






 , Xianghui Fu 1, Yan Tian 1,*
, Xianghui Fu 1, Yan Tian 1,*
1 Department of Biotherapy, State Key Laboratory of Biotherapy, West China Hospital, Sichuan University, 610041 Chengdu, Sichuan, China
Abstract
Ankyrin G (ANK3), belonging to the ankyrin family, contributes to cellular structural integrity by linking the cytoskeleton to the plasma membrane. Abnormal ANK3 expression has been reported across several human malignancies, yet the regulatory mechanisms involved are still poorly understood. The process of dividing introns into several steps is referred to as recursive splicing (RS). RS can control the quality of transcripts produced by regulating the retention of the RS-exon. Hundreds of annotated RS-exons in human mRNAs are attributed to the inhibition of RS by the exon junction complex (EJC).
In this study, we demonstrated that ANK3 is reduced in hepatocellular carcinoma (HCC) and suppresses HCC metastasis. We then analyzed the multiple splicing methods of ANK3, confirming that RS exists in ANK3 transcript variant 4 (ANK3-TV4) and that RS was weakened in HCC.
Mechanistically, ANK3 inhibited HCC metastasis, which may be partly attributed to inhibition of the Wnt pathway. ANK3 binds to E-cadherin via its N-terminal ankyrin repeat domain to regulate E-cadherin expression. ANK3 knockdown activates the Wnt pathway, downregulates E-cadherin expression, and promotes its degradation. Conversely, ANK3-TV4 overexpression inhibited the Wnt signaling pathway, upregulated E-cadherin protein expression, and inhibited E-cadherin degradation. RBM8A, a core EJC factor, regulates the RS of ANK3-TV4.
Knockdown of RBM8A promoted RS of ANK3-TV4 and upregulated its expression. We investigate the role of RS in HCC, providing a novel therapeutic perspective and identifying potential targets for intervention.
Keywords
- ankyrin G
- recursive splicing
- hepatocellular carcinoma
Liver cancer ranks as the sixth most prevalent malignancy globally and is the third leading cause of cancer-related mortality [1]. Hepatocellular carcinoma (HCC) constitutes nearly 80% of primary liver cancer cases [2]. Patients with HCC generally face poor clinical outcomes and limited effective treatment options [3]. Therefore, elucidating the pathogenic mechanism of HCC and its key regulatory molecules is essential for developing novel therapeutic strategies aimed at its prevention and treatment. Several studies have highlighted alternative splicing (AS) as an important source of new prognostic biomarkers and therapies for HCC [4, 5, 6, 7, 8, 9, 10].
More than 90 percent of human genes generate different transcripts through AS, thereby forming diversified transmissions of genetic information [5, 11, 12, 13, 14]. Recursive splicing (RS) is a noncanonical splicing mechanism in which introns are cut in several steps. In general, an RS-exon exists in the introns of genes that produce RS. This exon contains a premature termination codon (PTC), which commonly activates the nonsense-mediated mRNA decay (NMD) and terminates translation. RS removes the RS-exon, which can prevent the production of abnormal N-terminal mutant or truncated proteins, thereby maintaining normal gene expression and cellular functions [15, 16]. Several studies have shown that the Exon Junction Complex (EJC) suppresses RS and is an important factor in abnormal RS [17, 18]. It is worth emphasizing that, as a rare AS pattern, RS has seldom been studied; moreover, its underlying mechanisms and implications in cancer remain poorly understood.
Cytoskeletal rearrangement is necessary for multiple pathological processes, such as the migration and invasion of tumor cells, and plays a key role in tumor metastasis [19]. Therefore, in-depth research on this topic is of great significance for identifying the intrinsic mechanisms and targets of tumor metastasis and developing new targeted therapies. Ankyrin 3 (ANK3), a member of the Ankyrin family, plays a crucial role in maintaining cell stability by linking the cytoskeleton to the plasma membrane [20, 21]. ANK3 typically comprises three major functional regions: an N-terminal membrane-interacting domain, a central domain that engages pectin/fodrin, and a regulatory C-terminal domain [20, 22]. Current studies have reported that there are multiple transcripts of ANK3 [16, 23, 24] and it is abnormally expressed across various tumor types [21]. Nonetheless, the association between ANK3 expression and AS in HCC patients has not yet been elucidated.
In our study, we first demonstrated that ANK3 was downregulated in HCC and inhibited HCC metastasis, confirming that RS exists in ANK3 transcript variant 4 (ANK3-TV4) and that RS is weakened in HCC. Mechanistically, ANK3 inhibited HCC metastasis, which may be partly attributed to inhibition of the Wnt pathway. ANK3 binds to E-cadherin via its N-terminal ankyrin repeat domain to regulate E-cadherin expression. ANK3 knockdown activates the Wnt pathway, downregulates E-cadherin expression, and promotes its degradation. Conversely, overexpression of ANK3-TV4 had opposite effects. RBM8A, a core EJC factor, regulates the RS of ANK3-TV4. RBM8A knockdown promotes the RS of ANK3-TV4. We explored the involvement of RS in HCC, offering a new strategy for HCC treatment and identifying promising targets.
SNU449, SK-Hep1, and Huh7 cells were purchased from the Type Culture Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Cells were cultured following the protocols provided by the supplier. All the cells in this study were purchased from the Type Culture Cell Bank of the Chinese Academy of Sciences (Shanghai, China). All cell lines were validated by STR profiling and tested negative for mycoplasma.
Small interfering RNAs (siRNAs) used in this study were commercially synthesized by GenePharma (Shanghai, China). Transfection was carried out using HiPerfect for siRNAs and Attractene for plasmids (Qiagen, Hilden, Germany), as per the supplier’s guidelines. Details of the siRNA sequences can be found in Supplementary Table 1.
Total RNA was isolated using Tri-Reagent (Molecular Research Center, Cincinnati, OH, USA) following the manufacturer’s standard protocol. We performed RT-PCR by first generating cDNA with the TransScript One-Step gDNA Removal and cDNA Synthesis SuperMix (TRAN, Beijing, China). The subsequent amplification step utilized the Golden Star T6 Super PCR Mix (TSINGKE, Beijing, China), as per the manufacturer’s instructions. Primer sequences are listed in Supplementary Table 2.
cDNA synthesis for real-time quantitative PCR (qRT-PCR) was carried out using
murine leukemia virus reverse transcriptase (Invitrogen, Life Technologies,
Carlsbad, CA, USA), followed by amplification with Power SYBR Green PCR Master
Mix (Applied Biosystems, Foster City, CA, USA). Gene expression levels were
normalized to
To assess cell proliferation, an 3-[4,5-dimethylthiazol-2-yl]-5-[3-carboxymethoxyphenyl]-2-[4-sulfophenyl]-2H-tetrazolium (MTS) assay was performed according to the established method [25]. Briefly, 2500 cells were inoculated in a 96-well plate, and cell proliferation rates were measured using the CellTiter 96 AQ Ueous Nonradioactive Cell Proliferation Assay (MTS) (Promega, Madison, WI, USA) according to the manufacturer’s instructions [26].
To knock down ANK3 or RBM8A, we utilized the pLKO.1 lentivirus vector (Addgene, Cambridge, Massachusetts, USA). Target cells were transduced with viral particles in the presence of polybrene (2 mg/mL; Sigma-Aldrich, St. Louis, MO, USA), and puromycin (Sigma-Aldrich) was applied for the selection of positive cells. Refer to Supplementary Table 1 for the short hairpin RNAs (shRNAs) sequences.
Cell migration and invasion experiments were performed as described previously
[25]. For the wound-healing experiment, cells were cultured in 6-well plates to
achieve a confluent monolayer. A sterile pipette tip was used to induce
scratches. Cell migration into the scratch area was then monitored and
photographed. To assess cell migration and invasion, transwell assays were
performed. 5
Western blot analysis was performed on total protein extracts using standard
protocols [27]. Respective primary antibodies against E-Cadherin (1:1000, 3195,
Cell Signaling Technology, Danvers, MA, USA),
We performed immunohistochemical (IHC) staining on formalin-fixed, paraffin-embedded tissue sections. The sections were incubated with an anti-ANK3 antibody (1:2000, 27980-AP, Proteintech, Wuhan, China) following the manufacturer’s instructions.
Paired clinical specimens, comprising liver tumors and their matched adjacent normal tissues, were obtained from West China Hospital of Sichuan University. Total RNA was extracted from these tissues with Tri-Reagent and subsequently analyzed via qRT-PCR. This study obtained ethical approval from the Ethics Committee of West China Hospital of Sichuan University (2020, no. 196) for the use of human samples. All participants provided written informed consent prior to inclusion.
