylogenetic tree was constructed using the Neighbor-Joining method (bootstrap setting = 1000) based on the results of GeneFamily analysis. MLY158 was found to cluster with GMI1000, indicating a close relationship, and also formed a branch with CMR15 (Fig. 5). PSI07, RS-T95 and RS-SL2064 were closely related to each other and clustered together. RS-CIAT078 and Po82 were closely related, and RS-UW251 and CFBP2957 were also closely related, with each of these pairs forming a cluster. However, RS-Rs5 was more distant from the other strains and formed a unique branch.
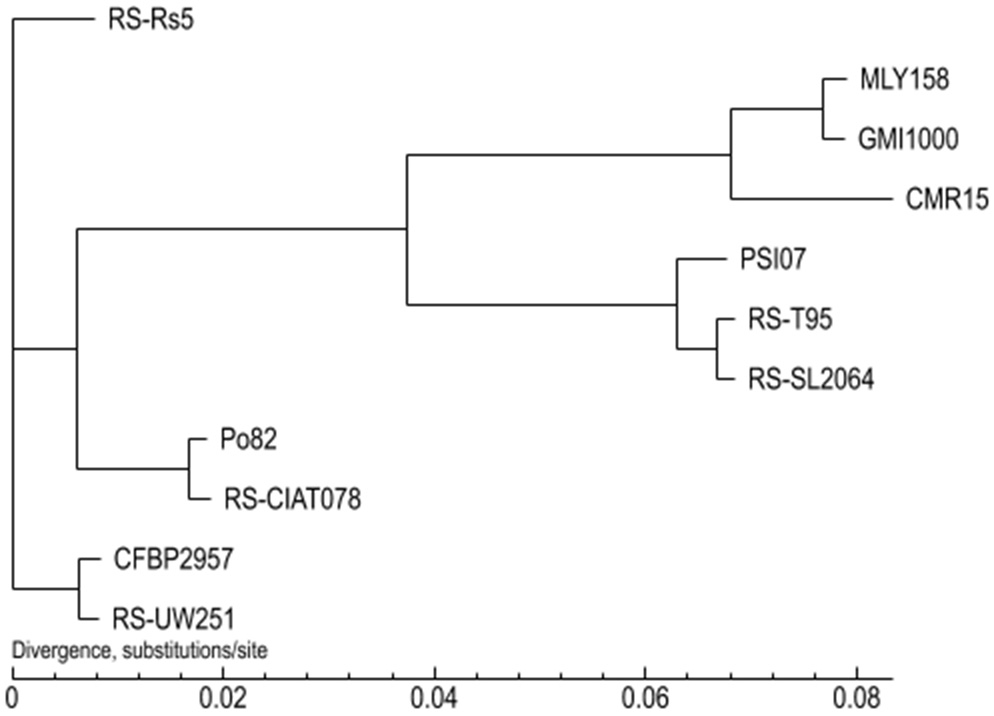 Fig. 5.
Fig. 5.
Phylogenetic tree between MLY158 and reference strains.
We subsequently performed BLAST alignment of MLY158 against the NCBI database
and selected 10 highly similar plasmid sequences (10 R.
pseudosolanacearum plasmid sequences). The comparative genomic circle is shown
in Fig. 6. The full-length MLY158 plasmid spans 2,056,450 bp, with a GC content
of 67.08%. Four antimicrobial resistance genes were identified: the efflux
pump-associated adeFGH gene adeF, the mdtABC gene
mdtB, and the OXA-type
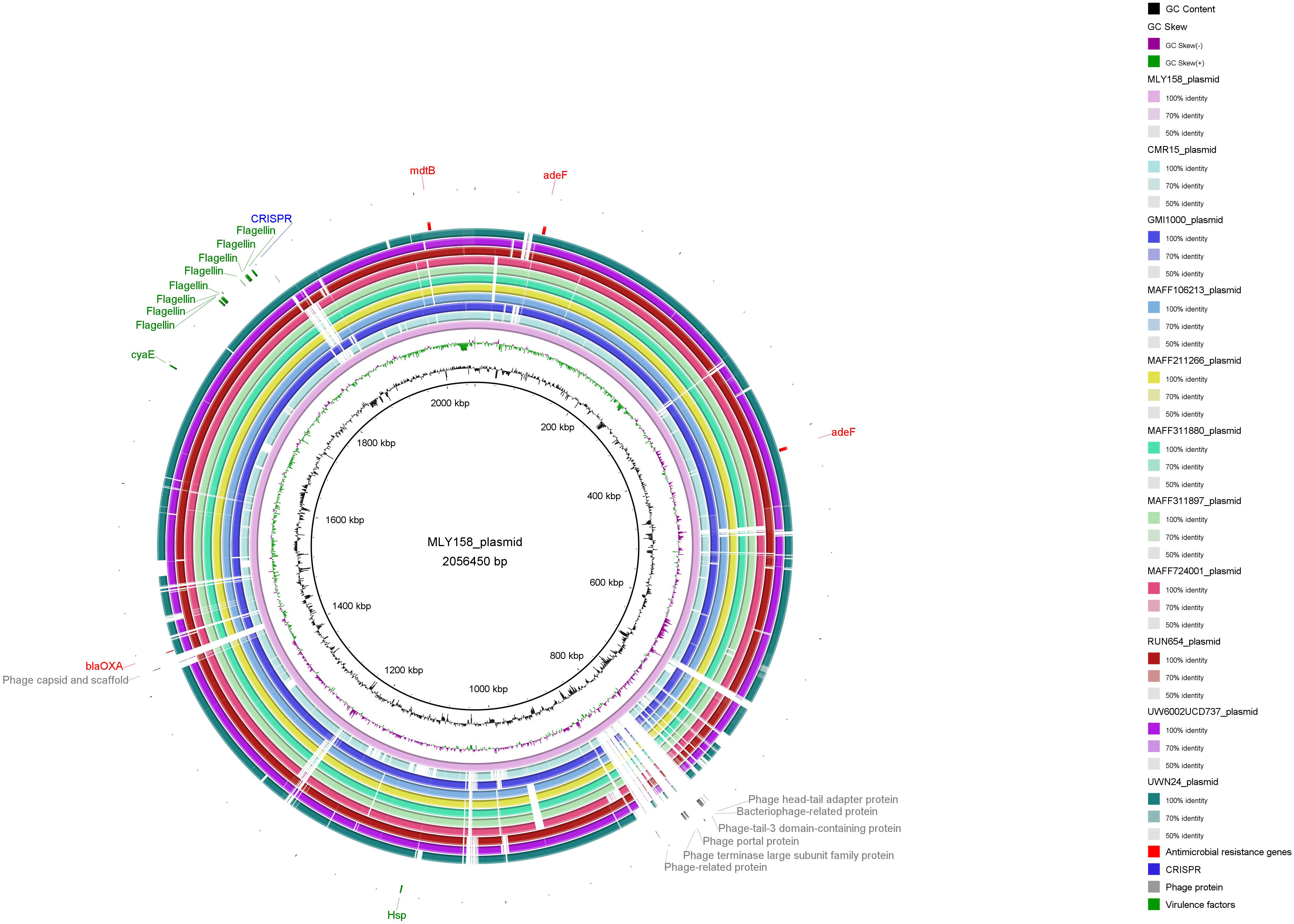 Fig. 6.
Fig. 6.
Comparison of genome circle analysis of MLY158.
The whole genome sequence of R. pseudosolanacearum GMI1000 isolated from tomato plants was reported in 2002. This was the first whole genome sequence of the species and laid a solid foundation for elucidating its pathogenic mechanisms and evolutionary history [17]. With the continuous improvements in microbial genome sequencing technology, an increasing number of strains have now been sequenced. The complete genome of R. pseudosolanacearum strain MLY158, isolated from diseased tobacco, was sequenced here using a combination of PacBio and DNBSEQ technologies. The results provide key insights into the genomic diversity of this pathogen.
