
- Academic Editors
†These authors contributed equally.
The C-X-C motif chemokine receptor 3 (CXCR3) antagonist AMG 487 has been shown to alleviate acute lung injury (ALI) in mice. Other CXCR3 antagonists, including NBI-74330, TAK-779, and SCH 546738, exhibit anti-inflammatory effects in various diseases, including apical periodontitis, arthritis, and acute respiratory distress syndrome (ARDS). However, with the exception of AMG 487, the roles of these antagonists in ALI remain poorly understood. Macrophages can differentiate into various phenotypes and play a crucial role in the progression of inflammatory and autoimmune diseases.
In this study, we demonstrate that the CXCR3 agonist C-X-C motif chemokine ligand 10 (CXCL10) enhances macrophage efferocytosis and polarizes inflammatory macrophages toward the M1 phenotype, thereby exacerbating ALI in mice. Conversely, nine CXCR3 antagonists were found to inhibit macrophage efferocytosis and promote the polarization of inflammatory macrophages toward the M2 phenotype, resulting in the alleviation of ALI in mice. Subsequently, molecular docking techniques were employed to analyze interactions between nine CXCR3 antagonists and the CXCR3 protein, with the aim of screening for superior antagonist structures and designing more effective compound configurations targeting the CXCL10-CXCR3 axis. Notably, TAK-779 exhibited the most stable binding affinity to the CXCR3 protein. Furthermore, two newly modified compounds—TAK-779 from imidazolium 1 and TAK-779, 2745583—demonstrated enhanced efficacy compared to the original TAK-779 compound.
All nine CXCR3 antagonists were shown to influence macrophage function to varying degrees and confer protective effects against ALI. These finding suggest that comparative evaluation of CXCR3 antagonists and the discovery of novel compounds may provide new therapeutic targets for the treatment of inflammatory diseases.
Acute lung injury (ALI) is characterized by increased pulmonary vascular permeability, pulmonary edema, diffuse alveolar damage, and the infiltration of inflammatory cells, including neutrophils and macrophages [1, 2]. Chemokines such as CXCL4, CXCL9, CXCL10, and CXCL11, along with their respective receptors, serve as critical mediators of immune cell trafficking and play a pivotal role in both the pathogenesis and resolution of ALI [3]. The overexpression of chemokines can trigger inflammatory diseases, including asthma and atherosclerosis [4, 5]. The chemokine receptor C-X-C motif chemokine receptor 3 (CXCR3), a G protein-coupled receptor for the cytokines CXCL9, CXCL10, and CXCL11 [6], participates in tumor progression by regulating tumor