Data are presented as mean
To elucidate the role of ANK3 in HCC, we used GSE20017 data from the NCBI to analyze the expression of ANK3, and found that ANK3 expression in patients with microvascular invasion was markedly lower than that in patients without microvascular invasion (Fig. 1a), suggesting that ANK3 may be involved in regulating HCC metastasis. We investigated ANK3 expression in human HCC tissues. ANK3-total mRNA levels were lower in HCC tissues (Fig. 1b), consistent with publicly available data (Fig. 1c). Western blot (WB) results showed that the patterns of the different sizes of ANK3 proteins were inconsistent. The protein, approximately 150 kDa (ANK3-TV2), was significantly downregulated in HCC tissues, while the expression pattern of the protein, approximately 250 kDa (ANK3-TV3/4/5), was uncertain (Fig. 1d). IHC analysis further confirmed reduced ANK3 levels in HCC samples (Fig. 1e).
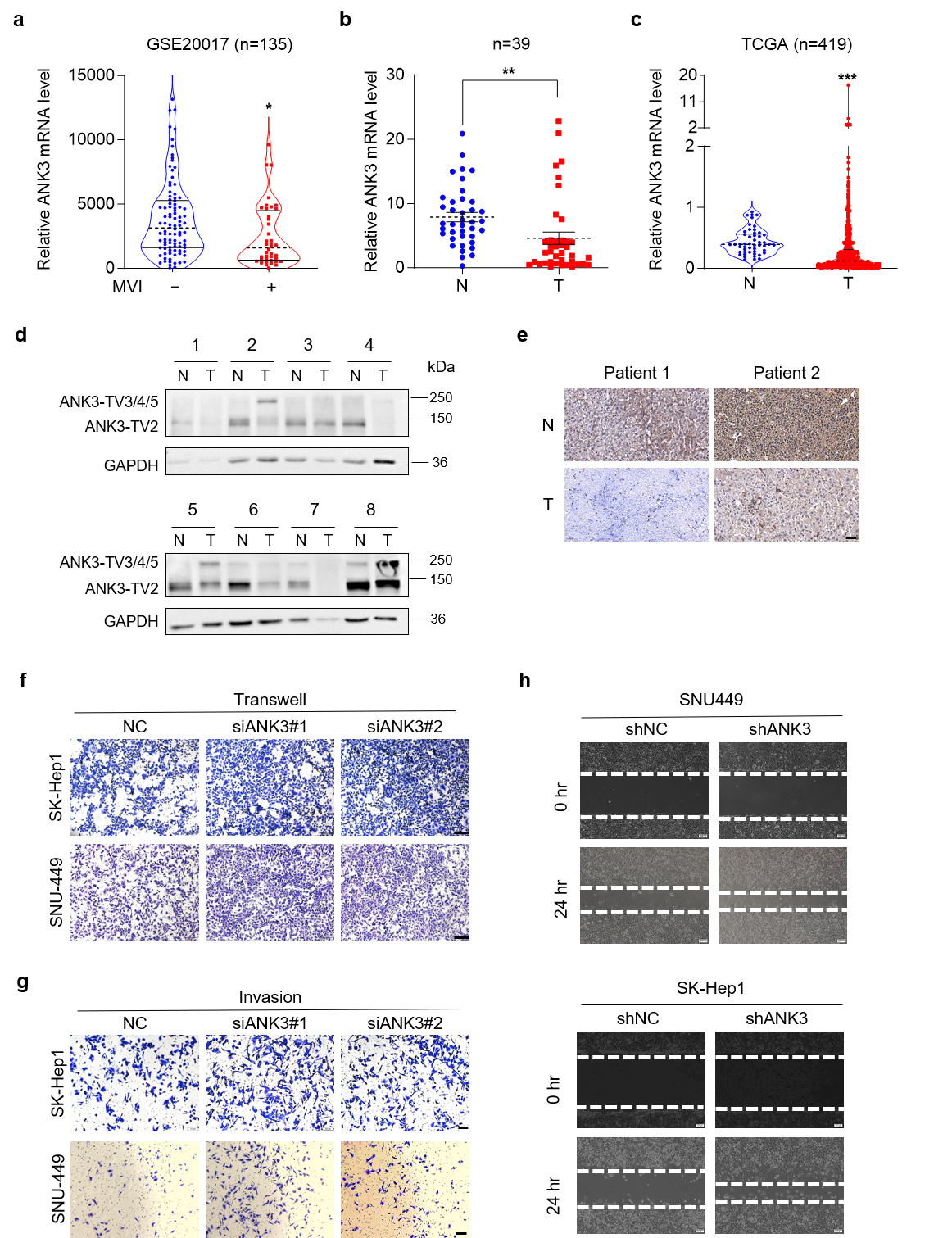 Fig. 1.
Fig. 1.
Ankyrin 3 (ANK3) is significantly
down-regulated in hepatocellular carcinoma (HCC). Knockdown of ANK3
promotes HCC metastasis. (a) Dot plots depicting ANK3
expression in HCC patients stratified by microvascular invasion (MVI) status
(GSE20017 dataset). (b) Total ANK3 mRNA levels in 39 paired HCC (T) and adjacent
non-tumorous (N) tissues.
(c) ANK3 expression levels in HCC (T) and
adjacent non-tumorous (N) tissues from the The Cancer Genome Atlas (TCGA) cohort. (d) Representative western blot (WB) images of
ANK3 protein in 8 paired HCC (T) and adjacent non-tumor (N) samples,
with quantitative analysis relative to Glyceraldehyde-3-phosphate dehydrogenase
(GAPDH) shown below. (e) IHC staining of ANK3 in representative
HCC and matched adjacent tissue. (f) Transwell assay of SK-Hep1 (top)
and SNU449 (bottom) cells transfected with negative control (NC) or siANK3. (g) Invasion assay of SK-Hep1 (top) and SNU449 (bottom) cells transfected
with NC or siANK3. (h) Wound-healing assay of SNU449 (top) and SK-Hep1 (bottom)
cells transfected with shNC or ANK3 shRNA (shANK3). Scale bar: 50 µm (e),
100 µm (f,g), 200 µm (h). Data are presented as mean
We next assessed whether ANK3 contributes to liver tumorigenesis. ANK3 knockdown by siRNA had no impact on the growth of SNU449 or SK-Hep1 cells (Supplementary Fig. 1), indicating that ANK3 does not regulate HCC proliferation. Conversely, ANK3 knockdown markedly promoted the migration ability of SNU449 and SK-Hep1 cells (Fig. 1f,g), which was confirmed by a wound-healing experiment (Fig. 1h). Collectively, these in vitro findings establish that ANK3 functions primarily to suppress the migratory and invasive capabilities of HCC cells.
