




 , Hai Zhang 2,†, Jianmin Gu 3, Minjie Ju 1,*
, Hai Zhang 2,†, Jianmin Gu 3, Minjie Ju 1,* , Chunbing Zhang 4,*
, Chunbing Zhang 4,*1 Department of Critical Care Medicine, Zhongshan Hospital, Fudan University, 200032 Shanghai, China
2 Department of Pulmonary and Critical Care Medicine, Shanghai Chest Hospital, Shanghai Jiao Tong University School of Medicine, 200030 Shanghai, China
3 Department of Thoracic Surgery, Zhongshan Hospital, Fudan University, 200032 Shanghai, China
4 Department of Geriatric, Renji Hospital, Shanghai Jiaotong University School of Medicine, 200127 Shanghai, China
†These authors contributed equally.
Abstract
Small ubiquitin-related modifier protein (SUMO)ylation is a reversible post-translational modification of proteins. SENP5, a SUMO-specific protease, plays key roles in a wide range of cellular processes. This study aims to investigate the potential involvement of SENP5 in lipopolysaccharide (LPS)-induced acute lung injury (ALI).
First, we established LPS-treated human normal lung epithelial cells (BEAS-2B) and a lung injury mouse model. SENP5 expression was then analyzed in vivo and in vitro using quantitative real-time PCR (qRT-PCR), Western blot, hematoxylin–eosin (H&E) staining, and immunohistochemistry. Then, CCK-8 assay and flow cytometry were employed to assess inflammatory response and apoptosis following SENP5 knockdown in LPS-induced BEAS-2B cells. Next, H&E, immunohistochemistry, and survival analysis were conducted to investigate apoptosis and proliferation in SENP5 conditional knockout (cKO) mice. Finally, RNA sequencing was used to screen for differentially expressed genes in SENP5 knockdown BEAS-2B cells. Downstream molecules and signaling pathways were analyzed using Western blot and qRT-PCR.
SENP5 was notably upregulated in both LPS-induced BEAS-2B cells and the lung injury mouse model. In vitro, SENP5 knockdown markedly exacerbated the LPS-induced suppression of BEAS-2B cell viability and promoted inflammatory response and apoptosis. Besides, the conditional knockout of SENP5 significantly increased apoptosis and inhibited proliferation in the lungs of mice. RNA sequencing indicated SENP5 deficiency inhibited solute carrier family 7 member 5/mechanistic target of rapamycin (SLC7A5/mTOR) signaling in LPS-induced BEAS-2B cells. Therefore, we confirmed that SENP5 might exert a protective effect against LPS-induced lung injury by inhibiting apoptosis of lung epithelial cells through the SLC7A5/mTOR signaling pathway.
SENP5 might play a protective role in LPS-induced lung injury by inhibiting apoptosis of lung epithelial cells through the SLC7A5/mTOR signaling pathway.
Keywords
- acute lung injury
- SUMOylation
- apoptosis
Acute lung injury (ALI) is a severe and heterogeneous respiratory syndrome characterized by abnormally increased vascular permeability, pulmonary edema, and plasma protein leakage, leading to high mortality among patients with pneumonia, aspiration pneumonitis, trauma, and sepsis [1, 2]. Although supportive interventions, including mechanical ventilation, antibiotics, and glucocorticoid therapy, could improve clinical outcomes in ALI patients, the mortality rate remains as high as 35–55% [3]. Therefore, it is critical to develop therapeutic approaches and identify specific biomarkers as well as molecular and pathophysiological mechanisms to alleviate the global burden of lung injury.
Small ubiquitin-related modifier protein (SUMO) modification is a highly dynamic and reversible post-translational modification that fine-tunes protein function and regulates various critical biological processes, including apoptosis, cell cycle progression, and DNA damage repair [4, 5]. Mounting evidence positions SUMOylation as a promising therapeutic target for lung diseases [6]. The inhibition of SUMOylation has been shown to alleviate pulmonary inflammation and oxidative stress in chronic obstructive pulmonary disease (COPD) and to induce cell cycle arrest and apoptosis in lung cancer models [7, 8].