growth, migration, invasion, and angiogenesis [7]. Additionally, CXCR3 contributes to the activation of macrophages in mice [8, 9]. The CXCL10-CXCR3 axis is pivotal in the immune system, regulating the differentiation, activation, and migration of immune cells [10]. The selective recruitment of cells via CXCL10-CXCR3 can result in tissue damage and exacerbate inflammation. The overexpression of CXCR3 and its ligands is closely linked to pulmonary inflammatory diseases [11, 12]. Notably, increased levels of CXCL10 and CXCR3 have been documented in the lung tissues of mice infected with severe acute respiratory syndrome (SARS), influenza virus, and SARS-coronavirus 2 (SARS-CoV-2) [12, 13, 14]. Therefore, inhibiting CXCR3 activation may serve as a viable therapeutic target for pulmonary inflammatory diseases.
Macrophages play a crucial role in various inflammatory and autoimmune diseases through both innate and adaptive immunity [15]. An imbalance between the activation and suppression of M1 and M2 macrophage phenotypes has been linked to the pathogenesis of numerous diseases [16]. CXCL10 is essential for macrophage recognition of inflammation. During the early stages of inflammation, CXCR3+ macrophages are recruited to the site of injury by the chemotactic effects of CXCL10, thereby inducing local inflammatory responses. CXCL10 facilitates macrophage migration via the extracellular signal-regulated kinase (ERK) and P38/mitogen-activated protein kinase (P38/MAPK) signaling pathways. The activation of the CXCL10-CXCR3 axis triggers intracellular signaling pathways, including Janus kinase (JAK) and signal transducer and activator of transcription 1 (STAT1), which promote M1 polarization of macrophages and induce inflammatory responses in lung tissue. Inhibition of CXCL10 or CXCR3 secretion can reduce the M1/M2 macrophage ratio and decrease the secretion of inflammatory factors, thereby exerting anti-inflammatory effects [17]. Regardless of whether macrophages are tissue-resident or inflammatory, the same stimulus can elicit distinct responses [18]. The regulation of macrophage polarization by the CXCL10-CXCR3 axis is contingent upon the inflammatory state of the macrophages. In cultured RAW264.7 cells, CXCL10 promotes M2 polarization, whereas the CXCR3 antagonist AMG 487 induces M1 polarization. Conversely, in RAW264.7 cells treated with Poly(I:C), a reagent that simulates viral infection, CXCL10 and AMG 487 induce M1 and M2 polarization, respectively [19]. Previous study has demonstrated that the CXCR3-CXCL10 axis plays a critical regulatory role in both the phagocytic function and polarization of macrophages [20]. CXCL10 enhances the efferocytosis capability of macrophages, while AMG 487 inhibits this function by blocking CXCR3 signaling.