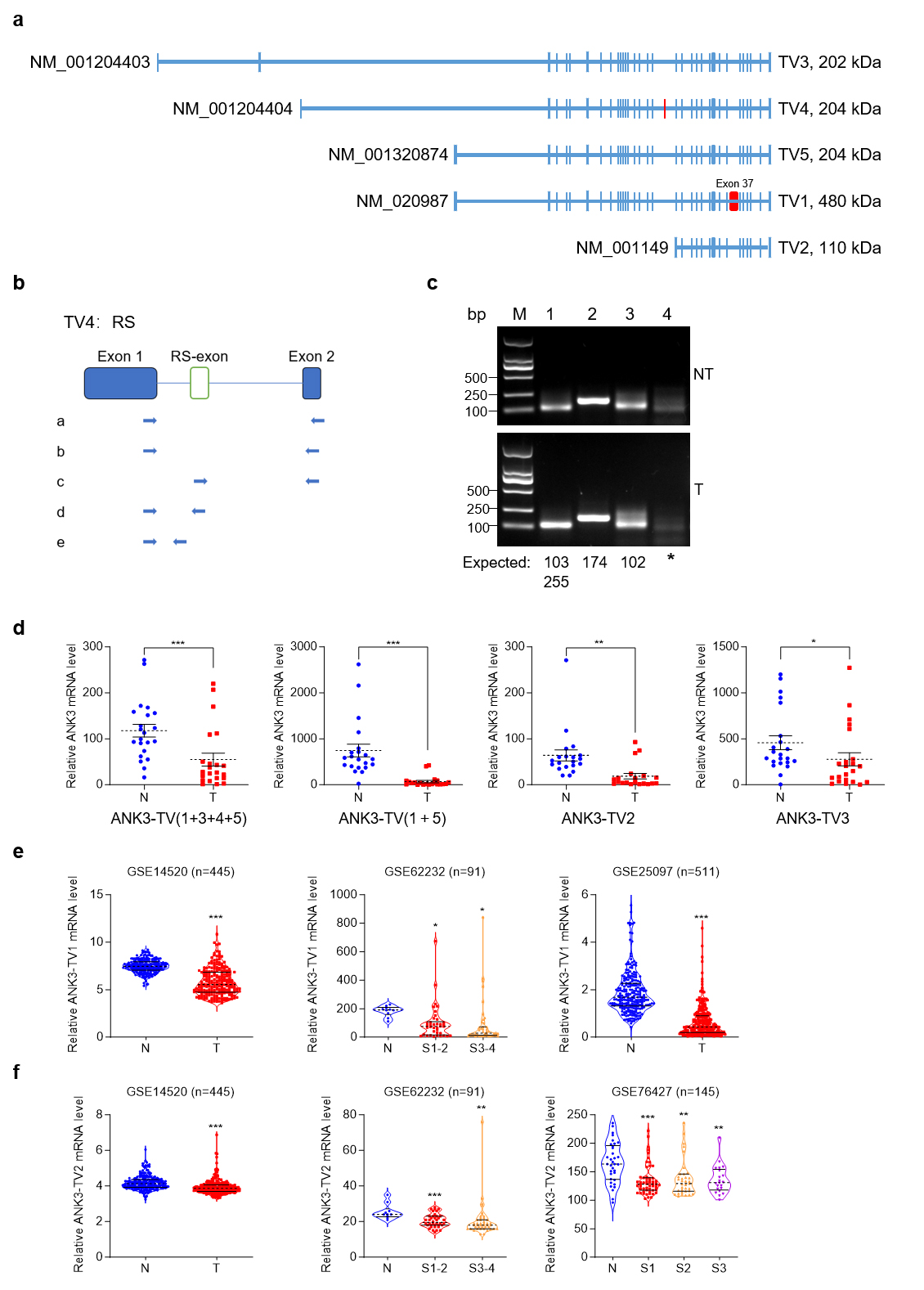
With reference to NCBI, we analyzed the splicing methods existing in ANK3 (Fig. 2a), including alternative promoters and exon jumping, which resulted in ANK3 producing five transcripts. The five transcriptional variants (TVs) of human ANK3 in the NCBI database encoded three proteins of different sizes. First, through an alternative promoter, ANK3 produces four transcription start sites that produce four transcripts: transcript variant 3 (TV3) (NM_001204403), transcript variant 4 (TV4) (NM_001204404), transcript variant 1 (TV1) (NM_020987), and transcript variant 2 (TV2) (NM_001149). As a result of exon jumping, TV1 produces another transcription variant named transcript variant 5 (TV5) (NM_001320874), which lacks exon 37. Another study reported that TV1 also underwent alternative 5’ splicing and retained a part of exon 37 [28]. In addition, RS has been reported in ANK3 [16]. Sequence alignment and analysis revealed that RS may exist in ANK3-TV4. We first used primers from the literature for RT-PCR [16] but failed to detect RS-exon in human liver tissue. We then redesigned the primers and performed a nested PCR. Primer a was used for the first round of PCR, b/c/d/e primers were used for the second round of PCR in lanes 1/2/3/4, respectively (Fig. 2b,c). Theoretically, there were two bands in lane 1 at 103 bp (RS exon skipping) and 255 bp (RS exon retention). However, in lane 1, only the 103 bp band was visible to the naked eye, suggesting that only a small proportion of transcripts retained the RS-exon. The product sizes in lanes 2 and 3 (using primers c and d, respectively) were the same as expected. Collectively, our results demonstrate that the RS and RS-exon exist in ANK3-TV4 in the human liver and hepatoma tissues.
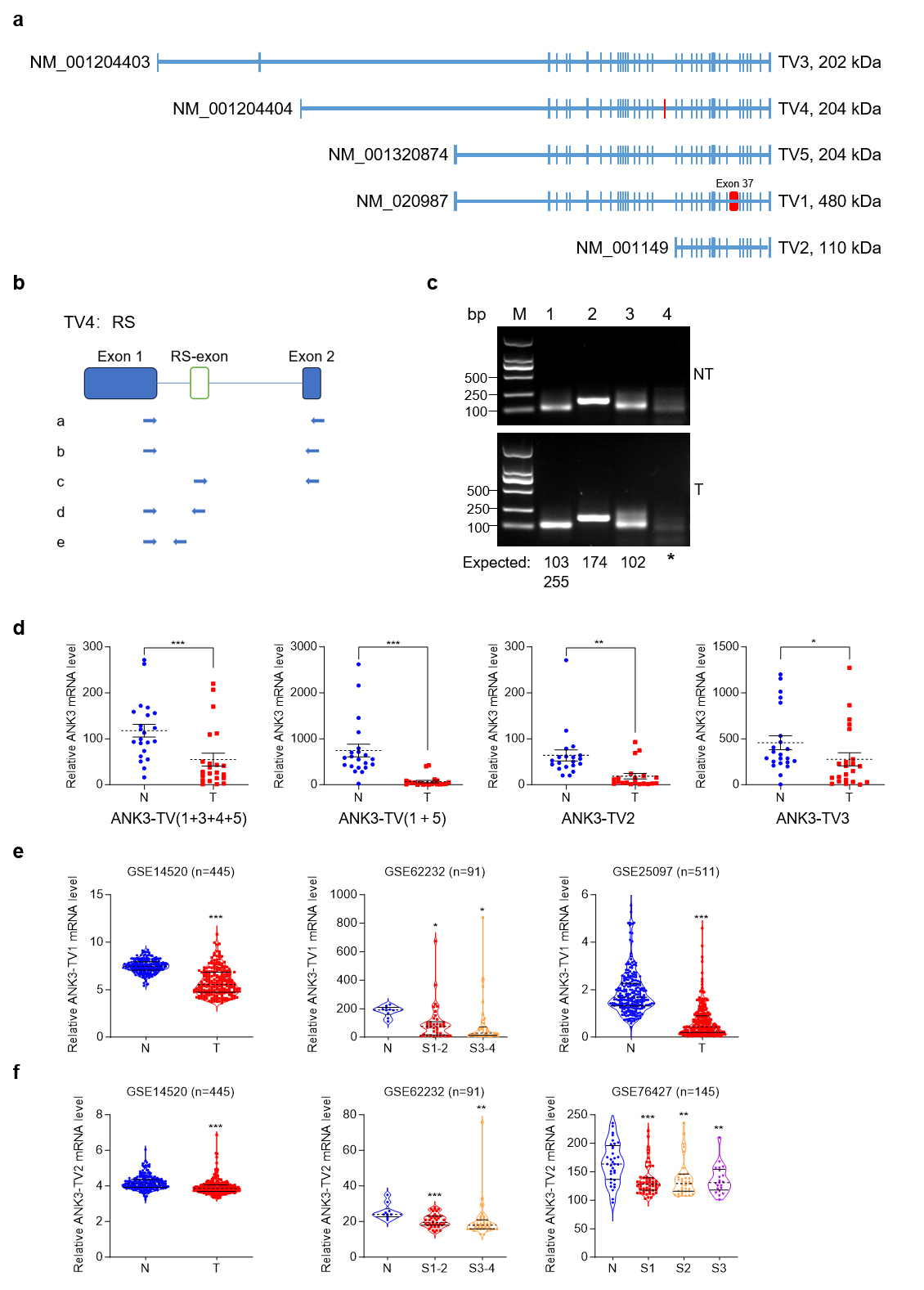 Fig. 2.
Fig. 2.