One of the complexities of RSSC infestation of plants is its ability to utilize different carbon sources as nutrients. The strain adapts to the xylem of plants in relation to bacterial metabolism. KEGG annotation identified the gene scrA, which is related to sucrose degradation metabolism, but which was not found in the CFBP2957 strain. The biochemical results for MLY158 showed that it could utilize dGLU, SAC and dTRE as carbon sources. Sucrose serves as an important carbon source in the xylem sap of plants, providing nutrients for R. pseudosolanacearum to colonize the xylem after infection [41]. Additionally, MLY158 carries the envelope-associated genes murI (glutamate racemase), tatC (component of the Tat secretion system) and dsbA (catalyzes disulfide bond formation in periplasmic proteins), which significantly enhance its adaptability in the xylem. Biochemical characterization of Gram-negative bacteria with the GN card significantly reduces the incubation time required for traditional biochemical testing (24–48 h). Biochemical test reports can be generated within 6–24 h, enabling rapid analysis of antibiotic susceptibility for multidrug-resistant Gram-negative bacteria (P. aeruginosa, E. coli, and E. cloacae) in clinical settings. This is critical for patients with severe infections [16]. Studies have shown that the GMI1000 and PSI07 strains require the auxiliary biosynthetic genes thiC (thiamine biosynthesis gene) and purU (putative formyltetrahydrofolate deformylase) to allow full growth in xylem sap [42]. MLY158 also possesses these two genes. R. pseudosolanacearum may consume SAC and dTRE during the infestation of plants by R. pseudosolanacearum. Moreover, R. pseudosolanacearum infestation has been shown to reduce soil ammonium nitrogen and total plant nitrogen, thereby severely damaging crops [43].