To further elucidate the functions of chemokines and to design therapeutic
interventions for ALI, it is crucial to investigate the structural basis of the
interactions between chemokines and their receptors [21]. Numerous small-molecule
CXCR3 antagonists have been reported, and related medicinal chemistry studies
have revealed the molecular properties of these antagonists. For instance, AMG
487 selectively blocks CXCR3 with high efficacy in vitro and
demonstrates therapeutic effects in a bleomycin-induced murine model of pulmonary
inflammation [22]. AMG 487 ameliorated inflammatory changes and pelvic pain in
mice with experimental autoimmune prostatitis by reducing Th1 cell
differentiation and suppressing M1 macrophage activation [23]. The selective
CXCR3 antagonist NBI-74330 reduced neuropathic pain-related behaviors in rats
subjected to chronic constriction injury and enhanced the analgesic efficacy of
morphine [24]. In low-density lipoprotein receptor-deficient mice treated with
NBI-74330, the antagonism of CXCR3 significantly reduced the migration of
CD4+ T cells and macrophages to the peritoneal cavity, thereby attenuating
the formation of atherosclerotic plaques [25]. The broad-spectrum antagonist
TAK-779 effectively blocked the binding of CXCR3 to CXCL10 and inhibited
CXCL10-induced cell adhesion and chemotaxis in vitro [26]. In a mouse
model of SARS-CoV-2-associated acute respiratory distress syndrome (ARDS),
TAK-779 suppressed the development of cytokine storms, resulting in a 3–5-fold
downregulation of two C-C motif chemokine receptor 5 (CCR5) ligands and
macrophage inflammatory proteins (MIP-1
| Compound | Structure | MW | HBA | HBD | RB | AR | Reference |
| AMG 487 | 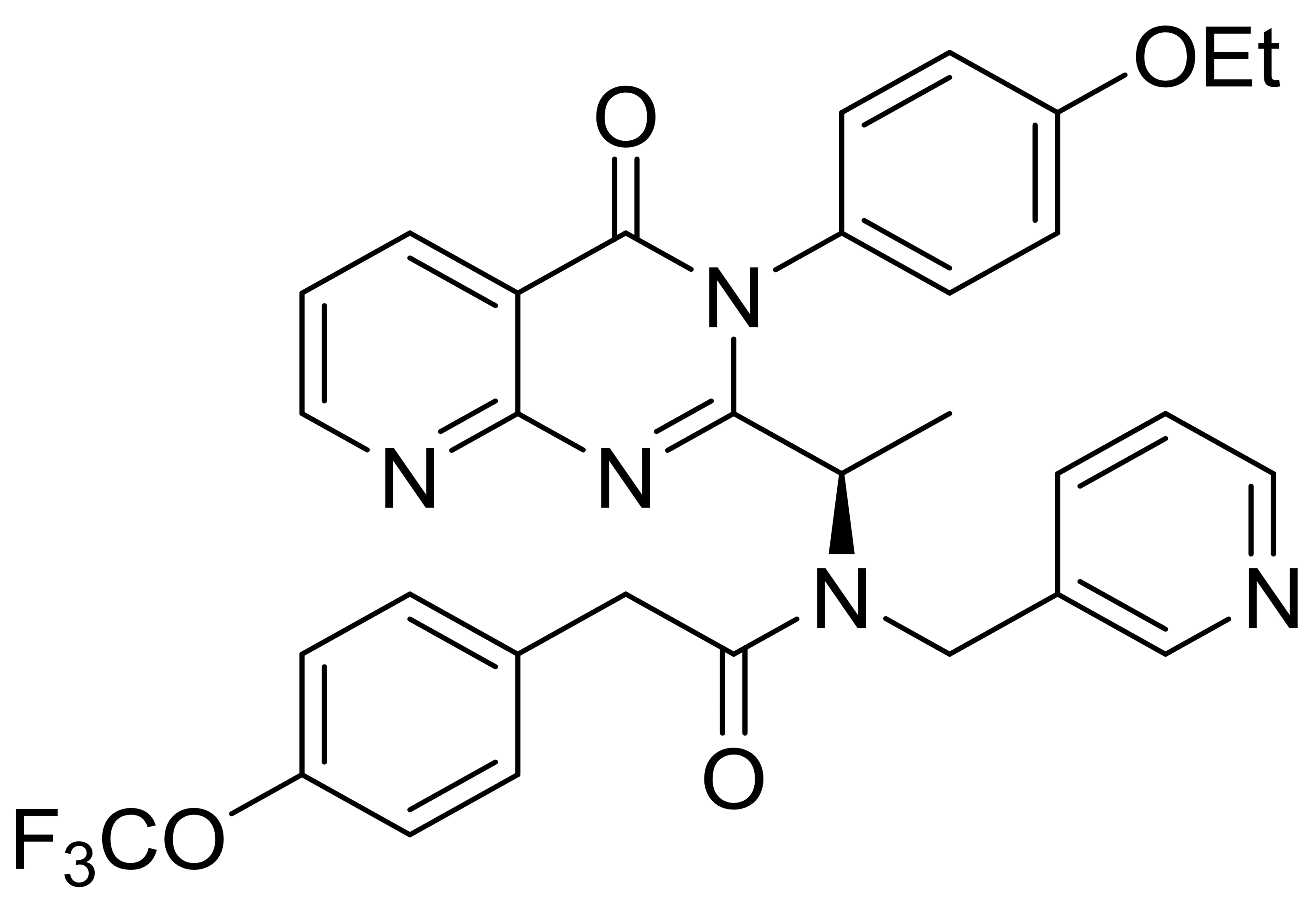 |
603.6 | 10 | 0 | 10 | 4 | [7, 38, 39] |
| NBI-74330 | 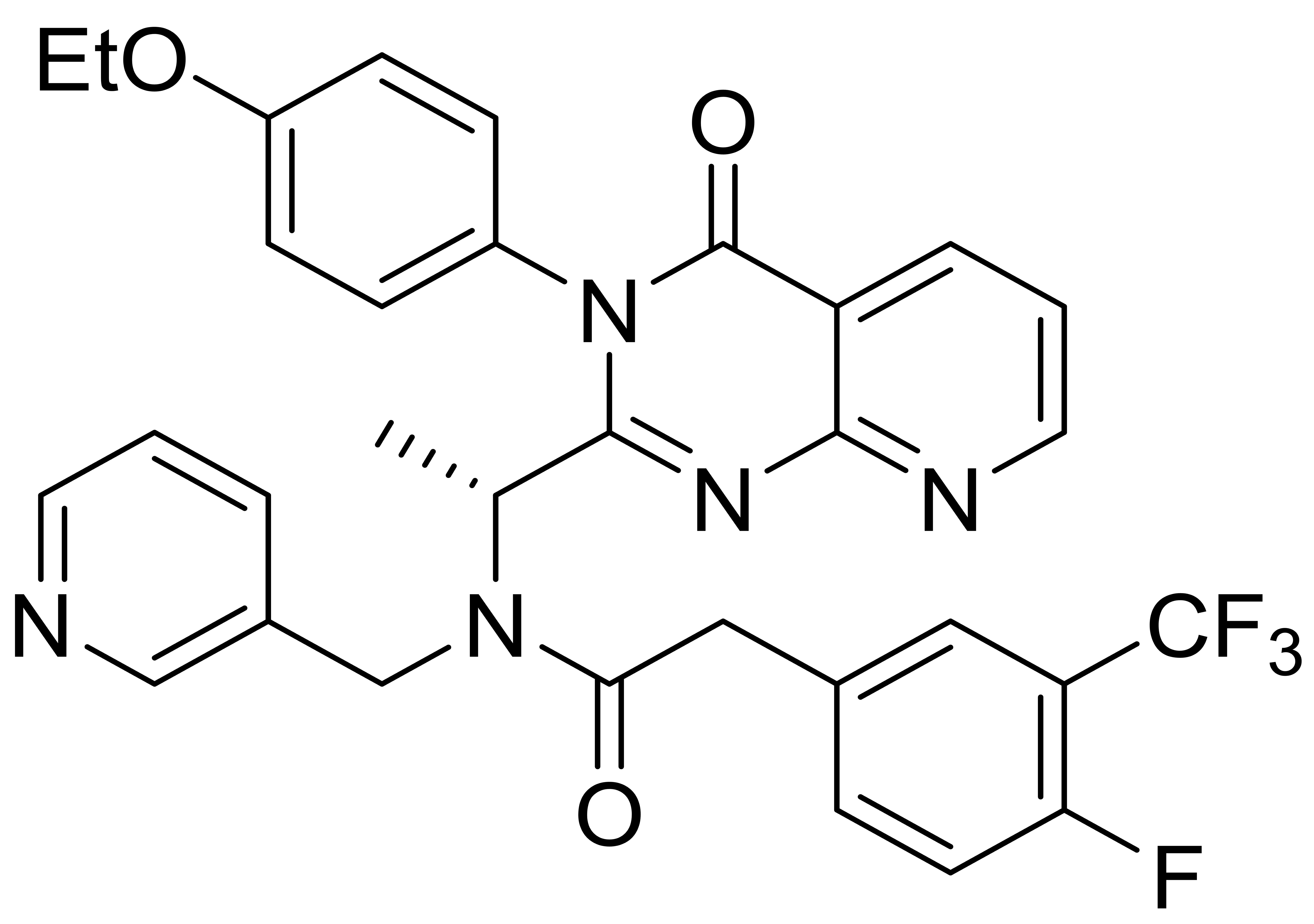 |
605.6 | 10 | 0 | 9 | 4 | [40, 41] |
| TAK-779 | 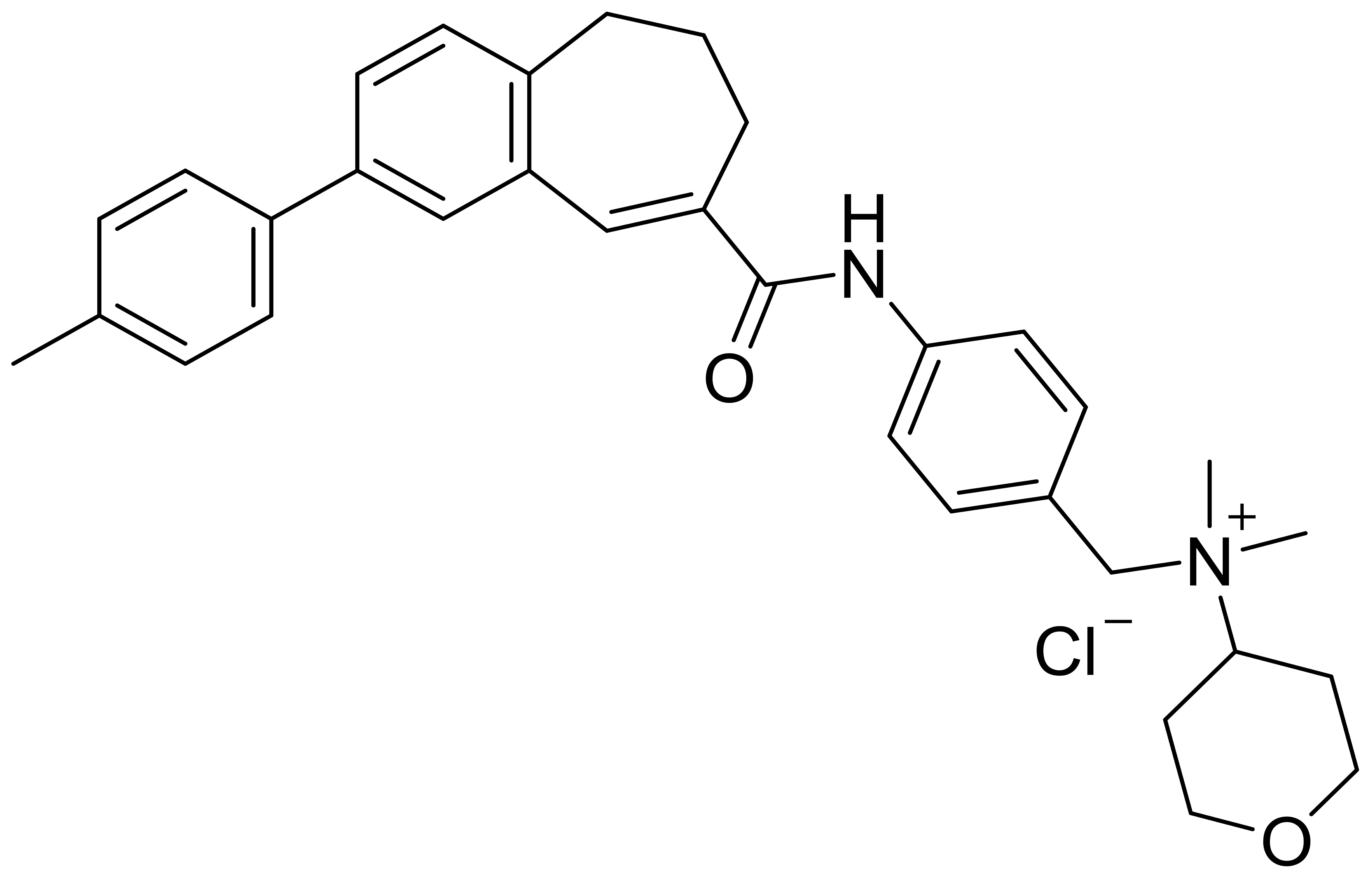 |
531.1 | 3 | 1 | 6 | 3 | [42, 43, 44, 45] |
| SCH 546738 | 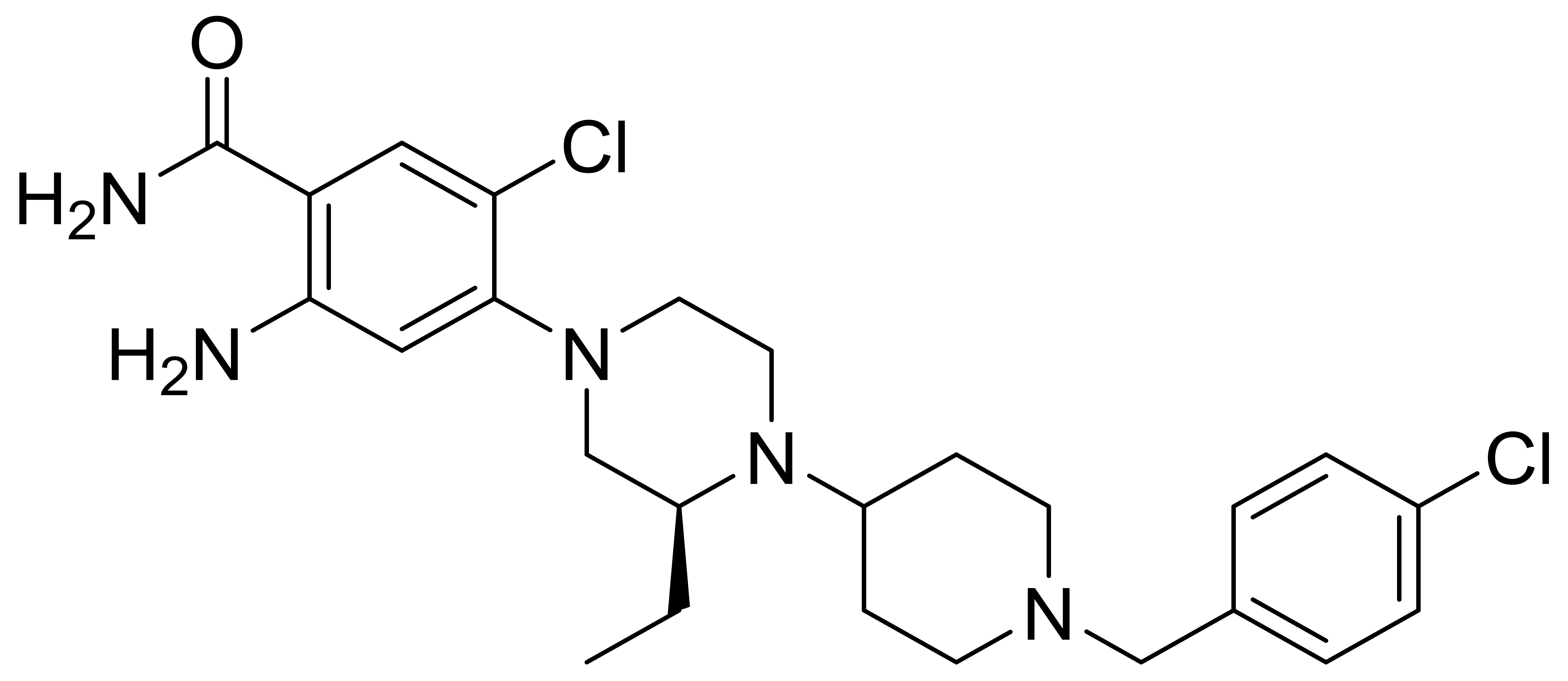 |
492.4 | 7 | 2 | 6 | 2 | [40, 46] |
| C1 | 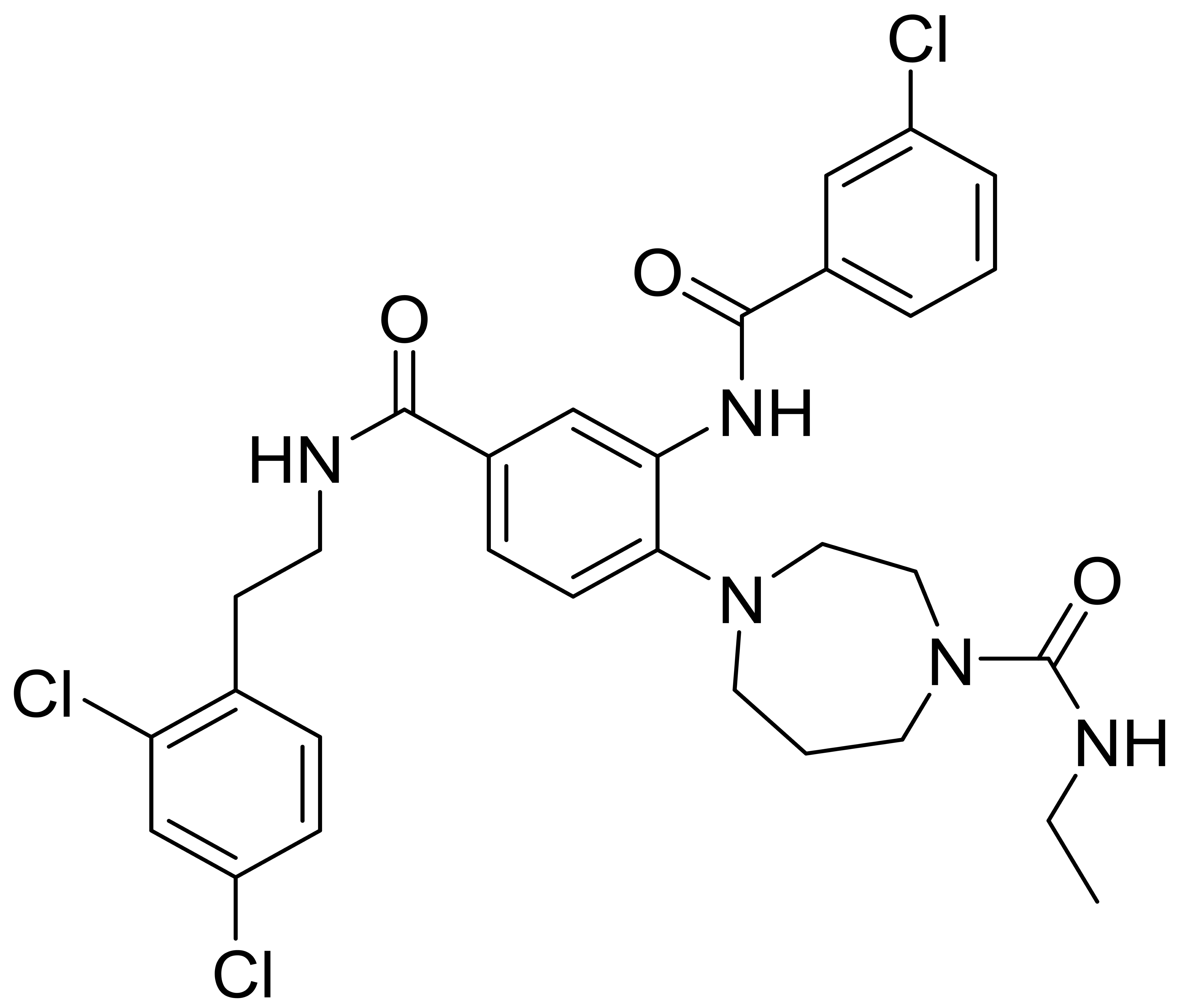 |
617.0 | 4 | 3 | 8 | 3 | [33] |
| C2 | 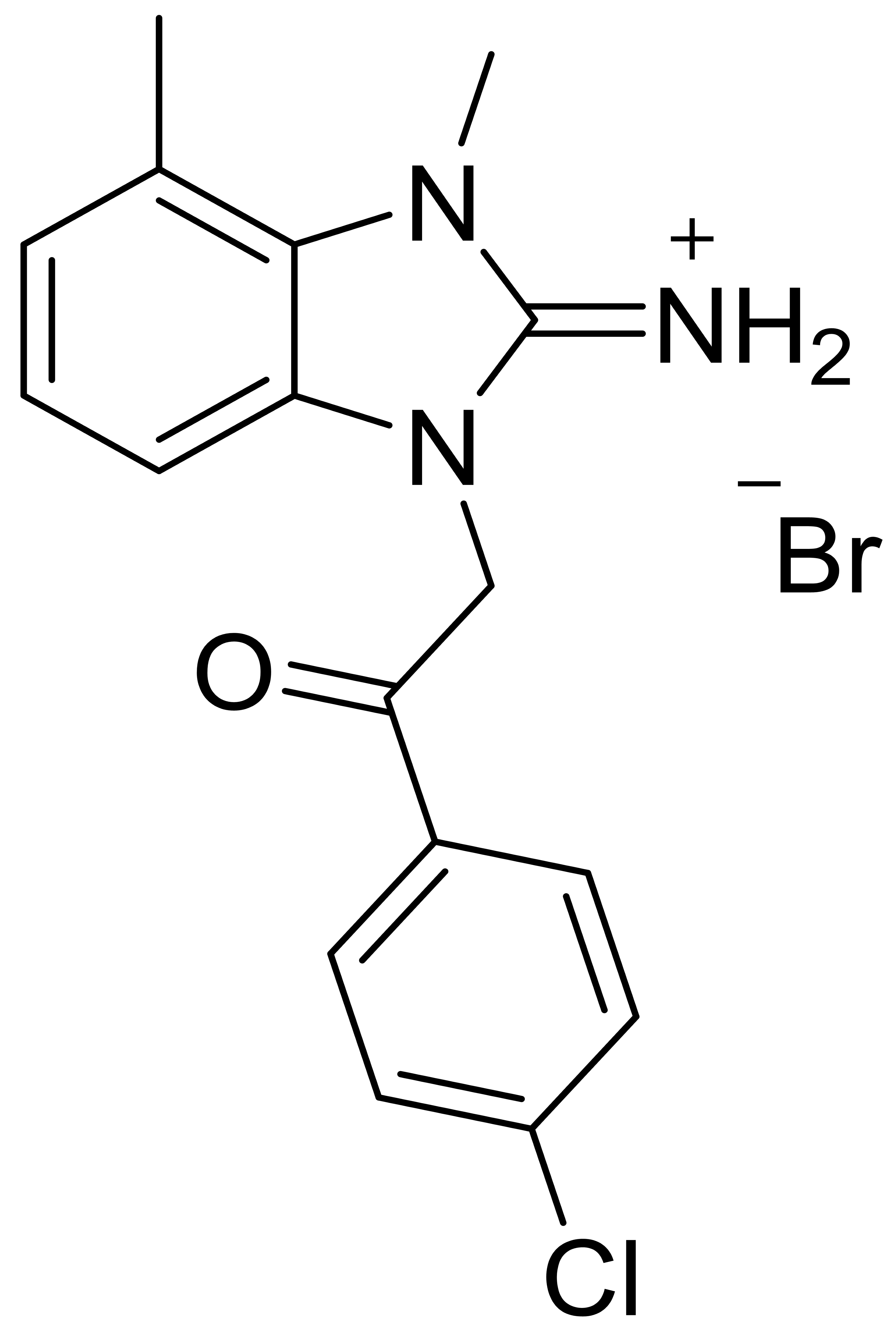 |
394.7 | - | 1 | - | 2 | [34] |
| C3 | 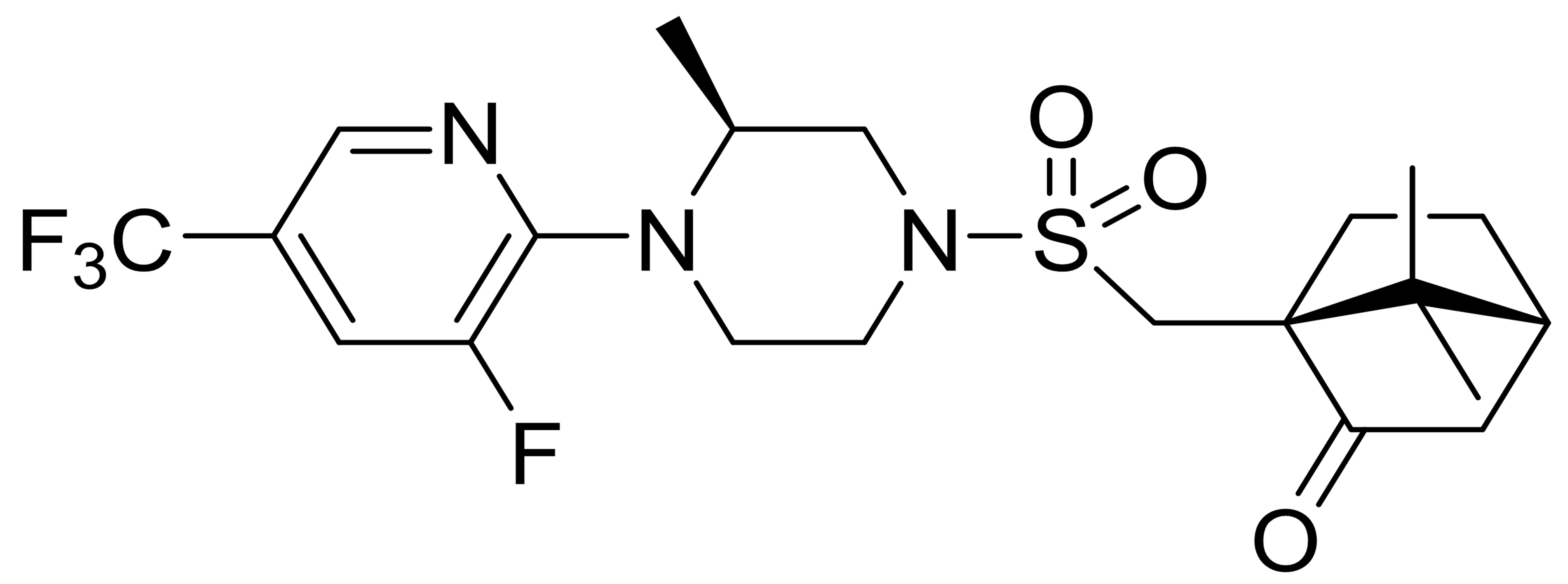 |
477.5 | - | 1 | - | 1 | [35] |
| C4 | 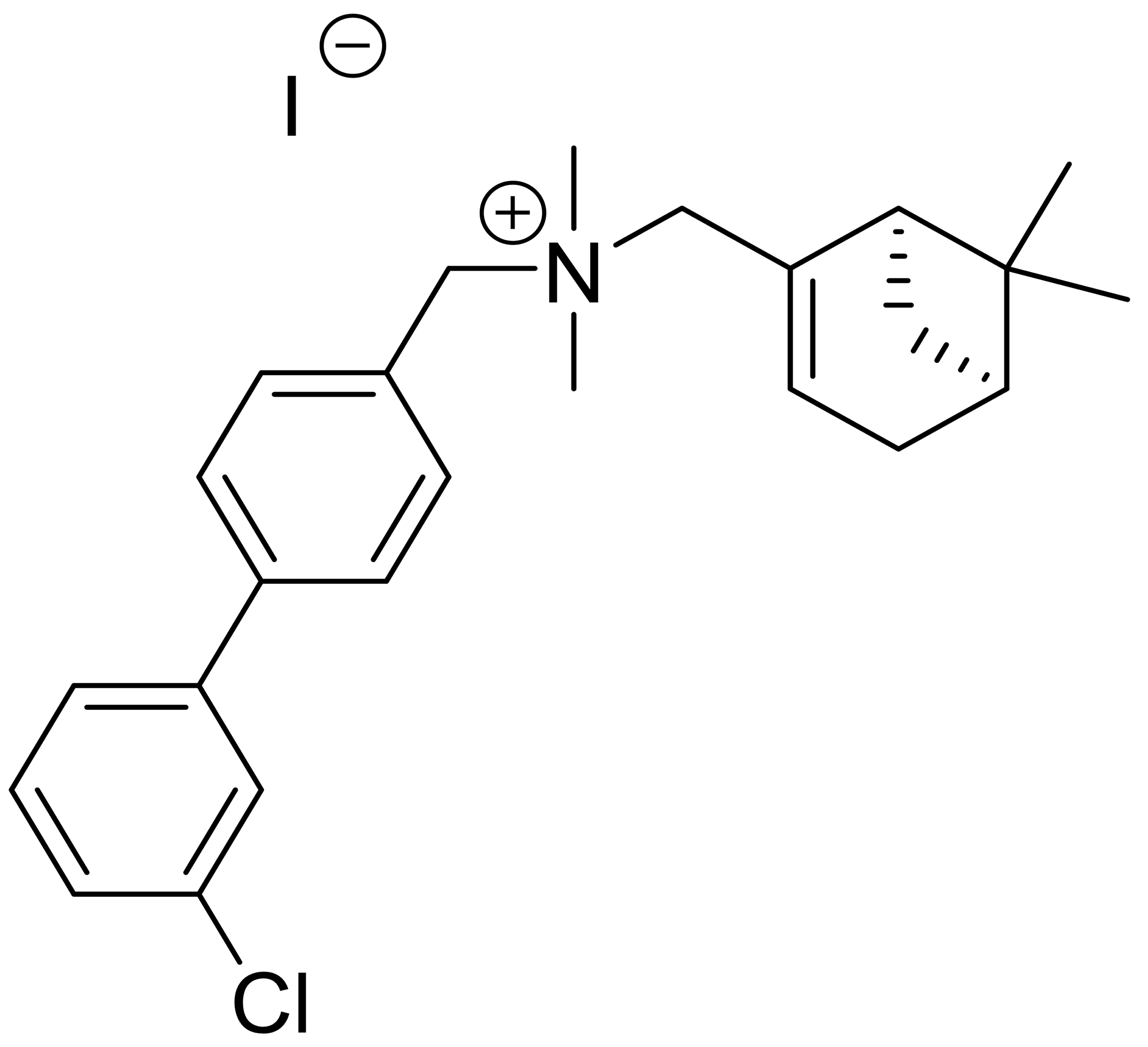 |
507.9 | - | 0 | - | 2 | [36] |
| C5 | 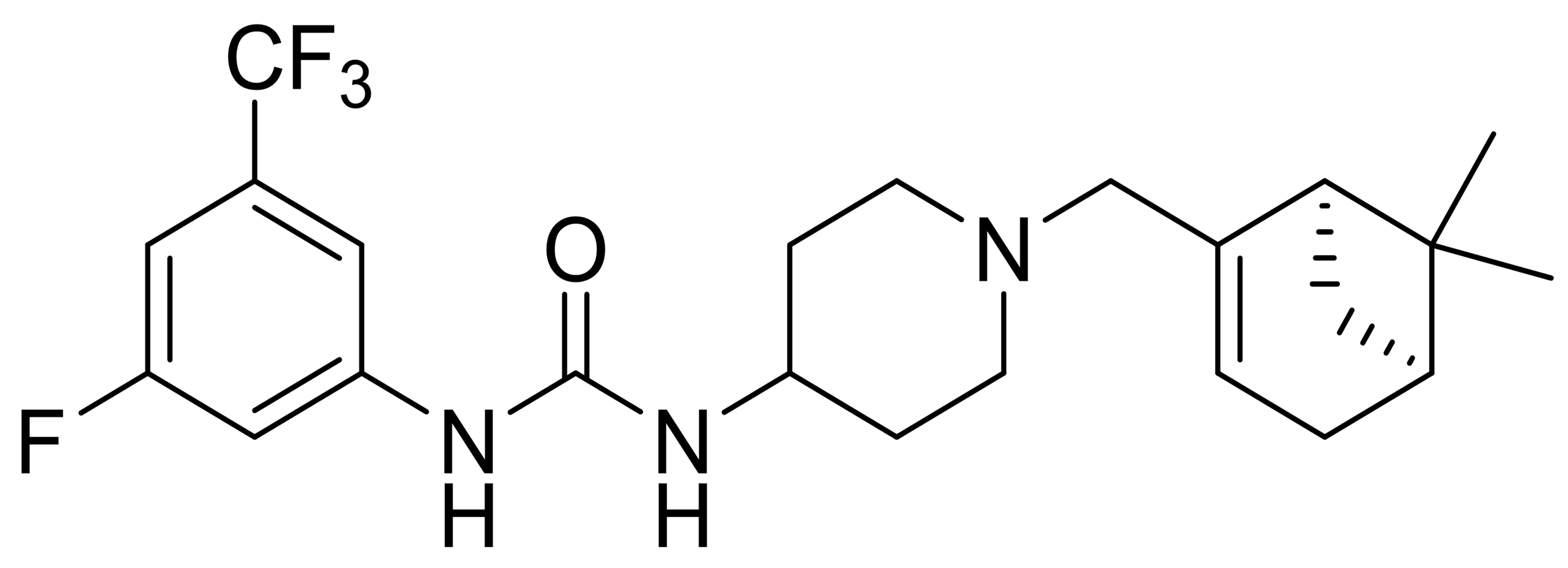 |
439.5 | - | 2 | - | 1 | [37] |
MW, Molecular weight; HBA, hydrogen bond acceptor; HBD, hydrogen bond donor; RB, rotatable bond; AR, aromatic rin.