Transcriptome structure diagram of ANK3. ANK3
transcript variant 4 (TV4) undergoes recursive splicing (RS). (a)
ANK3 from the NCBI database underwent alternative promoter and exon
jumps, producing five TVs. The blue boxes represent exons. The red boxes
represent the unique exons of TV1 and TV4,
respectively. (b) Schematic diagram of the nested PCR primers. (c)
ANK3-TV4 undergoes RS. Using the nest-PCR assay, ANK3-TV4 was
recursively spliced in both HCC tumours (T, down) and adjacent non-tumor (NT,
up). Primer a was used for the first round of PCR. b/c/d/e primers were used for
the second round of PCR in lanes 1/2/3/4, respectively. PCR product sizes are
annotated below each lane. (d) mRNA levels of different transcripts in T
and N tissues. (e) mRNA level of ANK3-TV1 in available HCC
databases. (f) mRNA level of ANK3-TV2 in available HCC
databases. Data are presented as mean
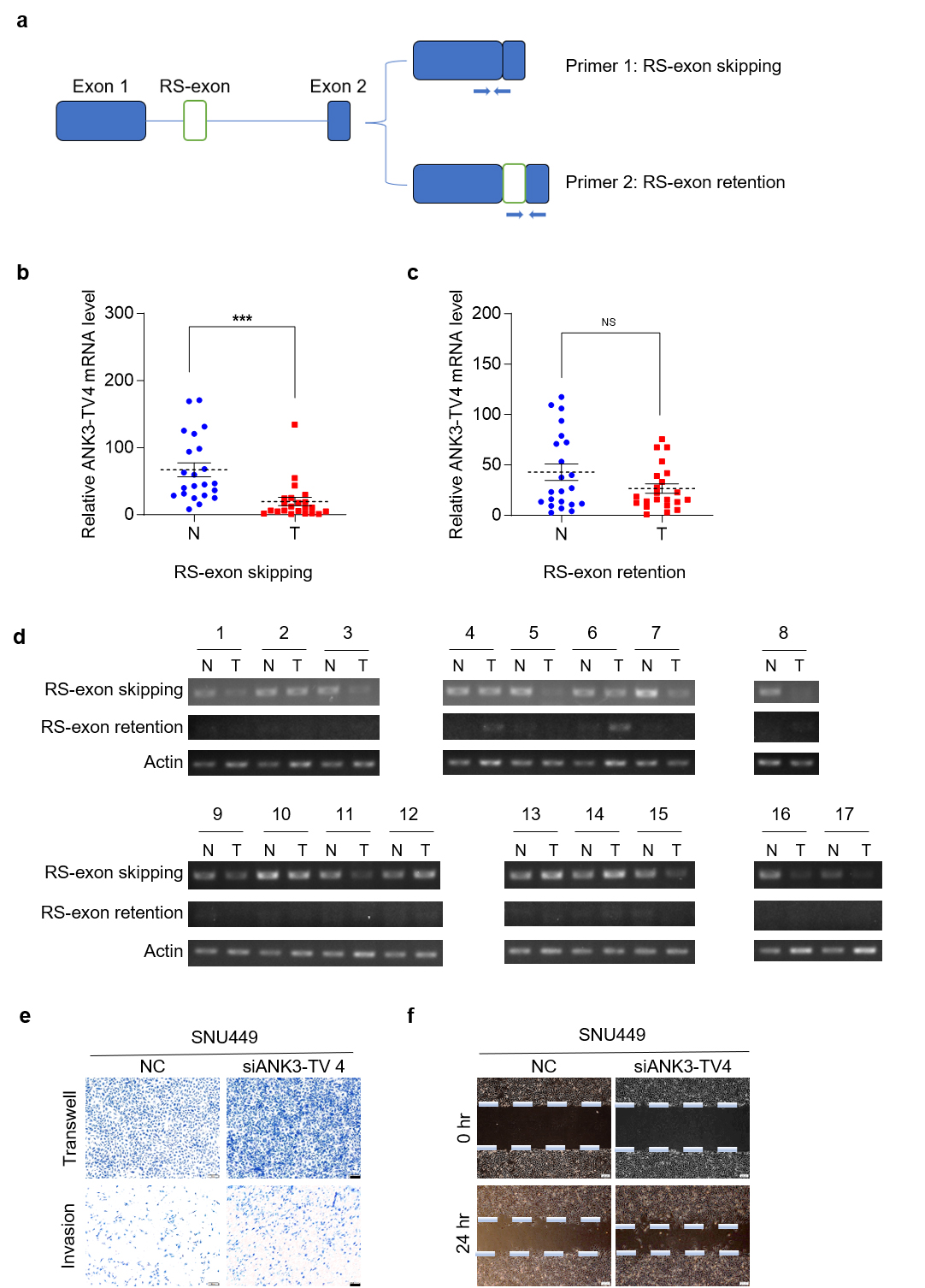
As illustrated in Fig. 1, ANK3 expression was markedly reduced in HCC. We designed primers specific to different ANK3 transcripts to detect their expression in human HCC tissues. All transcripts were significantly downregulated in HCC (Fig. 2d), which was consistent with the publicly available data (Fig. 2e,f). In addition, the results showed that in ANK3-TV4, the transcript with RS-exon skipping was significantly downregulated in HCC (Fig. 3a,b), whereas the transcript with RS-exon retention showed no significant pattern (Fig. 3c). Similar results were obtained by semi-quantitative PCR (Fig. 3d). This suggests that the RS of ANK3-TV4 was weakened in HCC.
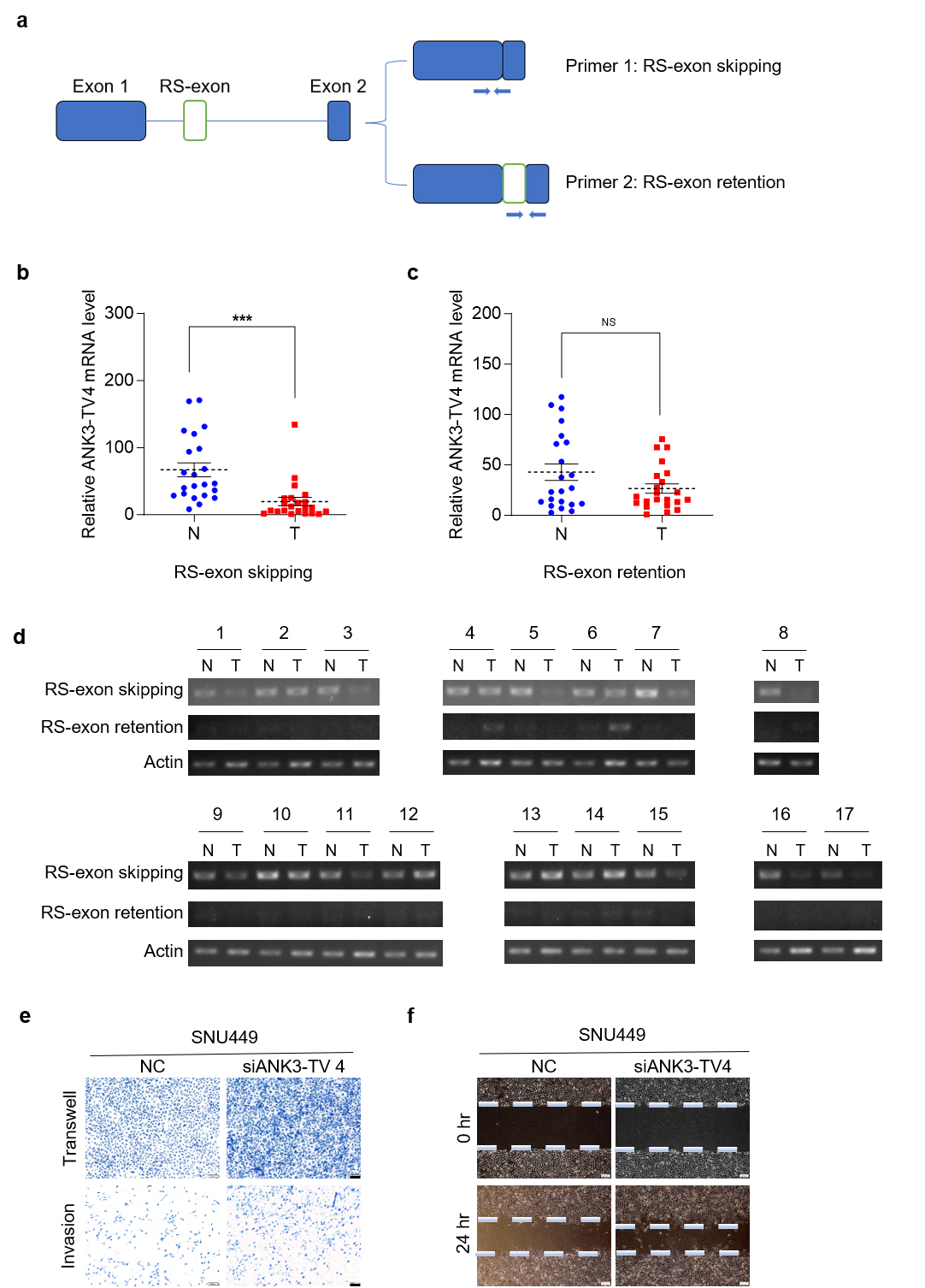 Fig. 3.
Fig. 3.
RS of ANK3-TV4 is weakened in HCC, and
ANK3-TV4 inhibits the metastasis of HCC cells. (a) Primer 1 was
designed across the primary/secondary exon of ANK3-TV4, specifically
quantifying the transcripts with RS (RS-exon skipping). Primer 2 was designed in
the ANK3-TV4 RS-exon and the second exon, respectively, specifically
quantifying the transcripts with RS-exon retention. (b) Quantification of
ANK3-TV4 (RS-exon skipping) mRNA in HCC (T) and paired
non-cancerous tissues (N). (c) mRNA abundance of ANK3-TV4
(RS-exon retention) in HCC samples (T) relative to adjacent controls
(N). (d) Semi-quantitative PCR analysis of both ANK3-TV4 splicing variants in 24
clinical sample pairs (Cycle parameters: Actin-25, ANK3-TV4-40). (e)
Transwell and invasion assays of SNU449 cells following
ANK3-TV4 knockdown. (f) Wound-healing assay of SNU449 cells transfected
with NC or siANK3-TV4. Scale bar: 100 µm (e), 200 µm (f). Data are
presented as mean
Next, we explored the role of ANK3-TV4 in liver tumorigenesis. Because of the particularity of the ANK3-TV4 sequence, only one pair of siRNAs could be specifically designed. Specific knockdown of ANK3-TV4 by siRNAs markedly promoted the migration ability of SNU449 cells, as demonstrated by transwell and invasion assays as well as a wound-healing assay (Fig. 3e,f), suggesting that cell migration and invasion are suppressed by ANK3-TV4 in vitro.