The SUMO-specific protease (SENP) family comprises cysteine proteases that catalyze the deconjugation of SUMO from modified substrates and maintain the dynamic balance of cellular SUMOylation [9]. In mammalian cells, the SENP family primarily comprises SENP1–3 and SENP5–8 [10]. SUMO-specific peptidase 5, known as SENP5, is predominantly located in the nucleus. During interphase, SENP5 predominantly localizes to nucleoli while translocating to the mitochondrial surface upon G2/M transition, indicating its regulative role in mitochondrial dynamics, morphology, and function [11]. Recent studies showed that SENP5 was required to maintain normal mitochondrial morphology and to control intracellular levels of reactive oxygen species (ROS). Moreover, SENP5 inhibition could lead to decreased osteosarcoma cell proliferation, accompanied by elevated apoptosis. Our team has been engaged in the SENPs-mediated SUMOylation, its regulatory mechanism in cell signaling transduction, as well as their roles and significance in various diseases. However, the biological role of SENP5 in inflammatory diseases such as lung injury remains poorly understood.
Although SENP5 had been implicated in apoptotic processes in certain cancer contexts, its specific role in inflammatory lung diseases, particularly in ALI, remains largely unexplored. Our research remained the first to demonstrate the significant upregulation of SENP5 in lipopolysaccharide (LPS)-induced ALI model, suggesting its potential involvement in the pathogenesis of lung injury. Furthermore, we identified the solute carrier family 7 member 5/mechanistic target of rapamycin (SLC7A5/mTOR) signaling pathway as a key downstream mechanism mediated by SENP5 in the progression of lung injury through RNA sequencing. Collectively, our findings elucidated the important role of SENP5 in ALI and offered novel insights into the molecular mechanisms involving its regulation of lung epithelial cell apoptosis, underscoring its potential as a therapeutic target.
Male C57BL/6 wild-type mice (8 weeks old) were purchased from GemPharmatech (Nanjing, China). All mice were housed under controlled environmental conditions with a 12-hour light/dark cycle and provided with water and food prior to experiments. The study protocol was approved by the Animal Review Committee of Zhongshan Hospital, Fudan University (2022/02/01).
SENP5flox/flox mice and Sftpc-Cre mice were generated by GemPharmatech (Nanjing, China) via the CRISPR-Cas9 system. To generate alveolar epithelial cell-specific SENP5-deficient mice, Sftpc-Cre mice were crossed with SENP5flox/flox mice to obtain Sftpc-Cre-SENP5flox/flox mice (SENP5 conditional knockout (cKO)). Littermate SENP5flox/flox mice were used as controls.
The LPS-induced ALI model was established using 8-week-old male C57BL/6 mice. The mice were randomly divided into control and ALI groups. Briefly, mice were anesthetized with isoflurane, and a midline neck incision was made to expose the trachea. The isoflurane concentration was titrated between 3% and 5% to maintain an appropriate depth of anesthesia. A total of 100 µL lipopolysaccharide (LPS) (0111: B4, L2630, Sigma, St. Louis, MO, USA) was slowly administered intratracheally at a dose of 1 mg/kg. This dosage was selected based on well-established protocols, which have been demonstrated to induce robust and reproducible acute lung injury characterized by significant inflammatory cell infiltration, pulmonary edema, and alveolar damage within 24–48 hours [12, 13, 14]. The control group received an equal volume (100 µL) of sterile PBS. 24 h or 48 h after LPS inhalation, the mice were euthanized via intraperitoneal injection 1% pentobarbital (150 mg/kg), followed by cervical dislocation, and the mice’s lung tissues were collected for further analysis.
Total RNA was extracted from control (NC) and SENP5 knockdown BEAS-2B cells
using Trizol reagent (Thermo Fisher Scientific, Waltham, MA, USA). RNA quality
was verified on an Agilent 2100 bioanalyzer (Agilent Technologies, Santa Clara,
CA, USA), and sequencing libraries were prepared with the
NEBNext® Ultra™ RNA Library Prep Kit (NEB,
Ipswich, MA, USA). The libraries were sequenced on an Illumina NovaSeq 6000
platform (Illumina, San Diego, CA, USA) by Sangon Biotech (Shanghai, China).