Multiple CXCR3 antagonists have shown promising therapeutic potential in mitigating inflammatory responses. However, there is currently a lack of relevant research elucidating the mechanisms of action of these CXCR3 antagonists in ALI or conducting a comparative analysis of their effects. This study selected nine CXCR3 antagonists for comparative research, systematically investigating the regulatory effects of CXCL10 and these nine antagonists on macrophage function, while also evaluating their intervention effects on lipopolysaccharide LPS induced ALI in mice. Furthermore, molecular docking technology was employed to analyze the binding interactions between the nine CXCR3 antagonists and the CXCR3 protein, predicting novel structures with more stable binding affinities. This approach may provide new insights and intervention strategies for the clinical treatment of ALI and ARDS.
The CXCR3 antagonists AMG 487, NBI-74330, TAK-779, and SCH 546738 used in this
study were purchased from (MedChemExpress, Monmouth Junction, NJ, USA). The
compounds C1, C2, C3, C4, and C5 were synthesized based on the literature
references provided in Table 1, with specific synthetic routes detailed in
Supplementary Table 1. Solvents, reagents, and deuterated solvents were
purchased from Energy Chemical (Shanghai, China), Aladdin (Shanghai, China),
Adamas (Shanghai, China), Innochem (Beijing, China), and were used without
further purification. Chemical shifts are expressed in ppm in the
C57BL/6J mice (Qingyuan Biotech, Anhui, China) aged 6–8 weeks and weighing 18–25 g were used in this study. The mice were randomly divided into several groups: a blank control group, a lipopolysaccharide (LPS) group (Sigma, St. Louis, MO, USA), and treatment groups consisting of LPS combined with AMG 487, NBI-74330, TAK-779, SCH 546738, C1, C2, C3, C4, and C5. The number of mice receiving individual drugs or drug combinations was in each case 6 (n = 6 per group). This study was performed following the China Council on Animal Care and Protocol guidelines. All animal studies were conducted in accordance with the guidelines approved by the Ethics Committee for Animal Experiments of Bengbu Medical University (approval number 2021-003) and all applicable institutional and governmental regulations concerning the ethical use of animals were followed.
The control and experimental groups received airway injections of phosphate-buffered saline (PBS) (Servicebio, Hubei, China) and lipopolysaccharide (LPS) (10 ng/g) (Sigma, MO, USA), respectively. After 30 h, the mice were anesthetized with tribromoethanol (20 µL/g) (Shanghai Dowobio Biotechnology, Shanghai, China). The mice were subsequently euthanized via cervical dislocation, and lung tissues were harvested. The other experimental groups were administered LPS followed by AMG 487 (5 mg/kg), NBI-74330 (5 mg/kg), TAK-779 (10 mg/kg), SCH 546738 (10 mg/kg), C1 (5 mg/kg), C2 (5 mg/kg), C3 (5 mg/kg), C4 (5 mg/kg), and C5 (5 mg/kg). After 24 h, these mice were also euthanized by cervical dislocation, and their lung tissues were collected.
The mouse ALI model and treatment group were established as previously described. Mice were deeply anesthetized with tribromoethanol (20 µL/g) (Shanghai Dowobio Biotechnology, Shanghai, China), after which the trachea was incised, and a tracheal tube was inserted. Lung function changes in the mice were assessed using a lung function ventilator (SCIREQ Scientific Respiratory Equipment Inc., Montreal, Quebec, Canada) and Flexware version 7.6.4 software.
Following thoracotomy, the mouse trachea was incised, and the BALF was collected
by repeatedly rinsing the lungs with PBS. The samples were centrifuged at 1000
Mouse mononuclear macrophages (RAW264.7) were cultured in Gibco Dulbecco’s Modified Eagle’s Medium (DMEM) (Thermo Fisher Scientific, Waltham, MA, USA), supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin solution (Biosharp, Anhui, China). The cells were incubated at 37 °C in a 5% CO2 atmosphere for 48 h. All cell lines were validated by STR profiling and tested negative for mycoplasma.
RAW264.7 cells were seeded into 96-well plates (1
The RAW264.7 cells were stained with 1% neutral red staining solution (Solarbio, Beijing, China). RAW264.7 cells were stained, incubated for 2 h and washed thrice using PBS solution. Cell decolorization solution was prepared by glacial acetic acid and anhydrous ethanol in the ratio of 1:1 to decolorize the stained cells, and the cells were shaken at 4 °C overnight. The absorbance at 540 nm wavelength was detected using an enzyme labeler (Thermo Fisher Scientific, Waltham, MA, USA).
Mouse lung tissues and cells were lysed using a radioimmunoprecipitation assay
(RIPA) lysis solution (Beyotime, Shanghai, China), and the lung tissues were
ground with a grinder (Servicebio, Wuhan, China). After sufficient lysis,
centrifugation was performed at 4 °C for 10 min at 12,000 rpm, and the
supernatant was collected. Protein quantification was conducted using a
bicinchoninic acid (BCA) protein concentration assay kit (Beyotime, Shanghai,
China). Proteins were separated via 7.5% sodium dodecyl sulfate-polyacrylamide
gel electrophoresis and subsequently transferred onto a polyvinylidene difluoride
membrane. Blocking was performed with 5% skimmed milk for 2 h. The membranes
were then incubated with primary antibodies at 4 °C overnight. After
washing three times with tris-buffered saline containing Tween 20 (TBST) buffer
(15 min per wash), the membranes were incubated with secondary antibodies at room
temperature for 2 h. Following another three washes with TBST buffer (15 min per
wash), the membranes were visualized using the Extremely Ultrasensitive ECL
Chemiluminescence Kit (Beyotime, Shanghai, China), and protein signals were
detected using the Tanon 5200 (Shanghai Tanon Life Science, Shanghai, China).
ImageJ software was utilized to analyze the grayscale images. The primary
antibodies used were glyceraldehyde-3-phosphate dehydrogenase (GAPDH),
Total RNA was extracted from cells using TRIZOL (Biosharp, Anhui, China) and quantified with a Nanodrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). Subsequently, 1 µL of total RNA was reverse transcribed into single-stranded cDNA at 25 °C for 10 min, followed by 42 °C for 60 min, 80 °C for 10 min, and finally held at 4 °C for 10 min, using a first-strand cDNA synthesis kit (Beyotime, Shanghai, China). PCR was conducted using SYBR-Green PCR Master Mix (Biosharp, Anhui, China), along with specific primers, on a Light Cycler 480 real-time PCR system (Roche Applied Science, Mannheim, Germany). A three-step cycling scheme was employed for gene amplification: 95 °C for 2 min, followed by 95 °C for 10 sec, 58 °C for 30 sec, and 72 °C for 10 sec. The expression levels of specific genes were normalized to that of GAPDH (internal reference), and the fold change was calculated using the 2-ΔΔCt method. The sequences of the gene primers are presented in Table 2.
| Gene | Forward sequence (5′-3′) | Reverse sequence (5′-3′) |
| GAPDH | GGCCTTCCGTGTTCCTAC | TGTCATCATATCTGGCAGGTT |
| TNF- |
CATCTTCTCAAAATTCGAGTGAC | TGGGAGTAGACAAGGTACAACCC |
| IL-1 |
TGGAAAAGCGGTTTGTCTTC | TACCAGTTGGGGAACTCTGC |
| IL-6 | TAGTCCTTCCTACCCCAATTTCC | TTGGTCCTTAGCCACTCCTTC |
| CD86 | TGTTTCCGTGGAGACGCAAG | TTGAGCCTTTGTAAATGGGCA |
| CD206 | CTTCGGGCCTTTGGAATAAT | TAGAAGAGCCCTTGGGTTGA |
| Arg1 | CTCCAAGCCAAAGTCCTTAGAG | AGGAGCTGTCATTAGGGACATC |
| Mmp9 | GCCGACTTTTGTGGTCTTCC | GGTACAAGTATGCCTCTGCCA |
| Axl | TGGTGAGGGAGGAGCATGTT | AAAAGAAGGGGAGCTTGCTGA |
| Pros1 | CGCCGTGCAAATACCTTGTT | AATGAGCCAACACGGAATGC |
| GAS6 | TGCTGGCTTCCGAGTCTTC | CGGGGTCGTTCTCGAACAC |
RAW264.7 cells were inoculated onto cell crawlers and incubated for 24 h following 6 h of treatment according to the experimental groups. After three washes with PBS, the cells were fixed in a 4% paraformaldehyde solution for 15 min and subsequently washed three times with PBS. The cells were permeabilized with 0.5% Triton at room temperature for 15 min, followed by three washes in PBS. The cells were incubated for 6 h, and then for an additional 24 h as per the experimental groupings. Blocking was performed using a blocking solution (Beyotime, Shanghai, China) for 30 min, after which the cells were incubated with primary antibodies at 4 °C overnight. The cells were then washed three times with PBS containing Tween 20. Following this, the cells were incubated with a fluorescent secondary antibody (Thermo Fisher Scientific, Waltham, MA, USA) at room temperature in the dark for 2 h, and subsequently washed three times with PBS containing Tween 20. The slices were blocked with a blocking agent containing 4’,6-diamidino-2-phenylindole, and fluorescence images were captured using a Zeiss Observer Z1 inverted microscope (ZEISS, Göttingen, Germany) to obtain cell images. The average fluorescence intensity was analyzed using ImageJ software. Primary antibodies (CD86 and CD206) were sourced from AiFang Biological (Hunan, China).