Structurally, ANK3 is organized into three functional domains: the N-terminal membrane domain (including 24 anchoring protein repeat sequences responsible for binding the entire membrane protein), a central spectrum protein/fodrin-binding domain that connects the anchoring protein to the actin-based cytoskeleton through spectrum protein isomers, and a C-terminal regulatory domain [20]. All other transcripts contained the three domains, except for ANK3-TV2, which completely lacked the ankyrin repeat domain. To explore the influence of various ANK3 domains on HCC migration, ANK3-TV2 was initially overexpressed in stable ANK3-total knockdown cells to assess its impact on metastatic behavior. Overexpression of ANK3-TV2 did not markedly restore the promoting effect of ANK3 knockdown on cell migration (Fig. 4a,b). Since ANK3-TV2 lacks while ANK3-TV4 contains an ankyrin repeat domain, we compared the effects of overexpressing ANK3-TV2 and the ankyrin repeat-containing ANK3-TV4 (Supplementary Fig. 2a–c). The results clearly showed that only ANK3-TV4 significantly inhibited HCC cell invasion and migration (Fig. 4c–e), suggesting that ANK3 significantly inhibits HCC metastasis through its ankyrin repeat domain.
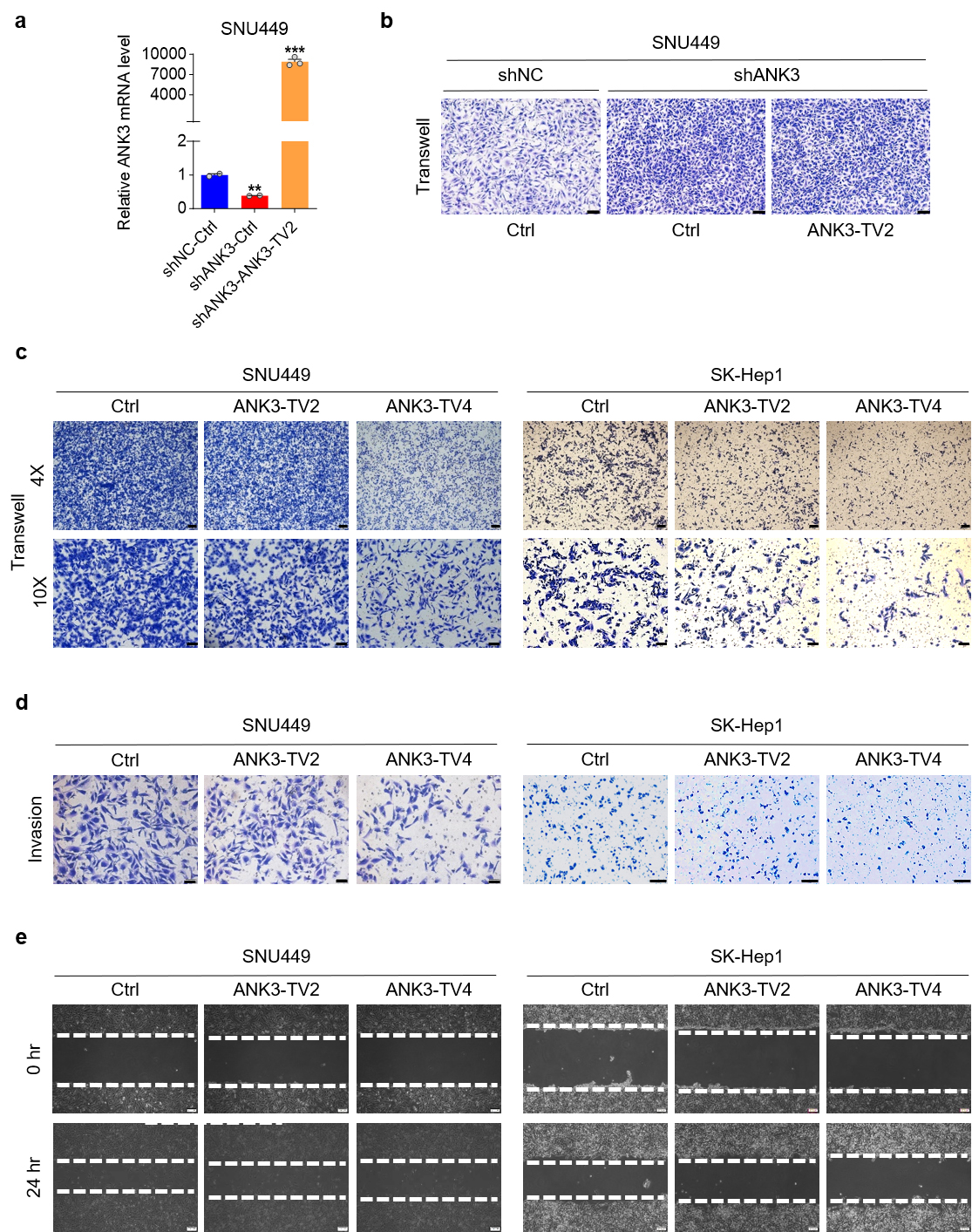 Fig. 4.
Fig. 4.
Ankyrin repeats of ANK3 are critical in cell migration.
(a) ANK3 mRNA expression in SNU449 cells with stable ANK3-total
knockdown, following transfection with an empty vector (Ctrl) or an
ANK3-TV2 overexpression vector. (b) Transwell assay of
ANK3-TV2 overexpression in stable cell lines with ANK3-total
knockdown. (c) Transwell assay of SNU449 (left) or SK-Hep1 (right) cells
transfected with ctrl (Empty Vector) or ANK3-TV2/ANK3-TV4 overexpression
vectors. (d) Invasion assay of SNU449 (left) or SK-Hep1 (right) cells transfected
with ctrl (Empty Vector) or ANK3-TV2/ANK3-TV4 overexpression vectors.
(e) Wound-healing assay of SNU449 (left) or SK-Hep1 cells (right) transfected
with ctrl (Empty Vector) or ANK3-TV2/ANK3-TV4 overexpression vectors.
Scale bar: 100 µm (b), 200 µm (c 4
To explore the molecular mechanism of ANK3 regulation of cell
migration, transcriptomic sequencing was performed on RNA extracted from stable
ANK3 knockdown SNU449 cells. Gene Ontology Enrichment Analysis (GO)
showed that ANK3 participates in the regulation of the Wnt signaling
pathway (Fig. 5a–d). We initially searched for proteins that might interact with
ANK3 in the BioGrid database and found that ANK3 might interact
with 151 proteins (Supplementary Fig. 3a). We screened the proteins
associated with metastasis and summarized them in Supplementary Fig. 3b.