Subsequent bioinformatic analysis was performed as follows: Raw reads were
quality-controlled using FastQC and processed with fastp to remove adapters and
low-quality bases. The clean reads were then aligned to the reference genome
using HISAT2, and gene expression was quantified using featureCounts.
Differential expression analysis was conducted using the DESeq2 version 1.40.2 (Bioconductor, Boston, MA, USA) in R Studio 2024.12.0+467 (Posit Software, PBC, Boston, MA, USA) with normalization based on the median of ratios method. Genes with
BEAS-2B human bronchial epithelial cells were purchased from American Type Culture Collection (ATCC) and cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (Gibco, Grand Island, NY, USA), 100 U/mL penicillin, and 100 µg/mL streptomycin (Gibco, USA) at 37 °C with 5% CO2. To induce an inflammatory response, BEAS-2B cells were treated with LPS (10 µg/mL) (0111:B4, L4391, Sigma, USA). The cell line used in this study was routinely tested for mycoplasma contamination and was confirmed to be negative. Besides, the cell line was validated by STR profiling.To knockdown SENP5 expression, lentivirus targeting SENP5 (Genepharma, Shanghai, China) and empty vectors (NC) were used to infect the BEAS-2B cells. Six hours after infection, the medium was replaced. Cells were then cultured for an additional 48 h before harvesting for subsequent experiments. All cell experiments were conducted with three biological replicates per group.
Cell viability was assessed using the CCK-8 assay. For CCK-8, BEAS-2B cells were
seeded in 96-well plates at a density of 1
The apoptosis rate in BEAS-2B cells was determined by flow cytometry (CytoFLEX LX, Beckman Coulter, Brea, CA, USA) using an Annexin V-APC/PI kit (Vazyme, Nanjing, China). Data analysis was conducted with FlowJo software 10.8.1 (TreeStar, Ashland, OR, USA).
Mice’s left lung tissues were fixed in 4% paraformaldehyde at room temperature overnight and then embedded in paraffin. Sections were cut at 4 µm thickness for staining. Then sections were deparaffinized in xylene twice and rehydrated through a graded ethanol series (100%, 90%, and 70%). The sections were washed in distilled water before hematoxylin and eosin staining. Subsequently, sections were sequentially dehydrated through 95% and 100% ethanol solutions and cleared in xylene. Finally, lung sections were sealed with neutral gum for microscopic examination of the pathological changes of lung injury.
The mouse lung tissues were embedded in paraffin and then deparaffinized and rehydrated before antigen retrieval. Later, these sections were subjected to 3% H2O2 and 10% goat serum at room temperature for 30 min. After cleaning, the SENP5 primary antibody was added and incubated overnight at 4 °C. The following day, a horseradish peroxidase (HRP)-conjugated secondary antibody (1:1000 dilution) was applied for 15 minutes at room temperature and a diaminobenzidine (DAB) substrate was used for chromogenic development. The immunohistochemical detection of lung samples was conducted through the GeneTech kit (Shanghai, China). All experiments were conducted independently three times.
Total RNA was extracted from BEAS-2B cells and lung tissue using Trizol reagent (Thermo Fisher Scientific, USA). cDNA was synthesized using SuperRT III All-in-one RT Mix for qPCR (with gDNA Remover) (Yeasen, Shanghai, China). Quantitative PCR was performed using the Hieff® qPCR SYBR Green Master Mix (Yeasen, Shanghai, China) using the CFX-96 real-time PCR detection machine (Bio-Rad, Hercules, CA, USA) according to the manufacturer’s instructions: 95 °C for 5 min, 45 cycles of 95 °C for 10 s, 60 °C for 30 s, and 72 °C for 20 s. The primers were synthesized by Sangon Biotech (Shanghai, China). Relative mRNA expression levels were analyzed through the 2-ΔΔCt method. The primer sequences are listed in Table 1. All experiments are representative of three independent experiments.