Mouse lung tissues were fixed in 5% paraformaldehyde, processed, and embedded
in paraffin blocks according to a standard protocol. Tissue sections, each with a
thickness of 5 µm, were stained with hematoxylin and eosin. Lung injury was
evaluated based on the following criteria: alveolar hemorrhage and congestion,
alveolar edema, infiltration or aggregation of neutrophils in the alveolar or
vascular regions, and thickening of the alveolar septa. A five-point scale was
utilized for scoring: 0 (no lesions or very mild lesions) to 1 (75% lesion
extent). The scores were aggregated and reported as the median
The corresponding crystal structure of the CXCR3 protein was obtained from the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB ID: 8HNM). The protein crystals underwent preprocessing, which included regenerating ligand states, optimizing hydrogen bond assignments, minimizing protein energy, and removing water, all performed using the Protein Preparation Wizard module of Schrödinger software (New York, NY, USA). The two-dimensional (2D) structural data files for AMG 487, NBI-74330, TAK-779, SCH 546738, C1, C2, C3, C4, and C5 were processed using the LigPrep module in Schrödinger software, resulting in the generation of all their three-dimensional (3D) chiral conformations. The Receptor Grid Generation module in Schrödinger software was employed to define the most appropriate enclosing box that perfectly encapsulates the ligand molecules within the 8HNM crystals. Consequently, the active site of the CXCR3 protein was identified. Extra Precision (XP) docking was utilized to molecularly dock the nine processed ligand compounds with the CXCR3 protein’s active site. A lower XP score indicates a reduced free energy of binding between the compound and the protein, which correlates with increased binding stability. The XP docking results are evaluated based on the XP GScore, where a value less than –6 signifies stable binding between the ligand and the protein. The active sites of the nine ligand compounds and the CXCR3 protein were further analyzed using molecular mechanics with Generalized Born and Surface Area Solvation (MM-GBSA) calculations, referencing MM-GBSA dG Bind. A value less than –30 kcal/mol suggests that the binding free energy of small molecules to proteins is low, indicating stable binding between the ligands and proteins.
The 2D structural data file of the TAK-779 compound was processed, and all its 3D chiral conformations were generated using the LigPrep module in Schrödinger software. The R-Group Enumeration module was employed to structurally modify one site of the TAK-779 compound using 13 structural modification libraries (Table 3). The best binding site was predicted using the SiteMap module in Schrödinger software, and subsequently, the Receptor Grid Generation module was utilized to deifne the most suitable Enclosing box to perfectly wrap the predicted binding site. Based on this, the active pocket of the protein was identified. Standard precision (SP) and XP were empolyed to molecularly dock each modified structure with the active site of the CXCR3 protein, with the docking precision being incrementally enhanced. A lower score indicates a higher likelihood of binding between the compound and the protein. The screened modified structures were analyzed in relation to the active site of the CXCR3 protein using MM-GBSA calculations.
| Modification sites | Structural modifier library | |
 |
eMolecules R-groups (2098) | R-groups to Create Cation-Pi interaction (96) |
| Diverse R-groups (43) | R-groups to Create a Hydrogen Bond (148) | |
| Solubilizing R-groups (33) | R-groups to Create Pi-Cation Interaction (86) | |
| Ring Decorations (24) | R-groups to Create Pi-Pi Interaction (86) | |
| Aliphatic Monocyclic Rings (38) | R-groups to Create Salt Bridges (107) | |
| Aromatic Monocyclic Rings (73) | R-groups to Replace a Water (139) | |
| R-groups to Displace a Water (111) | ||
All values are expressed as mean
The IC50 values indicating the effects of nine CXCR3 antagonists on macrophages were determined using the CCK-8 assay (Fig. 1A–I). Treatment of RAW264.7 cells with CXCL10 (300 ng/mL) for 24 h resulted in increased in absorbance and a rise in intracellular neutral red particles within the macrophages. CXCL10 enhanced macrophage phagocytosis (Fig. 2A). The addition of each of the nine CXCR3 antagonists resulted in a reduction in macrophage absorbance, with all treatments significantly suppressing macrophage phagocytosis by over 50%. Among these antagonists, C5 exhibited the most potent inhibition, achieving a 69% reduction (Fig. 2B). CXCL10 upregulated the mRNA expression of efferocytosis-associated molecules Axl, Pros1, and GAS6 (Fig. 2C), while also promoting the protein expression of Axl and Pros1 (Fig. 2D). All nine antagonists significantly downregulated the mRNA expression of Axl and Pros1 (Fig. 2E). Western blot analysis confirmed that these antagonists inhibited the protein expression of both Axl and Pros1 (Fig. 2F,G).
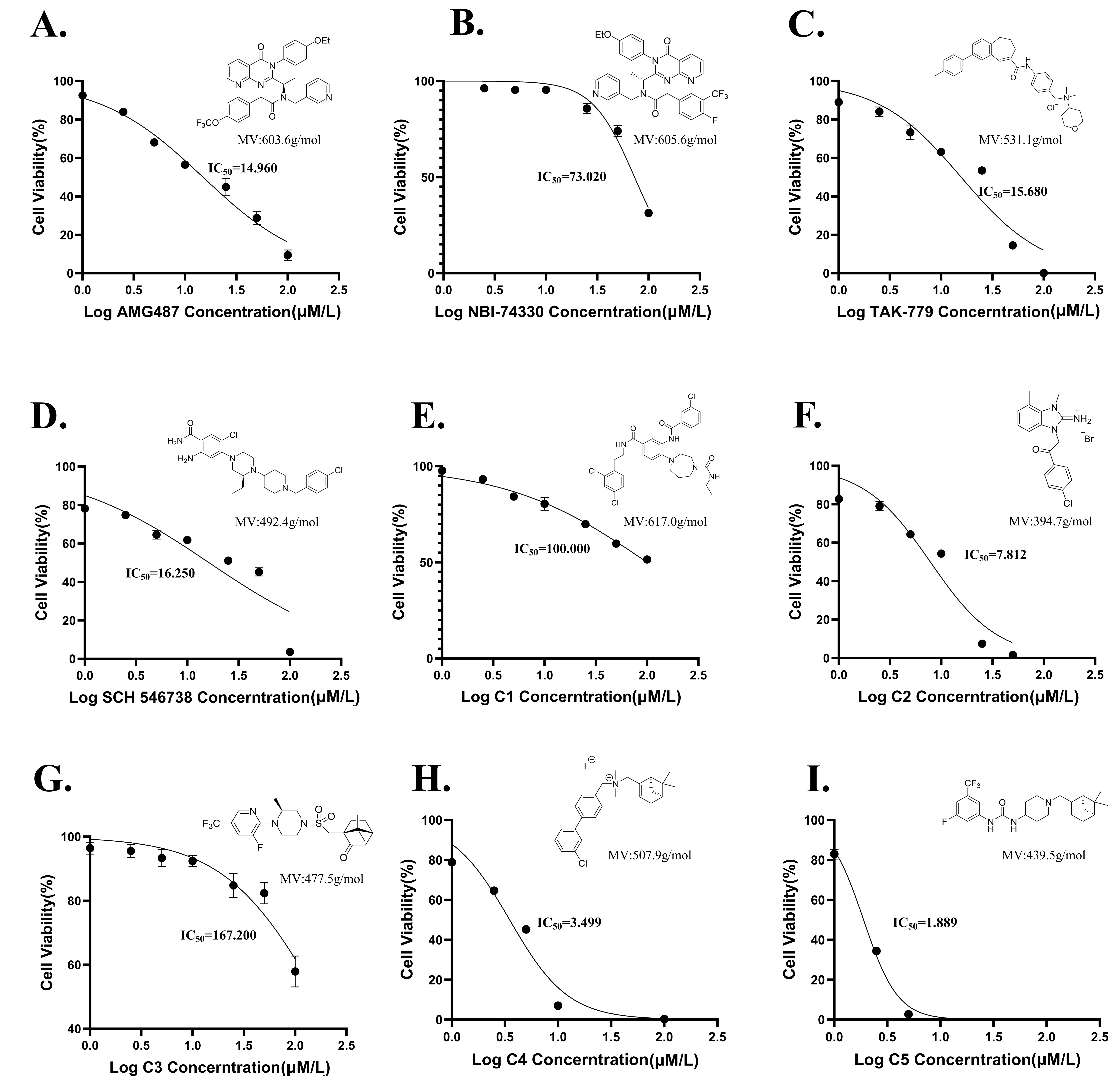 Fig. 1.
Fig. 1.
IC50 values indicating the effects of nine CXCR3 antagonists. (A–I) CCK-8 assay detected the IC50 values indicating the effects of nine CXCR3 antagonists in RAW264.7 cells. Treatment with 14.960 µM AMG 487, 73.020 µM NBI-74330, 15.680 µM TAK-779, 16.250 µM SCH 546738, 100.000 µM C1, 7.812 µM C2, 167.200 µM C3, 3.499 µM C4, and 1.889 µM C5 induced 50% growth inhibition in RAW264.7 cells. CXCR3, C-X-C motif chemokine receptor 3; CCK-8, Cell Counting Kit-8.
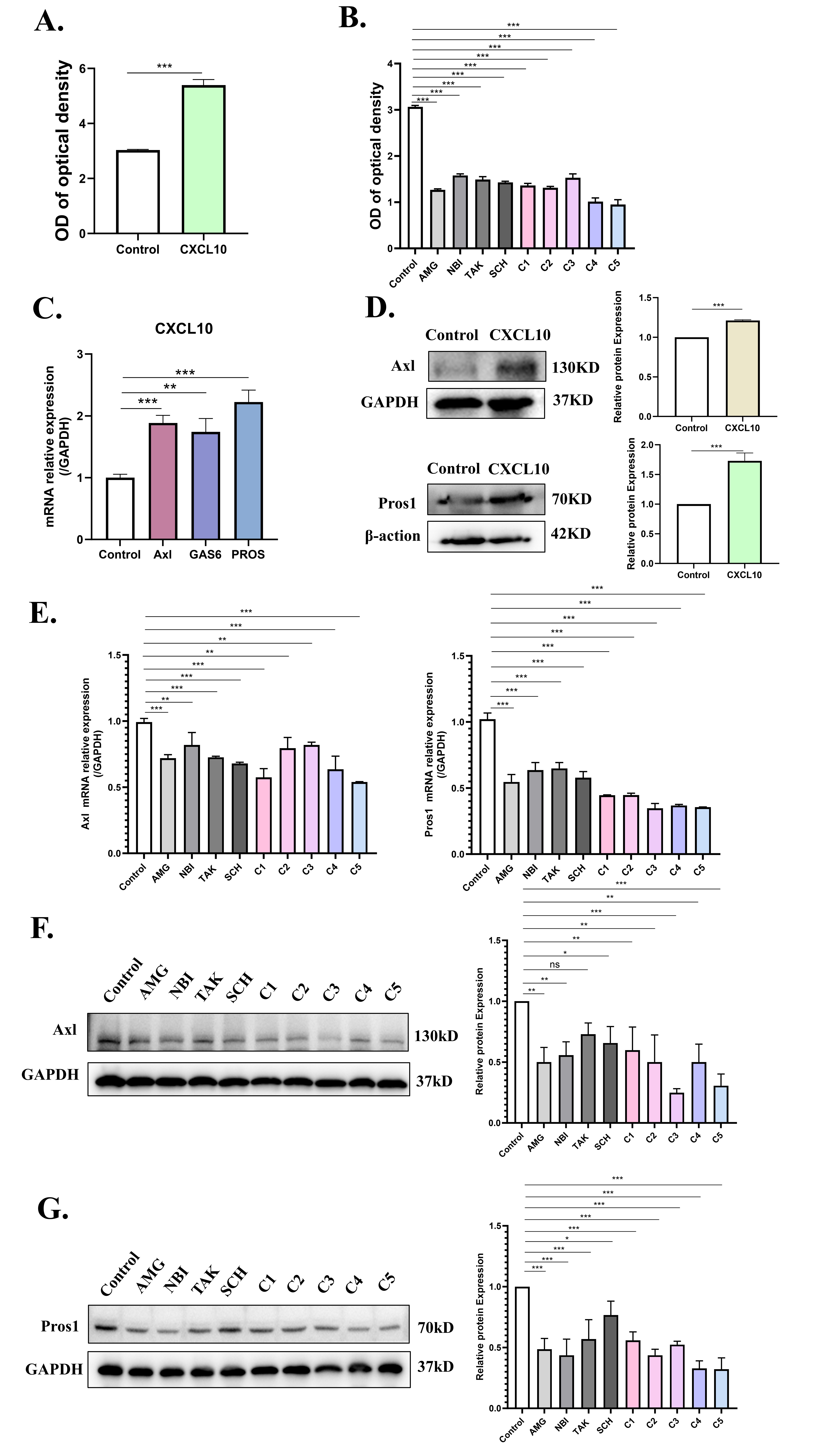 Fig. 2.
Fig. 2.