At the same time, we reviewed the literature on these proteins. E-cadherin,
located in the cytomembrane, has been reported to interact with ANK3 and
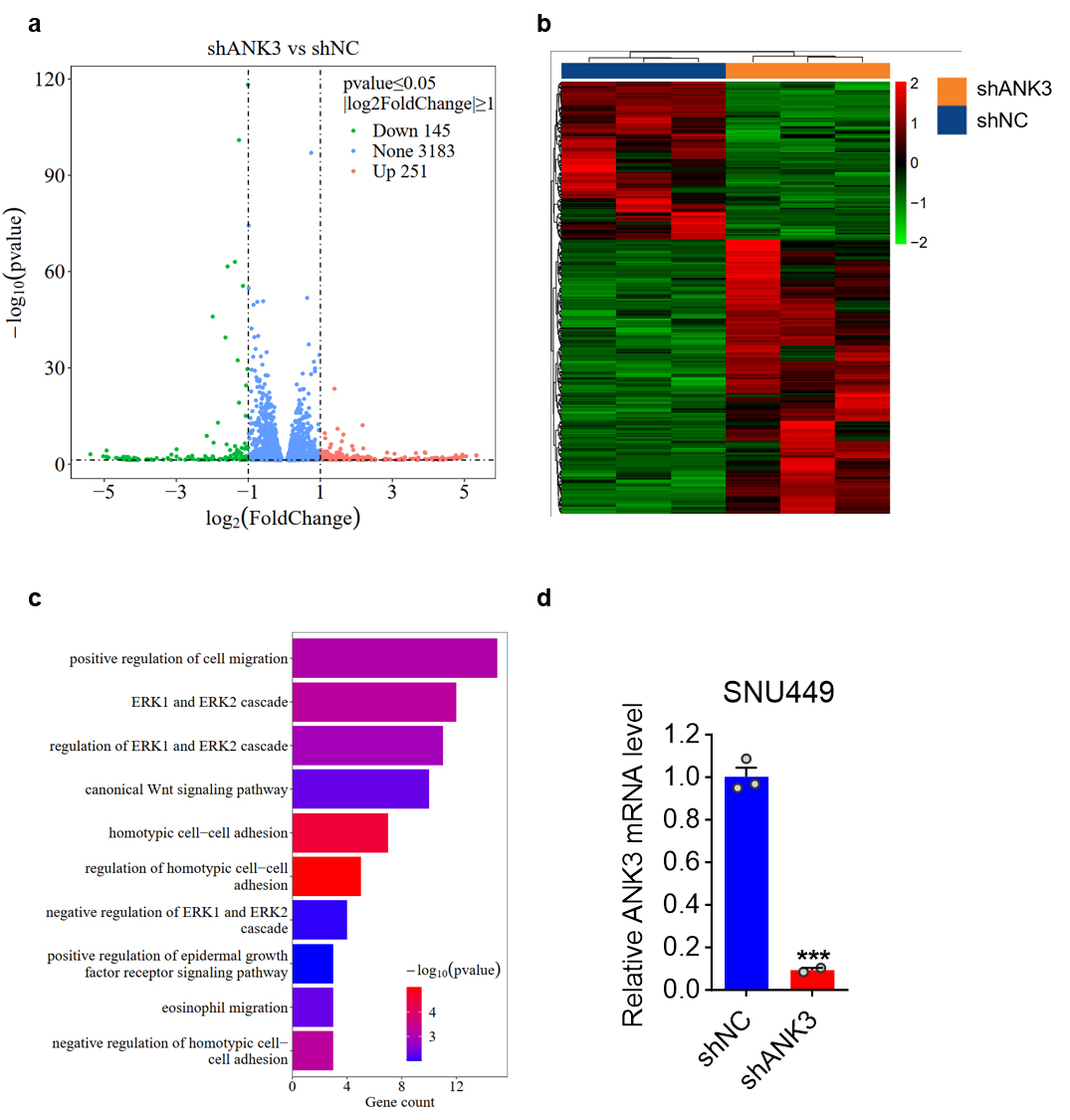 Fig. 5.
Fig. 5.
Enrichment analysis of differentially expressed protein-coding
genes in SNU449 cells with stable ANK3 knockdown. (a) A volcano plot
visualization of the data. The X-axis represents the magnitude of expression
change (log2 fold change), while the Y-axis indicates the statistical
significance (–log10 p-value) of that change. Genes are
highlighted as down-regulated (green), up-regulated (red), or unchanged (blue).
(b) Cluster heat maps: Each column represents a sample, and each group has 3
biologically duplicated samples. Each line represents a protein-coding gene.
Fragments Per Kilobase Million (FPKM) values were used for clustering, with red
representing high-expression genes and green representing low-expression genes,
and the darker the color, the greater the degree of up-regulation or
down-regulation. (c) Gene Ontology Enrichment Analysis (GO): The vertical axis is
the name of the GO pathway, and the horizontal axis represents the number of
genes enriched into that pathway. The darker the color, the smaller the p value,
indicating that the enrichment significance of differentially expressed genes in
this pathway is more reliable. (d) qRT-PCR analysis of ANK3 expression
in SNU449 cells infected with shNC or shANK3 lentivirus. Data are presented as
mean
Based on the transcriptome sequencing results, BioGrid database search, and literature review, we selected E-cadherin as the research object and conducted subsequent experimental verification. The schematic diagram of the ANK3 domain is shown in Fig. 6a. The expression of HA-tagged control and E-cadherin plasmid in 293T cells is shown in Fig. 6b. Western Blot assay showed that the E-cadherin antibody could precipitate ANK3 containing an ankyrin repeat domain with a size of approximately 240 kDa and E-cadherin, but could not precipitate ANK3-TV2 lacking the ankyrin repeat domain with a size of 110 kDa (Fig. 6c), indicating that endogenous E-cadherin interacts with the ankyrin repeat domain of ANK3. A co-immunoprecipitation (Co-IP) assay confirmed that ANK3-TV4 could, while ANK3-TV2 couldn’t, interact with E-cadherin (Fig. 6d,e). ANK3-TV4 overexpression significantly upregulated E-cadherin expression in Huh7 cells (Fig. 6f).
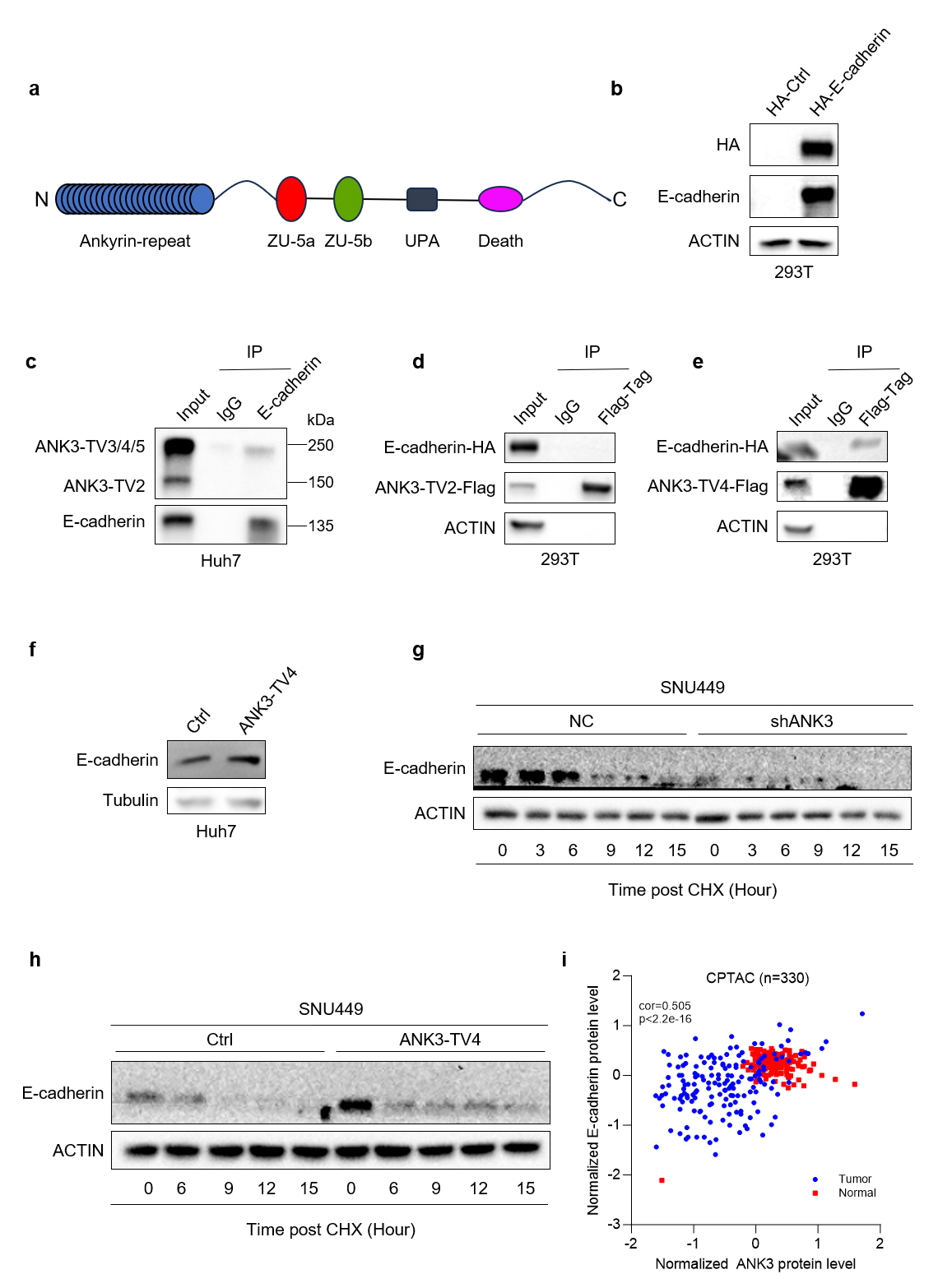 Fig. 6.
Fig. 6.
ANK3 interacts with E-cadherin, and knockdown of ANK3 promoted E-cadherin protein degradation. (a) Schematic diagram of the ANK3 domain. (b) Validation of HA-tagged control and E-cadherin plasmid expression in 293T cells by WB. (c) Endogenous interaction between ANK3 and E-cadherin in Huh7 cells, confirmed by co-immunoprecipitation. (d,e) Exogenous interaction of E-cadherin with ANK3-TV2 (d) or ANK3-TV4 (e) in 293T cells, assessed by co-immunoprecipitation. (f) Overexpression of ANK3-TV4 significantly upregulated E-cadherin. (g) Knockdown of ANK3 promoted E-cadherin protein degradation. (h) Overexpression of ANK3-TV4 inhibited E-cadherin protein degradation. (i) E-cadherin protein and ANK3 protein were positively correlated in HCC in the CPTAC database.