| Gene name | Forward primer (5′→3′) | Reverse primer (5′→3′) |
| SENP5 | AAGATGGACGAAGAAGGTGGACTTG | CGGCTGGAGAGTGTCACAGTAATG |
| IL-6 | GGTGTTGCCTGCTGCCTTCC | GTTCTGAAGAGGTGAGTGGCTGTC |
| IL-1 |
CCACCTCCAGGGACAGGATA | TCAACACGCAGGACAGGTAC |
| TNF- |
GCCTCTTCTCATTCCTGCTTGTGG | GTGGTTTGTGAGTGTGAGGGTCTG |
| SLC7A5 | GGCATCGGCTTCACCATCATCC | ACAGGACGGTCGTGGAGAAGATG |
Total protein was extracted from BEAS-2B cells and lung tissues by RIPA buffer
(Yeasen, Shanghai, China) supplemented with 1 mM PMSF and protease inhibitor
cocktail at 4 ℃. Protein concentrations were determined using a BCA protein assay
kit (Epizyme, Shanghai, China). Samples (40 µg) were separated by 8–12%
SDS-PAGE gels and transferred to PVDF membranes (Millipore, Burlington, MA, USA).
Membranes were then incubated with the primary antibodies at 4 ℃ overnight,
followed by incubation with HRP-conjugated anti-mouse and anti-rabbit IgG
antibodies at room temperature for 1 h. The following antibodies were used in
experiments: SENP5-Specific Polyclonal antibody (1:1000, 19529-1-AP, Proteintech,
Wuhan, China);
In this study, all data were presented as mean
To validate the role of SENP5 in lung injury, we first established an LPS-induced BEAS-2B human epithelial cell injury model. SENP5 expression level was upregulated at both mRNA and protein levels in BEAS-2B cells at 24 h and 48 h after LPS stimulation (Fig. 1A,B), accompanied by elevated pro-inflammatory cytokine expression (Fig. 1C). In addition, we further established an LPS-stimulated lung injury mouse model. For this, C57BL/6 mice were intratracheally administered with PBS or LPS (1 mg/kg). Western blot analysis revealed that a marked upregulation of SENP5 in mouse lung tissues following LPS challenge (Fig. 1D). Compared with PBS-treated mice, LPS-treated lung injury mice exhibited significant lung damage, including pulmonary edema, inflammatory cell infiltration, and alveolar wall thickening in a time-dependent manner (Fig. 1E). Consistently, the immunohistochemistry staining results from pulmonary tissues further confirmed that SENP5 significantly increased at 24 h and 48 h after LPS treatment (Fig. 1F). These findings suggest that SENP5 was highly expressed during LPS-induced lung injury.