CXCL10-CXCR3 axis regulates phagocytosis in RAW264.7 cells. (A)
Neutral red phagocytosis test detected the phagocytic ability of RAW264.7 cells
after CXCL10 stimulation. (B) Neutral red phagocytosis test detected the
phagocytic ability of RAW264.7 cells treatment with nine CXCR3 antagonists. (C)
The mRNA expression of Axl, GAS6 and Pros1 in RAW264.7 cells after CXCL10
treatment was detected by RT-PCR. (D) Western blotting analysis of Axl and Pros1
proteins in RAW264.7 cells after CXCL10 treatment. (E) The mRNA expression of Axl
and Pros1 in RAW264.7 cells after treatment with nine CXCR3 antagonists was
detected by RT-PCR. (F) Western blotting analysis of Axl proteins in RAW264.7
cells after treatment with nine CXCR3 antagonists. (G) Western blotting analysis
of Pros1 proteins in RAW264.7 cells after treatment with nine CXCR3 antagonists.
ns, not significantly different; *p
In LPS-induced M1 macrophages responding to CXCL10, mRNA expression levels of M1
polarization markers (TNF-
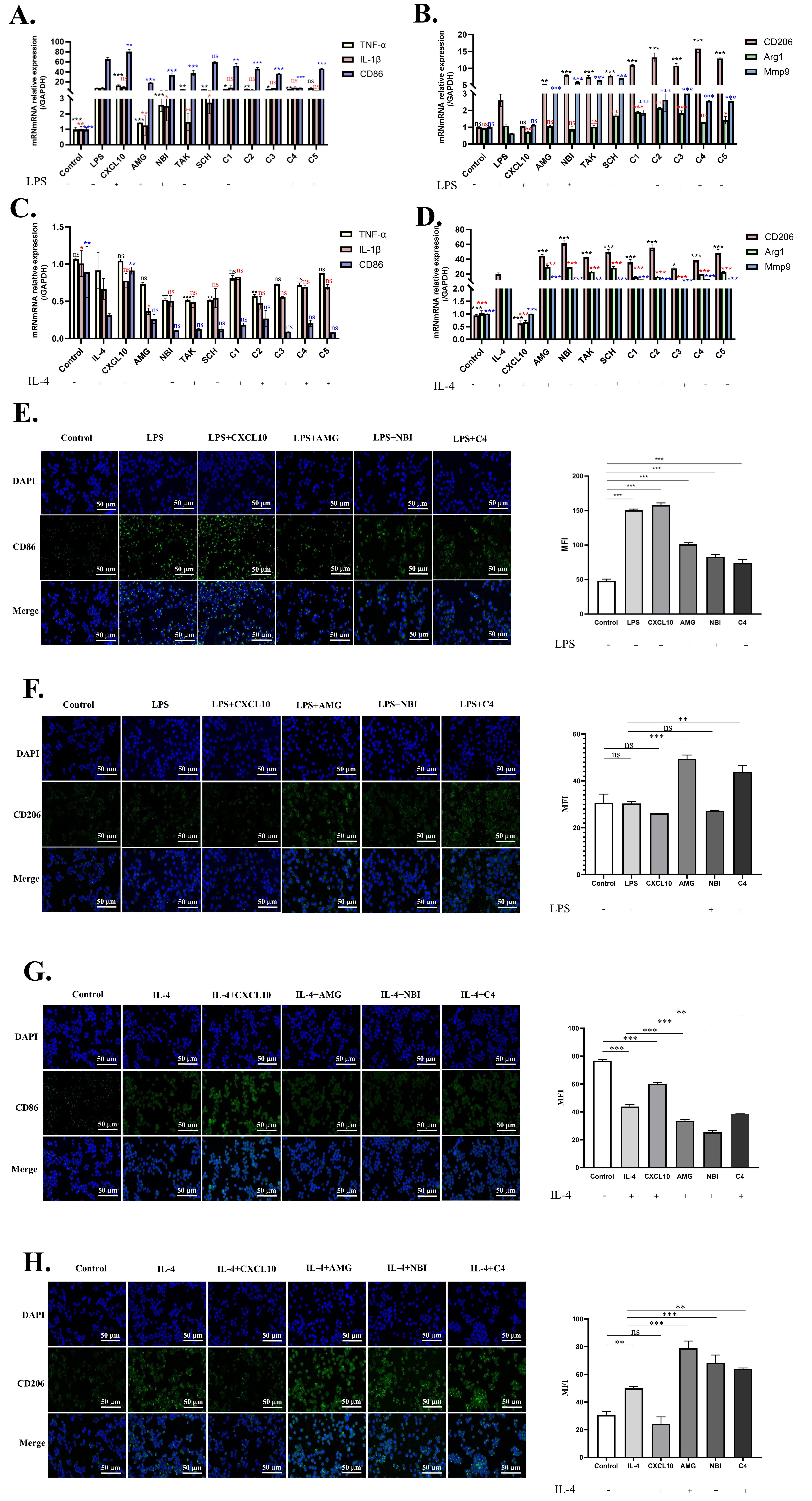 Fig. 3.
Fig. 3.
CXCL10-CXCR3 axis regulates macrophage polarization. (A) LPS
(100 ng/mL) treated with RAW264.7 cells, TNF-
In mice with LPS induced ALI, hematoxylin and eosin staining revealed
significant structural damage to lung tissue, characterized by thickening of the
alveolar walls and diaphragm, as well as pronounced alveolar congestion
accompanied by marked infiltration of inflammatory cells and airway hemorrhage.
Following the administration of CXCL10, alveolar congestion intensified, and the
infiltration of inflammatory cells continued to increase. All nine CXCR3
antagonists effectively mitigated LPS-induced pathological damage in ALI,
evidenced by a reduction in inflammatory cell infiltration, improvement in
pulmonary hemorrhage and congestion, and preservation of damaged alveolar
structures (Fig. 4A). The wet-to-dry (W/D) ratios of mouse lung tissue were
utilized to assess the extent of pulmonary edema. The W/D ratio was significantly
elevated in the LPS group and was subsequently reduced by all nine CXCR3
antagonists (Fig. 4B). BALF analysis revealed that the administration of LPS
resulted in an increase in the total protein content of alveolar lavage fluid in
mice, while treatment with the nine CXCR3 antagonists led to a decrease in this
total protein content (Fig. 4C). Additionally, RT-PCR results indicated that LPS
exposure elevated the mRNA expression levels of inflammatory cytokines
TNF-
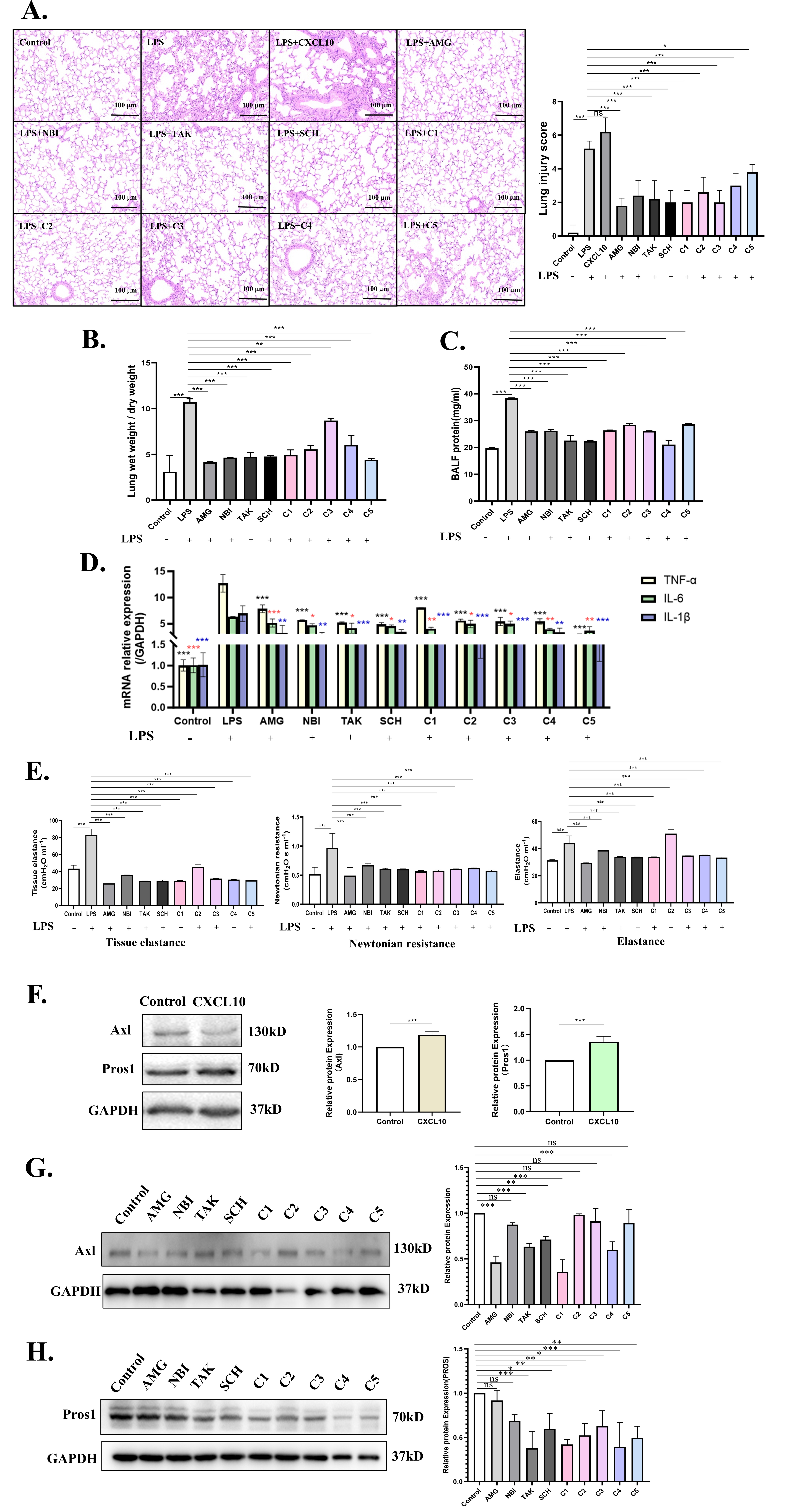 Fig. 4.
Fig. 4.
Nine CXCR3 antagonists alleviate LPS-induced acute lung injury.
(A) LPS and treated with CXCL10 and nine CXCR3 antagonists for 24 h. Mouse lung
tissues collected for HE stains and lung injury score. scale bar = 100 µm.
(B,C) Mouse lung tissues examined for lung wet-to-dry weight ratio and BALF
protein. (D) TNF-
The molecular docking scores of the antagonists TAK-779, C4, C1, NBI-74330, and C5 with CXCR3 were found to be –11.110, –10.351, –10.040, –9.713, and –8.499, respectively. Correspondingly, the results of the MM-GBSA analysis yielded values of –81.31, –74.73, –81.48, –52.34, and –56.79 kcal/mol. These low binding free energies and docking scores suggest a very stable binding of all five compounds to the CXCR3 protein. In contrast, the docking scores for the antagonists SCH 546738, AMG 487, and C2 with CXCR3 were –7.749, –7.323, and –6.406, respectively, with their corresponding MM-GBSA results being –39.89, –53.11, and –56.95 kcal/mol. These values also indicate stable binding interactions between these three compounds and the CXCR3 protein. The docking score of antagonist C3 to CXCR3 was found to be –4.596, and the MM-GBSA analysis yielded a result of –46.40 kcal/mol. These values indicate a low free energy of binding and a low docking score, suggesting that the binding of C3 to the CXCR3 protein is relatively unstable. The results of the XP and MM-GBSA analyses are presented in Table 4.
| Compound | Target | XP Gscore | MM-GBSA dG Bind (kcal/mol)2 | MM-GBSA dG Bind Coulomb1 | MM-GBSA dG Bind Covalent1 | MM-GBSA dG Bind Hbond1 | MM-GBSA dG Bind Lipo1 | MM-GBSA dG Bind Packing1 | MM-GBSA dG Bind SelfCont1 | MM-GBSA dG Bind Solv GB1 | MM-GBSA dG Bind vdW1 |
| TAK779 | –11.110 | –81.31 | 26.10 | 2.55 | –0.60 | –49.72 | –6.09 | 0.00 | –2.48 | –51.07 | |
| C4 | –10.351 | –74.73 | 25.77 | 1.43 | 0.00 | –41.86 | –6.56 | 0.00 | –0.36 | –53.14 | |
| C1 | –10.040 | –81.48 | –15.25 | 12.39 | –1.30 | –35.31 | –3.88 | 0.00 | 37.86 | –75.99 | |
| NBI74330 | –9.713 | –52.34 | –14.50 | 7.87 | –0.39 | –29.64 | –6.29 | 0.00 | 48.86 | –57.73 | |
| C5 | CXCR3 | –8.499 | –56.79 | –10.36 | 3.29 | –0.10 | –31.41 | –2.43 | 0.00 | 31.49 | –47.272 |
| SCH546738 | –7.749 | –39.89 | 4.27 | 11.34 | –2.12 | –26.65 | –3.40 | 0.00 | 7.66 | –30.99 | |
| AMG487 | –7.323 | –53.11 | 0.49 | 3.98 | –0.34 | –22.90 | –3.80 | 0.00 | 30.90 | –61.42 | |
| C2 | –6.406 | –56.95 | 73.35 | 1.41 | –0.53 | –20.00 | –6.88 | 0.00 | –69.55 | –34.75 | |
| C3 | –4.592 | –46.40 | –8.19 | 2.83 | 0.80 | –17.34 | –0.11 | 0.00 | 19.03 | –41.82 |
1Coulomb energy, Covalent binding energy, Hydrogen-bonding energy,
Lipophilic energy,
2MM-GBSA dG Bind = MM-GBSA dG Bind (Coulomb + Covalent + Hbond + Lipo + Packing + SelfCont + Solv GB + vdW).