Transcriptomic sequencing suggests that knockdown of ANK3 can activate the Wnt pathway. We first examined the potential role of ANK3 in regulating the Wnt signaling pathway. Both qPCR and WB experiments confirmed that ANK3 knockdown significantly upregulated the expression of several Wnt pathway components (Fig. 7a), while ANK3-TV4 overexpression inhibited the expression of these genes (Fig. 7b).
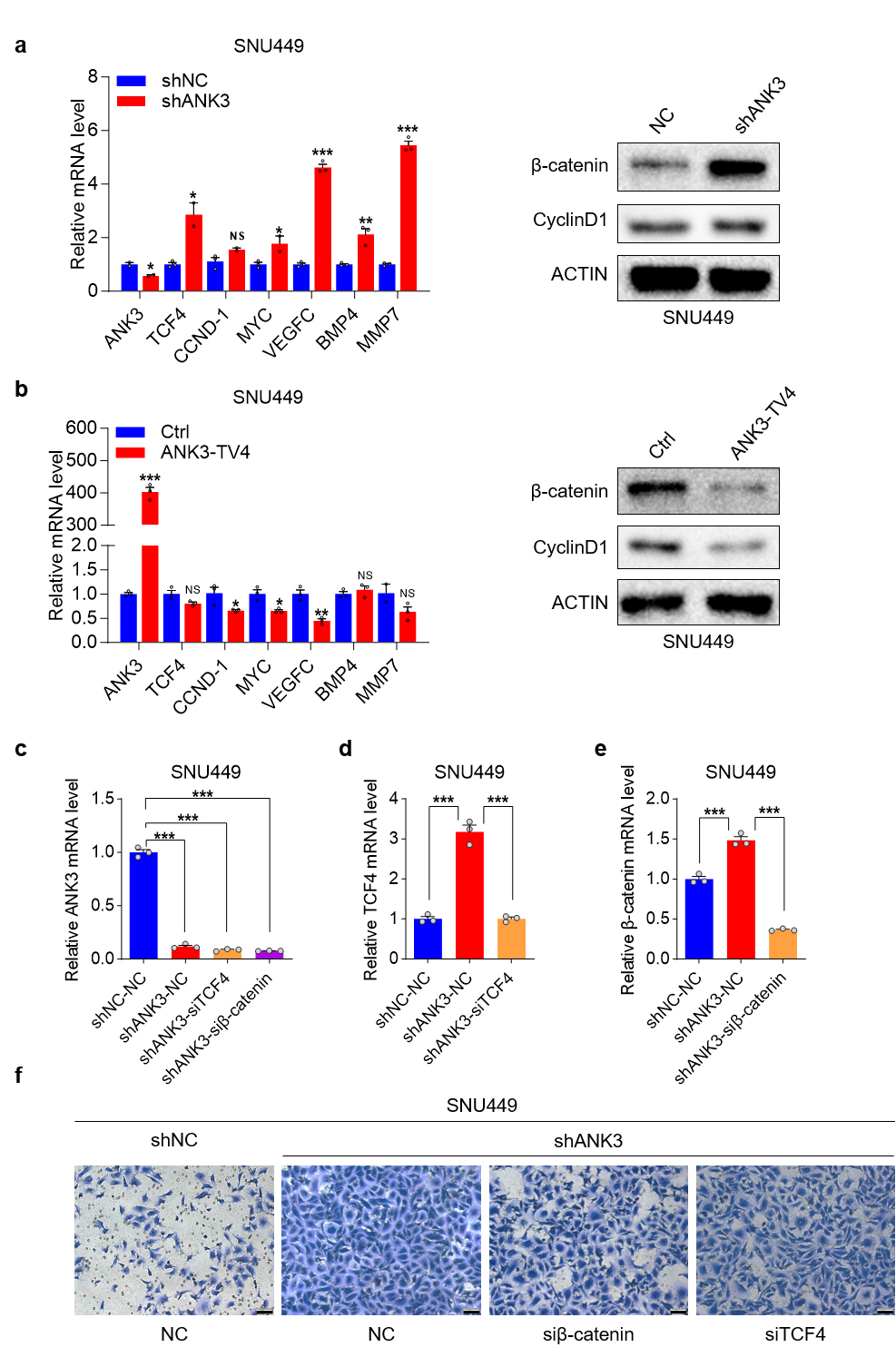 Fig. 7.
Fig. 7.
Knockdown of ANK3 activates the Wnt pathway. (a)
qRT-PCR and WB analysis of Wnt pathway factor expression upon ANK3
knockdown in SNU449 cells. (b) qRT-PCR and WB analysis of Wnt pathway components
in response to ANK3-TV4 overexpression in SNU449 cells. (c)
qRT-PCR analysis of ANK3 expression in response to transient
TCF4/
The pro-metastatic function of Wnt/
ANK3 binds to E-cadherin via its N-terminal ankyrin repeat domain to regulate E-cadherin expression. ANK3 knockdown activates the Wnt pathway, downregulates E-cadherin expression, and promotes its degradation. Conversely, ANK3-TV4 overexpression inhibited the Wnt pathway, upregulated E-cadherin protein expression, and inhibited its degradation (Fig. 6g,h). Finally, we analyzed the correlation between E-cadherin and ANK3 proteins in clinical samples from the Clinical Proteomic Tumor Analysis Consortium (CPTAC) database of The Cancer Genome Atlas (TCGA) and found that E-cadherin protein positively correlated with ANK3 protein in both liver cancer and para-cancer samples, which was consistent with our previous conclusions (Fig. 6i).
Multiple annotated RS-exons in human mRNAs can be attributed to the inhibition of RS by EJC. The EJC is generally composed of core EJC factors, including eIF4A3, MAGOH, and RBM8A, and peripheral factors such as PNN and RNPS1 [17]. Screening of candidate factors in clinical HCC samples revealed that RBM8A was the sole factor significantly overexpressed (Fig. 8a,b), consistent with publicly available data (Fig. 8c). Kaplan-Meier analysis of overall survival in the TCGA liver cancer cohort (n = 182) demonstrated a significant reduction in survival time for patients with high RBM8A expression (Fig. 8d). Elevated RBM8A expression was significantly correlated with reduced disease-free survival in HCC patients from the TCGA-LIHC cohort (Fig. 8e), underscoring its negative impact on patient prognosis.
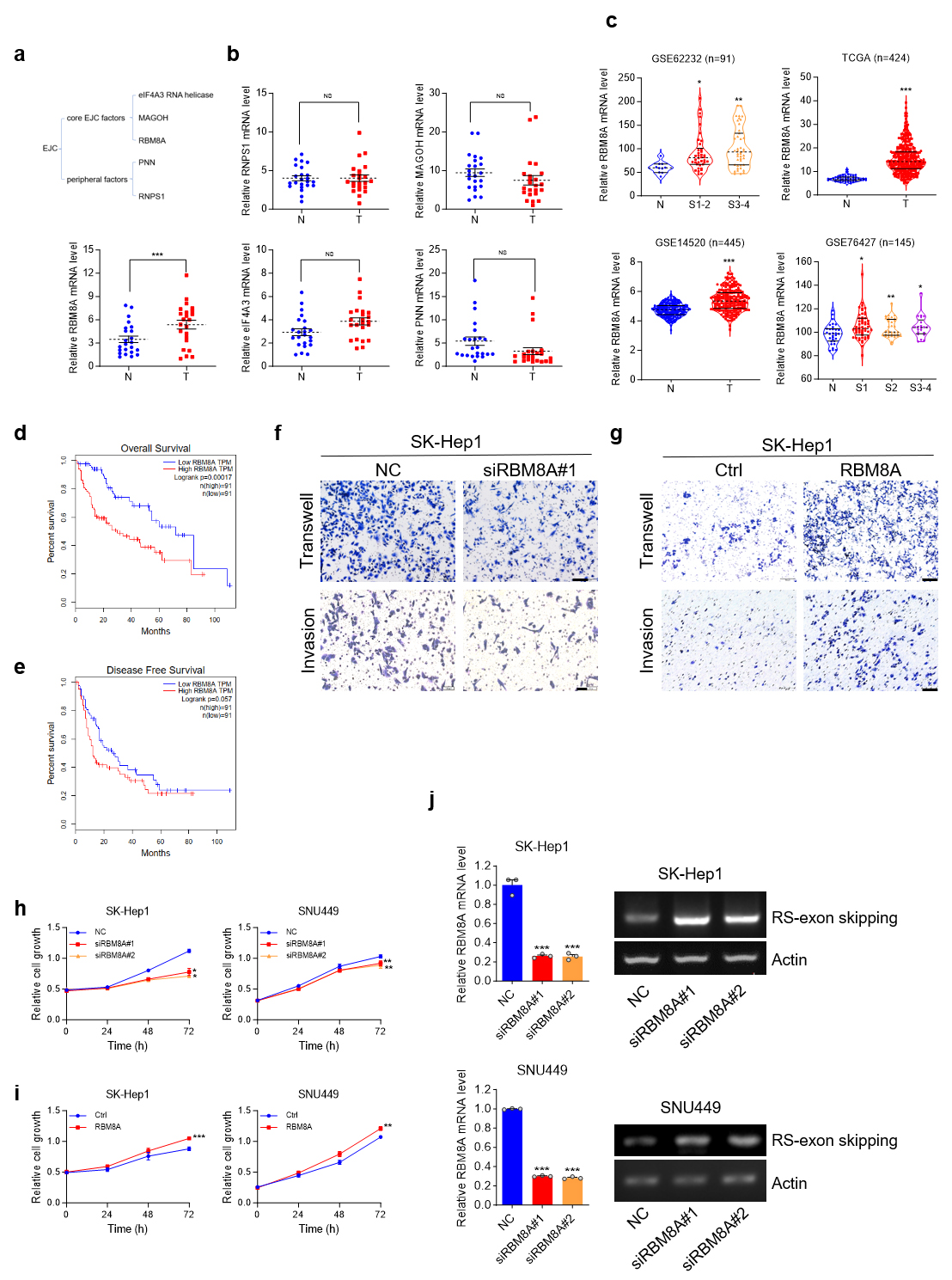 Fig. 8.