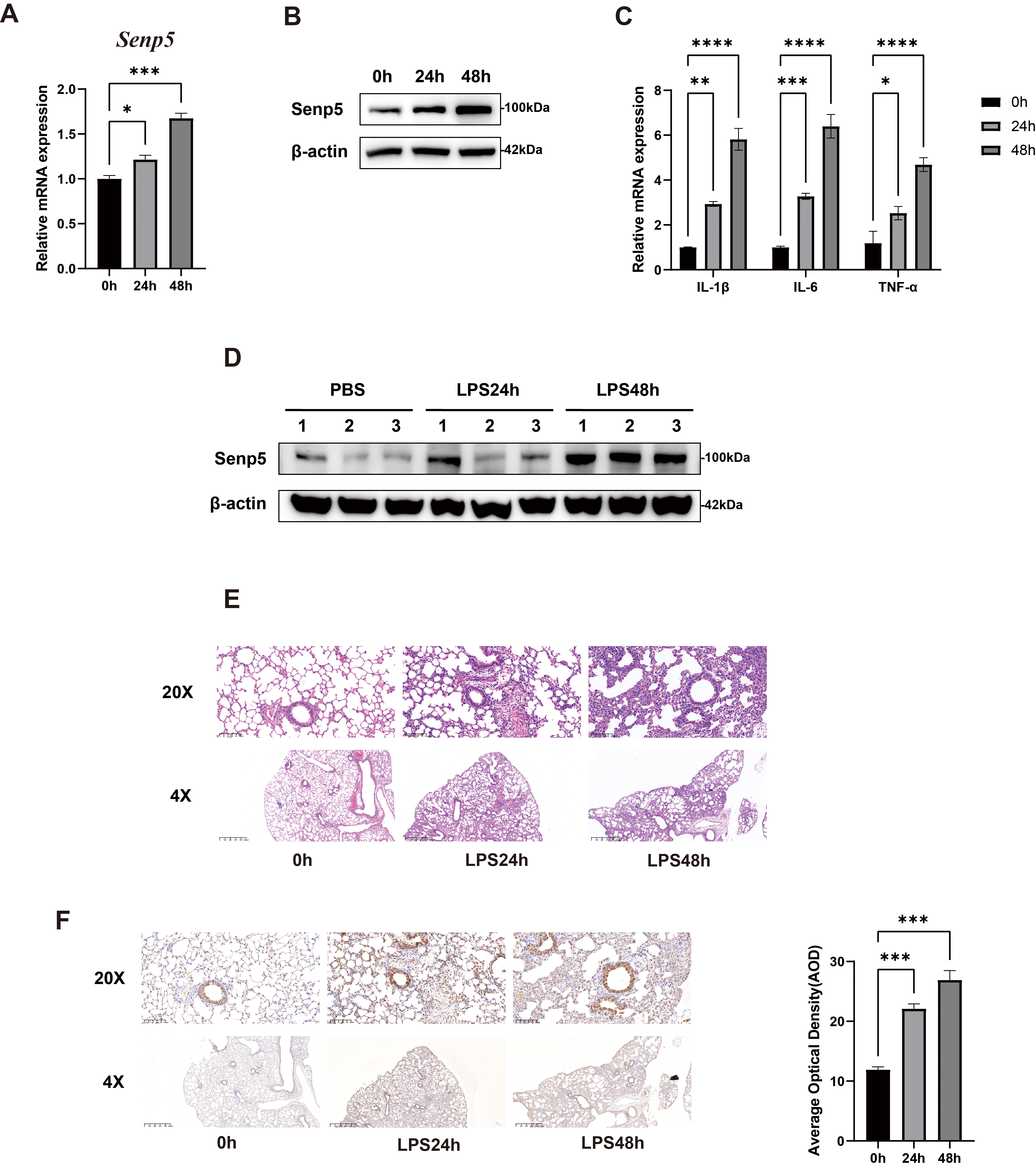 Fig. 1.
Fig. 1.
SENP5 was upregulated in LPS-stimulated BEAS-2B cells
and inflammatory mouse lung tissues. (A) Relative mRNA expression
levels of SENP5 in LPS-stimulated BEAS-2B cells at the indicated time
points (n = 3). (B) Representative Western blot images of SENP5 protein levels in
LPS-stimulated BEAS-2B cells at the indicated time points (n = 3). (C) Relative
mRNA expression levels of pro-inflammatory cytokines in LPS-stimulated BEAS-2B
cells at the indicated time points (n = 3). (D) Representative Western blot
images of SENP5 protein levels in mice lung tissues undergoing LPS-induced ALI at
the indicated time points (n = 3). (E) Representative H&E staining images of
mouse lung tissues undergoing LPS-induced ALI at the indicated time points
(n = 3). Upper scale bar, 100 µm; lower scale bar, 625 µm. (F)
Representative images of immunohistochemical staining of SENP5 in mice lung
tissues undergoing LPS-induced ALI at the indicated time points (n = 3). Upper
scale bar, 100 µm; lower scale bar, 625 µm. All the data are
expressed as the mean
To explore the underlying biological function of SENP5, we also analyzed
LPS-induced inflammatory response, cell proliferation, and apoptosis in BEAS-2B
cells transfected with SENP5 lentivirus shRNAs. Compared to the control (NC)
group, the #1 and #2 SENP5 shRNA group exhibited a significant decrease in
expression of up to 70% to 80% at mRNA and protein levels (Fig. 2A,B). Thus,
the #1 and #2 SENP5 shRNAs were utilized for further experiments. To explore
the effect of SENP5 knockdown on apoptosis and inflammatory cytokines, BEAS-2B
cells were treated with LPS for 24 h and 48 h after transfecting SENP5 shRNAs. As
illustrated in Fig. 2C, the knockdown of SENP5 distinctly caused a decrease in
cell viability when cotreated with LPS. Besides, the inflammatory response was
markedly exacerbated, with significantly elevated mRNA levels of IL-6,
TNF-
 Fig. 2.