The molecular docking 2D and 3D maps demonstrated that all nine CXCR3 antagonists penetrated deeply into the active pocket of the CXCR3 protein (Fig. 5A–I). The residues of the CXCR3 protein established hydrophobic interactions with the nine antagonists. Notably, residue Tyr308 contributed hydrophobic interactions with AMG 487 (Fig. 5A), NBI-74330 (Fig. 5B), C1 (Fig. 5E), C3 (Fig. 5G), and C5 (Fig. 5I). NBI-74330 formed a hydrogen bond with the residue asparagine (Asn) (Fig. 5B), while TAK-779 established a hydrogen bond with residue serine 304 (Ser304) (Fig. 5C). SCH 546738 formed a hydrogen bond with residue threonine 201 (Thr201) and two hydrogen bonds with residue aspartic acid (Asp) (Fig. 5D). C1 formed hydrogen bonds with residues Arg216 and cysteine 203 (Cys203) (Fig. 5E), C2 established a hydrogen bond with residue Ser304 (Fig. 5F), and C4 formed a hydrogen bond with residue glutamine 204 (Gln204) (Fig. 5G). The interpretation of the docking results is summarized in Table 5.
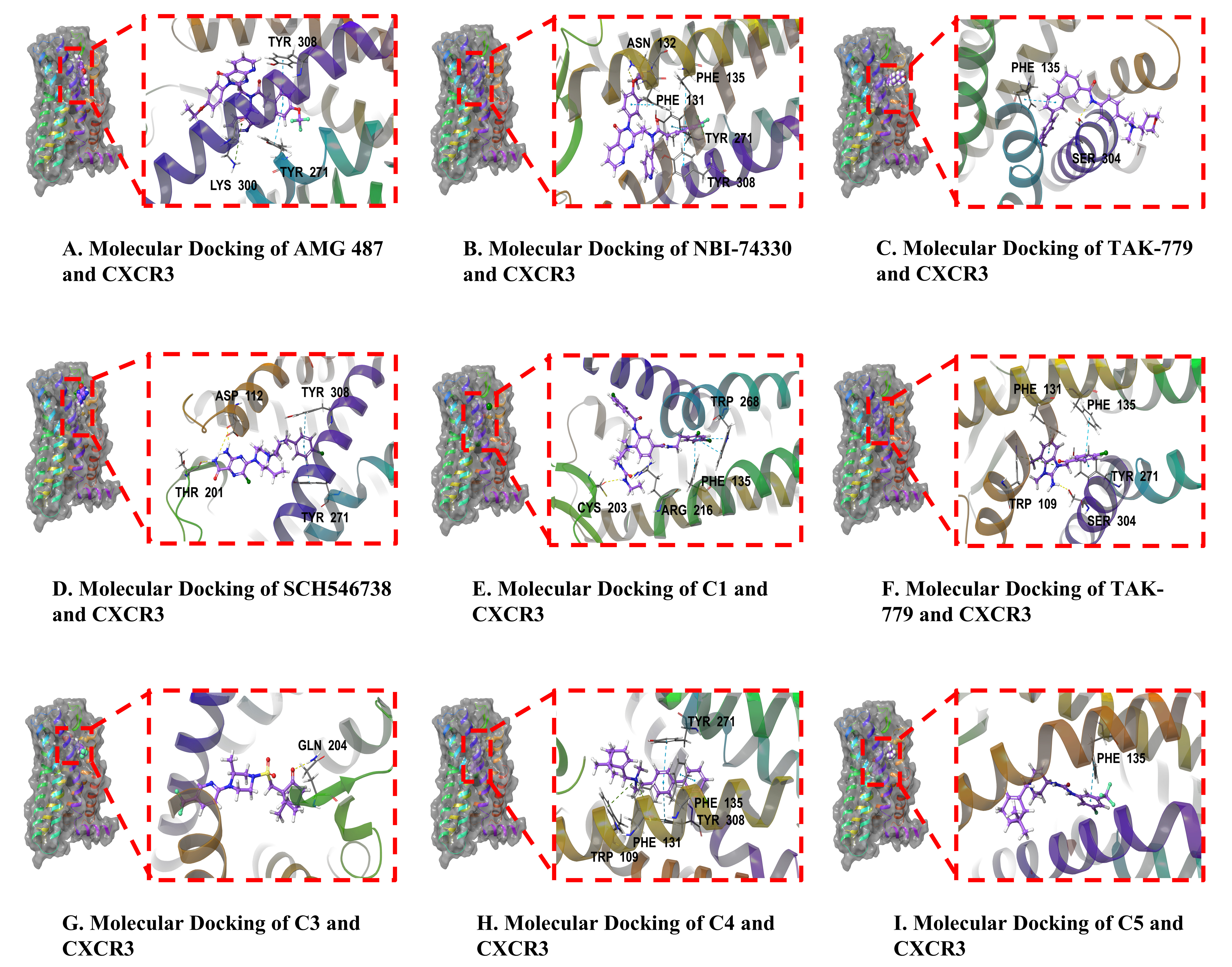 Fig. 5.
Fig. 5.
Binding mode of CXCR3 protein and nine CXCR3 antagonists. (A)
AMG 487. (B) NBI-74330. (C) TAK-779. (D) SCH 546738. (E) C1. (F) C2. (G) C3. (H)
C4. (I) C5. Yellow, blue, and green represent the hydrogen bond,
| CXCR3 antagonist | CXCR3 protein residue | Binding energy (Kcal/mol) | Hydrogen bonds | Hydrophobic interaction |
| AMG 487 | Tyr60 | –9.2 | 3.85 | Leu 56A (3.79 Å) |
| Leu102 | ||||
| Ala113 | ||||
| Tyr271 | ||||
| Lys300 | ||||
| Tyr308 | ||||
| NBI-74330 | Trp109 | –9.6 | 3.70 | Leu 56A (3.73 Å) |
| Asn132 | ||||
| Phe131 | ||||
| Phe135 | ||||
| Tyr-271 | ||||
| Tyr308 | ||||
| TAK-779 | Trp109 | –9.8 | 2.90 | Leu 49A (3.55 Å) |
| Ala110 | ||||
| Ala113 | ||||
| Ala114 | ||||
| Phe131 | ||||
| Phe135 | ||||
| Ser304 | ||||
| SCH 546738 | Trp109 | –8.7 | 3.22 | Trp 109A (3.42 Å) |
| Asp112 | ||||
| Ala113 | ||||
| Val115 | ||||
| Phe131 | ||||
| Thr207 | ||||
| Tyr271 | ||||
| Tyr308 | ||||
| C1 | Leu102 | –9.8 | 2.57 | Tyr 205A (3.55 Å) |
| Phe135 | ||||
| Cys203 | ||||
| Arg216 | ||||
| Trp268 | ||||
| Tyr308 | ||||
| Cys311 | ||||
| C2 | Trp109 | –8.7 | 2.40 | Trp 109A (3.48 Å) |
| Phe131 | ||||
| Phe135 | ||||
| Tyr271 | ||||
| Ser304 | ||||
| C3 | Tyr60 | –9.4 | 2.89 | Tyr 205A (3.55 Å) |
| Trp109 | ||||
| Ala110 | ||||
| Ala113 | ||||
| Gln204 | ||||
| Tyr308 | ||||
| C4 | Leu102 | –8.2 | - | Leu 56A (3.81 Å) |
| Trp109 | ||||
| Phe131 | ||||
| Phe135 | ||||
| Tyr271 | ||||
| Tyr308 | ||||
| Cys311 | ||||
| C5 | Leu106 | –8.9 | 1.98 | Leu 56A (3.99 Å) |
| TRP109 | ||||
| Phe-131 | ||||
| Phe135 | ||||
| Tyr-308 |
Based on the molecular docking results, TAK-779 exhibited the most stable
binding affinity to the CXCR3 protein among the nine antagonists evaluated. To
identify chemical structures that bind more stably to CXCR3, we modified a
specific site in the chemical structure of TAK-779, generating a total of 2128
new structures derived from a library of 13 structural modifications
(Supplementary Table 3). Of these, 2127 structures demonstrated the
ability to interact with the CXCR3 protein. According to the SP docking results,
270 compounds achieved a docking score of
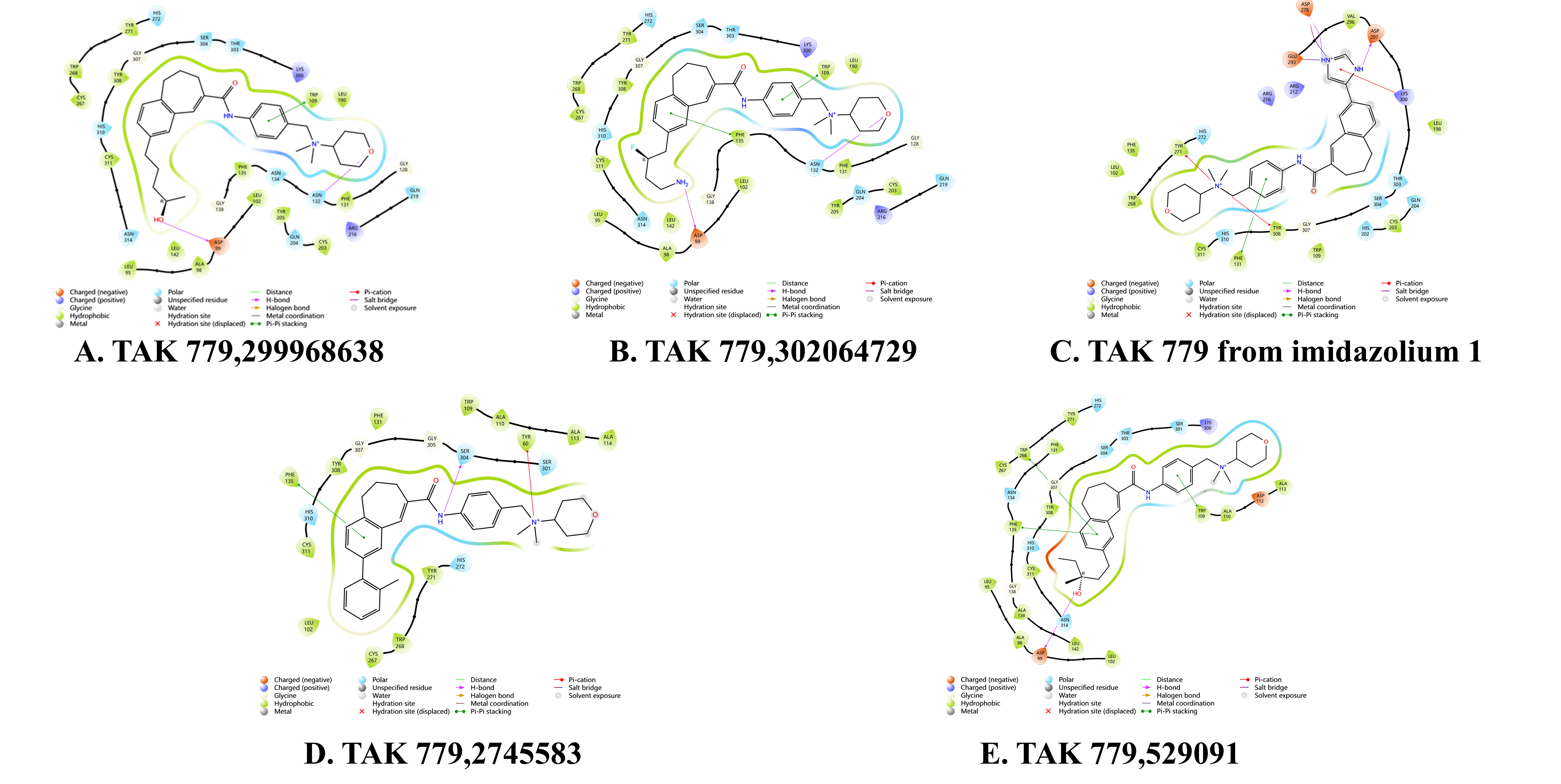 Fig. 6.