Genome sequencing can provide an important reference to understand plant-pathogen interactions at the genome level. The RSSC genome is approximately 5.9 MB in size and is composed primarily of a circular chromosome and a circular megaplasmid, with occasionally also a small plasmid (FJ1003 contains a 0.2 Mb small plasmid) [44]. Genome sequencing results revealed a smaller genome size for MLY158 compared to other strains isolated from tobacco such as gd-2 (5.93 Mb), CQPS-1 (5.89 Mb) and FJ1003 (5.90 Mb) [11]. However, the predicted genome of MLY158 (5485 genes) was larger than that of gd-2 (5074), CQPS-1 (5229) and FJ1003 (5010) [45]. In addition, strains isolated from different hosts each possess different genomic characteristics [44, 46], highlighting the complex diversity of R. pseudosolanacearum [47]. Comparative genetics identified candidate VFs and antibiotic resistance genes, including stress tolerance (bcp, acrA, acrB, and dps), motility (pilA and filC), cell wall-degrading enzymes (cbhA), and EPS biosynthesis (epsABCDEFP)-related virulence genes. In addition, we identified varG (from Vibrio cholerae) and adeF resistance genes similar to gd-2 [11]. This study enriches the available gene pool, expands our understanding of virulence mechanisms, and informs future drug design [46]. Unlike GMI1000 and PSI07, MLY158 lacks the SpeC gene, which is essential for growth in rich media. KEGG annotation revealed that MLY158 carries the phcA gene, which positively regulates the production of bacterial EPS under conditions of high cell density [48]. The earliest reported genome of R. pseudosolanacearum was model strain GMI1000, which laid the foundation for understanding the effector gene pool and the regulation of pathogenicity mechanisms. However, GMI1000 originated from diseased tomato plants in Madagascar and shows genetic differences with strains that are prevalent in Asia. MLY158 originates from Guizhou Province in China. Genomic sequencing revealed this strain carries multiple unique plasmid fragments and phage-associated elements, suggesting potentially novel characteristics in genomic plasticity and horizontal gene transfer. This discovery also provides new perspectives for investigating host-pathogen interactions and the ecological adaptation of this pathogen.
Comparison of strain MLY158 with 10 reference strains in this study revealed
that MLY158 was more similar (ANI
This study presented the complete genome sequence of R. pseudosolanacearum strain MLY158 isolated from infected tobacco plants. Strain MLY158 is able to utilize specific carbon sources, making it easier to infect plants. Functional annotation and comparative genomes provided evidence for the genetic diversity of R. pseudosolanacearum. In conclusion, these findings provide a foundation for understanding the underlying mechanisms of bacterial wilt in tobacco and for future studies on the R. pseudosolanacearum-tobacco interaction.
The MLY158 genome sequence was deposited in the DDBJ/ENA/GenBank database (BioProject/BioSample: PRJNA1271622/SAMN48881384). The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Conceptualization: JZ, GL, XC; Methodology: JZ, JG, DL, YC, YL; Formal analysis and investigation: BK, KX; Writing—original draft preparation: JZ, GL; Writing—review and editing: YL, XC; Funding acquisition: YL, XC. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The tobacco variety used for the experiment was Yunyan 85. The tobacco plants were supplied by the Guizhou Provincial Tobacco Company Zunyi Company Yuqing County Branch. According to China regulations and policies, this study does not involve human or animal subjects; therefore, ethical committee approval is not necessary.
We would also like to thank all the colleagues in our group who have supported us.
This research was supported by the R&S project of China Tobacco Hunan Industrial Co., Ltd. (grant no. KY2025JD0001).
All authors declare no conflicts of interest. Despite they received sponsorship from Guizhou Provincial Tobacco Company Zunyi Company Yuqing County Branch and China Tobacco Hunan Industrial Co., Ltd, the judgments in data interpretation and writing were not influenced by this relationship.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL46230.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.