Fig. 2.
SENP5 knockdown exacerbated LPS-induced inflammatory
response and apoptosis in BEAS-2B epithelial cells. (A) Relative mRNA
expression levels of SENP5 in BEAS-2B cells transfected with control
or SENP5-targeting shRNAs (n = 3). (B) Representative Western blot images of
SENP5 protein levels in BEAS-2B cells (n = 3). (C) Cell viability assessed by
CCK-8 assay in LPS-stimulated BEAS-2B cells following SENP5 knockdown (n = 3).
(D,E) Relative mRNA expression levels of pro-inflammatory cytokines in BEAS-2B
cells at 24 h and 48 h (n = 3). (F) Flow cytometry analysis and quantification of
apoptosis rate in BEAS-2B cells following SENP5 knockdown. All the data are
expressed as the mean
To validate the biological significance of SENP5, we established conditional SENP5 cKO mice by crossing floxed SENP5 mice (SENP5flox/flox) with Sftpc-cre mice and then validated the mice genotype (Supplementary Fig. 1). H&E staining results from mouse lung tissues indicated that SENP5 cKO mice exhibited disseminated thickening of alveolar septa, interstitial exudation, together with significant inflammatory cell infiltration after LPS administration (Fig. 3A). Immunohistochemistry staining results showed SENP5 deficiency further increased cell apoptosis and inhibited proliferation, as reflected by the expressions of caspase3 and ki67 (Fig. 3B,C). Survival analysis showed that SENP5 cKO mice had a limited probability of survival compared to SENP5flox/flox controls after LPS challenge (Fig. 3D). Taken together, these results revealed that SENP5 deficiency facilitated apoptosis and inhibited proliferation in vivo.
 Fig. 3.
Fig. 3.
SENP5 deficiency facilitated apoptosis and inhibited
proliferation in lung tissues. (A) Representative images of H&E
staining of lung tissues from SENP5flox/flox and SENP5 cKO mice (n = 3).
Left scale bar, 625 µm; Right scale bar, 100 µm. (B) Representative
images of immunohistochemical staining and quantification of caspase-3 in
SENP5flox/flox and SENP5 cKO mice lung tissues. Left scale bar, 625
µm; Right scale bar, 100 µm. (C) Representative images of
immunohistochemical staining and quantification of Ki-67 in SENP5flox/flox
and SENP5 cKO mice lung tissues. Left scale bar, 625 µm; Right scale
bar, 100 µm. (D) The survival curve of SENP5flox/flox and SENP5 cKO
mice undergoing LPS challenge (n = 10). All the data are expressed as the mean
To gain better insight into the molecular mechanism of SENP5, RNA sequencing was
performed in NC and SENP5-knockdown BEAS-2B cells. In total, 667 differentially
expressed genes (
 Fig. 4.
Fig. 4.
SENP5 deficiency facilitated apoptosis through
inhibiting SLC7A5/mTOR signaling pathway in BEAS-2B epithelial cells. (A) Volcano plot of differentially expressed genes between NC and
SENP5 knockdown BEAS-2B cells. DEGs were identified based on
SLC7A5 (LAT1), as an amino acid transporter, was reported to be linked with the activation of the mTOR pathway in cancers. Given the established role of SLC7A5 in amino acid transport and mTOR activation, we next investigated the downstream pathway. Western blot analysis showed that SENP5 deficiency could reduce LPS-induced phosphorylation of mTOR and its crucial downstream effector p70, indicating suppression of mTORC1 signaling (Fig. 4E,G). To summarize, SENP5 deficiency was suggested to induce apoptosis to aggravate LPS-induced ALI via inhibiting the SLC7A5/mTOR pathway in vitro.