Fig. 6.
Binding mode of CXCR3 protein and five TAK-779 modifiers. (A) TAK-779,299968638. (B) TAK-779,302064729. (C) TAK-779 from imidazolium 1. (D) TAK-779,2745583. (E) TAK-779,529091.
| ID | XP GScore | MM-GBSA dG Bind (kcal/mol) |
| TAK 779,299968638 | –11.390 | –66.22 |
| TAK 779,302064729 | –11.281 | –63.43 |
| TAK 779 from imidazolium 1 | –11.250 | –87.11 |
| TAK 779,2745583 | –11.440 | –84.17 |
| TAK 779,529091 | –11.138 | –54.44 |
| TAK 779 from benzenesulfonamide 2 | –11.094 | –77.70 |
| TAK 779,40295153 | –10.982 | –73.65 |
| TAK 779,53752591 | –10.944 | –47.00 |
| TAK 779,10872246 | –10.941 | –57.36 |
| TAK 779,537068 | –10.931 | –77.29 |
| TAK 779,477532 | –10.901 | –71.75 |
| TAK 779,24179700 | –10.898 | –71.25 |
| TAK 779 piperidine | –10.852 | –53.12 |
| TAK 779,490699 | –10.852 | –73.46 |
| TAK 779,50418945 | –10.837 | –78.29 |
| TAK 779,50418945 | –10.832 | –78.54 |
| TAK 779,724169 | –10.790 | –73.02 |
| TAK 779,724169 | –10.782 | –73.27 |
| TAK 779,37212260 | –10.761 | –43.00 |
| TAK 779,260220769 | –10.756 | –75.43 |
| TAK 779,44151685 | –10.744 | –70.03 |
| TAK 779,30485508 | –10.740 | –60.07 |
| TAK 779,46013797 | –10.733 | –57.79 |
| TAK 779,6885230 | –10.723 | –44.25 |
| TAK 779 from piperidinium 1 | –10.664 | –56.99 |
| TAK 779,7334276 | –10.660 | –40.54 |
| TAK 779,34736 | –10.658 | –70.84 |
| TAK 779,2745583 | –10.635 | –82.27 |
| TAK 779,106287920 | –10.629 | –78.36 |
| TAK 779 CF2 | –10.627 | –60.73 |
Chemokines are a class of small molecular proteins primarily responsible for regulating the migration and activation of immune cells. They play significant roles in both innate and adaptive immunity and are involved in processes such as inflammatory responses, immune surveillance, tissue development and repair [34]. The chemokine receptor CXCR3 and certain selective CXCR3 antagonists exhibit varying effects under inflammatory conditions. In lung tissues of pigs infected with Porcine Reproductive and Respiratory Syndrome Virus (PRRSV), the expression levels of CXCL10 and CXCR3 are elevated. Treatment with the CXCR3 antagonist AMG 487 results in reduced infiltration of inflammatory cells in the alveoli and alleviates lung injury [7]. CXCL10 enhances the mRNA expression of efferocytosis-related molecules, including GAS6, MFGE8, PROS, and Axl, in macrophages. Conversely, the CXCR3 antagonist AMG 487 suppresses the expression of these efferocytosis-related molecules [35]. In RAW264.7 cells treated with Poly(I:C), CXCL10 induces M1-type polarization, whereas AMG 487 promotes M2-type polarization [19]. Despite the discovery of multiple CXCR3 antagonists, there are few clinical trials focusing on these agents. Currently, only the small molecule antagonist AMG 487 has advanced to Phase II clinical trials, although its efficacy remains suboptimal. Various types of CXCR3 antagonists have demonstrated therapeutic effects in animal models of inflammatory diseases. The half-maximal inhibitory concentration (IC50) reflects a drug’s inhibitory capability against a specific target or cell type, with lower values indicating stronger inhibitory potency [36]. The varying IC50 values of different antagonists on the same cell primarily highlight the differences in their efficacy. These discrepancies arise from multiple factors, including the distinct mechanisms of action of the antagonists, cellular resistance mechanisms, and the influence of experimental conditions [37, 38]. By comparing the IC50 values of various antagonists, more potent drugs can be identified. This comparison of different antagonists’ activities allows for the exploration of their structure-activity relationships, which is essential for optimizing drug design and guiding subsequent chemical modifications. Furthermore, different experimental systems may exhibit varying responses to antagonists; thus, screening a diverse range of antagonists enables the selection of the most suitable compound for a specific system, thereby enhancing the reliability and reproducibility of experiments. In this study, we selected nine CXCR3 antagonist, AMG 487, NBI-74330, TAK-779, SCH 546738, C1, C2, C3, C4, and C5, based on prior findings and relevant literature to comparatively investigate their regulatory effects on macrophage function and their roles in ALI. The selection of various antagonists for the experiments aimed to comprehensively evaluate the biological functions of the CXCR3 target and identify the most effective CXCR3 antagonist. The CCK-8 assay demonstrated that the nine antagonists inhibited the growth of RAW264.7 cells in a dose-dependent manner, exhibiting varying IC50 values.
The process by which both professional and non-professional phagocytes clear apoptotic cells (AC) is referred to as efferocytosis [23]. Macrophages, a pivotal type of phagocyte, are essential for tissue remodeling under normal physiological conditions and for the resolution of inflammation following tissue injury [25]. Furthermore, there is a functional relationship between CXCL10 and the efferocytosis of macrophages. In chronic inflammation, CXCL10 may inhibit the efferocytic function of macrophages by recruiting pro-inflammatory immune cells. This recruitment leads to the accumulation of apoptotic cells and secondary necrosis, thereby exacerbating the inflammatory response. In a mouse model of atherosclerotic plaque formation, both mRNA and protein expression levels of Axl and Pros1 were found to be decreased [39]. Macrophages eliminate apoptotic alveolar interstitial neutrophils through GAS6-dependent expression, suppressing the production of inflammatory cytokines and pulmonary vascular leakage in ALI mice while significantly reducing mortality. This mechanism enables their protective role in inhibiting and resolving inflammation [40]. In this study, we investigated the effects of the CXCR3 agonist CXCL10 and nine CXCR3 antagonists on the efferocytosis function of macrophages. Our findings indicate that CXCL10 enhances macrophage phagocytosis and the expression of molecules associated with macrophage efferocytosis. Conversely, we observed that eight CXCR3 antagonists, NBI-74330, TAK-779, SCH 546738, C1, C2, C3, C4, and C5, suppress macrophage phagocytosis and the expression of efferocytosis-related molecules. This inhibition aligns with the effects of AMG 487 on macrophage phagocytosis and the expression of efferocytosis-related molecules, as documented in previous study [35]. Among the compounds studied, C5 exhibits the most pronounced inhibitory effect, exceeding that of AMG 487. This observation may be attributed to the presence of trifluoromethylphenyl and bicycloheptene structures in C5, which likely enhance its binding affinity to CXCR3. This hypothesis is further supported by molecular docking simulations.
LPS can enhance the expression of pro-inflammatory factors and M1 macrophage
markers by activating the TLR4 signaling pathway, including the NF-
ALI and ARDS represent clinical syndromes characterized by high morbidity and
mortality rates [27]. These conditions can arise from a variety of etiological
factors, including shock, severe sepsis, ischemia-reperfusion injury, pulmonary
contusion, and severe pneumonia [44]. Currently, there is no specific treatment
for ALI or ARDS, highlighting an urgent clinical need for safe and effective
therapeutic strategies. A study demonstrated that the combination therapy of
pseudoephedrine and emodin significantly inhibited the secretion of inflammatory
factors, including TNF-
In drug development and biomolecular research, understanding the interactions between ligands and receptors is crucial. Key factors influencing ligand-receptor binding include molecular-level aspects such as structure, affinity, and post-translational modifications; cellular-level factors such as receptor expression, internalization, and dimerization; and environmental-level conditions including pH, ion concentration, temperature, mechanical forces, competitive molecules, and extracellular matrix components [51]. These factors collectively determine the efficiency, specificity, and biological function of ligand-receptor binding. A systematic analysis of key regulatory factors elucidates the molecular interaction mechanisms of cellular signal transduction, providing theoretical support at the mechanistic level for the development of targeted drugs and the optimization of clinical treatment pathways. By employing molecular docking technology to analyze the interaction energy between nine antagonists and the CXCR3 protein, we found that TAK-779 exhibits the most stable binding with the CXCR3 protein. Based on the structural framework of TAK-779, we conducted novel structural modifications and subsequent screening for the first time. Our findings reveal that the modified compounds, TAK 779 and TAK 779,2745583, exhibit enhanced binding stability with CXCR3 in comparison to the original compound, TAK-779. Additionally, we reference alternative synthetic methods for the production of TAK-779 [52, 53]. We propose the following possible synthetic routes for the late stage synthesis of TAK-779 from imidazolium 1 and TAK-779,2745583 (Supplementary Fig. 2). In our subsequent work, we will further investigate the effects of these two compounds on macrophage function and their roles in LPS induced ALI in mice. We found that the results from macrophages and mouse lung tissues did not align well with the molecular docking results. This discrepancy may be attributed to several factors, including the inherent limitations of molecular docking, the complexity of animal experiments, potential target selection bias, and the properties of the drugs used [54, 55, 56]. Future studies could benefit from employing molecular dynamics simulations to further assess the binding stability of the nine CXCR3 antagonists and to refine molecular docking methodologies, thereby minimizing the discrepancies observed between molecular docking and both cellular and animal experiment results. This study revealed that all nine CXCR3 antagonists are capable of regulating macrophage function and alleviating ALI in mice, with AMG 487 exhibiting the most significant therapeutic efficacy. We hypothesize that the combined application of various CXCR3 antagonists may lead to improved therapeutic outcomes in clinical settings. By employing molecular docking technology to examine the interactions between different antagonists and the CXCR3 protein, two superior molecular configurations, TAK 779 from imidazolium 1 and TAK 779, 2745583, were identified through comparative screening. This finding offers new therapeutic insights and strategies for related clinical diseases.
CXCL10 enhances macrophage phagocytosis and promotes M1 polarization, whereas CXCR3 antagonists inhibit macrophage phagocytosis and concurrently encourage M2 polarization. Subsequent in vivo experiments indicated that all nine tested CXCR3 antagonists conferred protective effects against LPS-induced ALI in mice. Molecular docking analysis revealed that TAK-779 exhibited the most stable binding affinity to the CXCR3 protein. Two superior molecular configurations—TAK-779 from imidazolium 1 and TAK-779, 2745583—demonstrated increased binding stability to CXCR3 compared to the parent compound, suggesting significant optimization potential.
Data will be made available on request. The authors confirm that the data supporting the findings of this study are included within the manuscript and its supplementary materials. We adhere to the journal’s data sharing policy by making the data freely available. There are no ethical, confidentiality, or legal restrictions preventing the sharing of data from this study.
WG and GL developed the concept and designed the studies. MZ, ZW, ZZ and PW analyzed the in vivo and in vitro studies and performed statistical analysis. XX, TM and LL contributed to the experimental studies and data acquisition. FQ and GL contributed to the data analysis and statistical analysis. WG, MZ and ZW wrote and edited manuscript. All authors contributed to editorial changes in the manuscript. All the authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
This study was performed following the China Council on Animal Care and Protocol guidelines. All animal studies were conducted in accordance with the guidelines approved by the Ethics Committee for Animal Experiments of Bengbu Medical University (approval number 2021-003) and all applicable institutional and governmental regulations concerning the ethical use of animals were followed.
Not applicable.
This project was supported financially by the Natural Science Research Project of Anhui Educational Committee (2024AH030040), Young and Middle-aged Teacher Development Program of Anhui Educational Committee (JNFX2023029), and the National Natural Science Foundation of China (82104178). Longhu Talent Project of Bengbu Medical University (LH250103005). Innovation Training Program for College Students in Anhui Province, Grants S202410367155 (P. Wang).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL45931.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.