Fig. 8.
RBMBA is upregulated in HCC. Knockdown of RBM8A
inhibits the proliferation and metastasis of HCC cells, and knockdown of
RBM8A promotes RS of ANK3-TV4. (a) Schematic of exon junction
complex (EJC). (b) qRT-PCR analysis of EJC factors identifies RBM8A as
the sole significantly upregulated member in HCC. (c) RBM8A mRNA levels
in HCC and normal livers from published cohorts. (d) Kaplan-Meier analysis of
overall survival data from TCGA cohort (n = 182), stratified by RBM8A
expression. (e) Kaplan-Meier analysis of disease free survival in the TCGA-LIHC
cohort (n = 182). (f) Impact of RBM8A depletion on the migratory and
invasive capacity of SK-Hep1 cells. (g) Effect of RBM8A overexpression
on the migratory and invasive capacity of SK-Hep1 cells. (h) MTS analysis of
SK-Hep1 (left) and SNU449 (right) cells transfected with NC or siRBM8A. (i) MTS
analysis of SK-Hep1 (left) and SNU449 (right) cells transfected with ctrl (Empty
Vector) or RBM8A overexpression vector. (j) The semi-quantitative RT-PCR
proved that knocking down RBM8A promotes RS of ANK3-TV4 in
SK-Hep1 or SNU449 cells. Actin: 25 cycles; ANK3-TV4 with RS-exon
skipping: 42 cycles. Scale bar: 100 µm. Data are presented as mean
We next investigated the functional contribution of RBM8A to liver tumorigenesis. Functional interrogation of RBM8A in liver tumorigenesis revealed that its siRNA-mediated knockdown attenuated the proliferation of SK-Hep1 and SNU449 cells (Fig. 8h), and markedly impaired the migration and invasion of SK-Hep1 cells (Fig. 8f). Conversely, RBM8A overexpression enhanced these malignant properties, including migration, invasion, and growth (Fig. 8g,i). Collectively, these findings establish RBM8A as a promoter of HCC cell proliferation, migration, and invasion in vitro.
The role of RBM8A in modulating the RS of ANK3-TV4 was assessed. Transcripts with RS-exon skipping were quantified using primers crossing the first and second exons of ANK3-TV4. The data indicate that reducing RBM8A levels promotes RS of ANK3-TV4 (Fig. 8j), which promoted the jump of the RS-exon, reducing the NMD of ANK3-TV4. Thus, normal functional ANK3-TV4 was upregulated.
In 1998, Hatton et al. [32] first discovered the RS in Drosophila melanogaster Ubx and identified a new splicing site between the first and second exons of the Ubx gene. Researchers have conducted in-depth studies on the Ubx gene and found that the first intron of the gene is 73 kb long, and the removal of the intron involves three splicing steps [33]. In 2015, Sibley et al. [16] conducted deep RNA sequencing of vertebrate neurons and found that RS phenomena are widespread in vertebrate neurons, including in genes such as ANK3 and CADM2. Emmett [34] found that in the human brain tissue, seven genes associated with RS are closely related to neurological diseases. Given the established link between aberrant long-gene expression and neurological disorders [35, 36, 37], it is plausible that mutations or deletions at RS sites constitute another mechanism for human genetic diseases [16]. RS events, observed in diverse cell types including neurons [34] and endodermal cells [38], are associated with a spectrum of human diseases spanning neurological (e.g., Parkinson’s) and circulatory (e.g., retinal sclerosis, hypertension) disorders [39]. In addition, Srndic [40] detected over 12,000 RS events in multiple human tissues and found that RS might play a role in nonsense-mediated RNA decay.
ANK3 was originally identified in the axonal initial segment and nodes of Ranvier [41, 42]. It has been extensively characterized and is now recognized as an established risk gene for several neuropsychiatric disorders, including bipolar disorder, schizophrenia, and autism spectrum disorder [21, 43, 44, 45]. A relevant study has reported that ANK3 undergoes RS [16], but did not indicate the transcript in which the RS is present. ANK3 is perversely expressed in various human cancers; however, the underlying mechanism remains unclear. Currently, only studies have reported the function of ANK3 in prostate cancer [26], and colorectal cancer [46].
Referring to the study by Blazquez et al. [17], who found that the EJC suppressed hundreds of annotated RS, mainly constitutive RS-exons, and they investigated the role of EJC in regulating the RS of CADM2, mainly by RT-PCR. Given this context, we sought to determine whether the EJC, particularly its key components, regulates ANK3 in HCC. Systematic examination of EJC factors in clinical HCC samples revealed RBM8A as the sole significantly upregulated member. Subsequent functional characterization confirmed the oncogenic properties of RBM8A. Therefore, we explored the effect of RBM8A on ANK3 RS. However, we did not successfully detect RS by RT-PCR using the primers used in the study by Sibley et al. [16]; thus, we redesigned the primers to specifically detect the expression of RS transcripts by semi-quantitative PCR. Knockdown of RBM8A significantly promoted the RS of ANK3-TV4, which promoted a jump in the RS-exon, reducing the NMD of ANK3-TV4. Thus, normally functional ANK3-TV4 is upregulated, inhibiting cell migration. Of course, RBM8A’s regulation on the RS of the ANK3-TV4 is only slight, and this regulatory process must involve many other factors and more complex pathways. Our study merely preliminarily links RS to HCC. However, it is not yet clear whether the anti-metastasis effect of RBM8A depletion is specifically mediated by ANK3-TV4 RS.
Durak et al. [29] demonstrated that ANK3 regulates canonical
Wnt signaling by modulating the subcellular localization and availability of
The growing recognition of introns as critical regulators of gene expression, driven by advances in RNA sequencing, highlights the importance of deciphering RS in this process. HCC is the third most common cause of cancer mortality worldwide [47], creating an urgent need to define its molecular drivers, particularly for metastasis [26]. Investigating RS may therefore yield novel therapeutic targets for this lethal malignancy. We studied the effects of ANK3 on HCC development via its regulation by RS. This study only explored the regulation of RBM8A on the RS of ANK3-TV4 at the mRNA level, but there was no discussion on the level of ANK3 protein. Therefore, it is difficult to link it with the downstream mechanism of ANK3 regulation. In addition, the puzzling thing is that the expression pattern of the protein near 250 kDa (ANK3-TV3/4/5) in HCC is uncertain, which is not completely consistent with the tumor suppressive function of ANK3. Therefore, more complex regulatory mechanisms at the protein level need to be explored. Further in vivo experiments are required to assess the antitumor efficacy of ANK3.
In summary, we found that ANK3 was downregulated in HCC and inhibited HCC metastasis, confirming that RS exists in ANK3-TV4 and that RS is weakened in HCC. RBM8A, a core EJC factor, regulates the RS of ANK3-TV4. Mechanistically, ANK3 regulates HCC progression by simultaneously controlling the expression of E-cadherin and the Wnt signaling pathway. Our study on RS regulation of ANK3 is superficial, but it provides a new idea for introns to regulate gene expression and participate in disease, and an in-depth study of RS, ANK3, and EJC may pave the way for developing targeted therapies against HCC.
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
YG, XF and YT designed the research study. YG performed the research. YG analyzed the data. YG wrote the manuscript. YT provided help on the revision of the article. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The study was conducted in accordance with the Declaration of Helsinki. The research protocol was approved by the Ethics Committee of Institutional Review Committee of West China Hospital of Sichuan University (Ethic Approval Number: 2020, no. 196) and all of the participants provided signed informed consent.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL46013.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.