The lung functions as a crucial immune organ that harbors both innate and adaptive immune cells to induce a potent immune response against pathogens. However, excessive and irreversible pro-inflammatory responses can lead to severe tissue damage [15]. Over the past decade, pathogenesis and pathological changes of lung injury have become increasingly recognized, which is characterized by protein-rich hydrostatic pulmonary edema, refractory hypoxemia, and decreased lung compliance [16]. Identifying novel regulatory mechanisms, such as SUMOylation, will offer a promising target for therapeutic intervention.
LPS is a principal constituent of the outer membrane of Gram-negative bacteria [17]. To elucidate the role of SENP5 in ALI pathogenesis, we employed LPS-induced lung injury models. Our results first found that SENP5 expression was significantly upregulated in both LPS-stimulated BEAS-2B cells and murine ALI models. Functional experiments revealed specific knockdown of SENP5 exacerbated LPS-induced inflammatory cytokine production, increased the key apoptotic protein caspase-3 activity, and inhibited cell proliferation. Consistently, we established the SENP5 knockout mouse model, and in vivo experiments revealed that SENP5 deficiency exacerbated apoptosis protein infiltration and inhibited cell proliferation. SUMOylation, a reversible post-translational modification, has emerged as a key regulator of diverse cellular processes [18]. In particular, several SUMO-specific proteases that remove SUMO proteins from substrates have been identified. The human SENP family is composed of six members, including SENP1-3 and SENP5-7 [19]. They are mainly responsible for the initiation of the SUMO process and dissociation of SUMO proteins from substrates [20]. SENP5 has been found to be required for proper cell division due to its hydrolase and isopeptidase activities [21]. Although SENP5 has been implicated to play a crucial role in the process of apoptosis, the specific role of SENP5 in apoptosis remains to be explored across diverse diseases. Mechanistically, SENP5 knockdown was demonstrated to inhibit cell growth and trigger apoptosis in osteosarcoma cells; while SENP5 knockdown in colon cancer cells led to a significant reduction in proliferation and promoted apoptosis [22]. These findings collectively implicated SENP5 as an indispensable regulator of apoptotic processes.
To unravel the mechanistic basis of SENP5-mediated apoptosis, we performed RNA sequencing based on the SENP5 stably knockdown cells. Differential gene analysis revealed SLC7A5 was significantly decreased secondary to SENP5 knockdown in BEAS-2B cells. SLC7A5 (LAT1), a neutral amino acid transporter, mediates the transport of amino acids across cellular and organellar membranes to activate mTORC1 signaling [23]. Several studies have demonstrated that SLC7A5, a cancer-type amino acid transporter, is highly expressed in primary cancers originating from various tissues [24]. In vitro experiments confirmed that SLC7A5 was upregulated in LPS-induced BEAS-2B cells in a time-dependent manner. SLC7A5 is essential for mTORC1 signaling and the promotion of cell proliferation. Previous functional studies suggested that the SLC7A5/SLC3A2 complex uses intracellular L-glutamine as an efflux substrate to regulate the uptake of extracellular L-leucine, thus activating mammalian target of rapamycin complex 1 (mTORC1) [25]. The mTOR is a serine/threonine kinase that functions within two distinct multi-protein complexes, mTORC1 and mTORC2 [26]. Active mTORC1 phosphorylates and stimulates protein synthesis, metabolism, and cell proliferation via modulation of p70S6K1 [27]. Studies also report that knocking down SLC7A5 inhibited mTORC1 signaling, leucine-stimulated insulin secretion, and islet cell proliferation [28]. Our data further supported that SENP5 knockdown led to reduced SLC7A5 expression and subsequent suppression of mTOR and p70 phosphorylation upon LPS challenge.
Although our work demonstrated a strong functional link, elucidating the regulatory mechanism between SENP5 and SLC7A5 represents a critical direction for our future research. Based on the established role of SENPs in deSUMOylation, several plausible mechanisms could explain this regulatory relationship. First, as a SUMO-specific protease, SENP5 may directly deSUMOylate SLC7A5 or its upstream regulators, thereby modulating its stability and transcriptional expression. Alternatively, SENP5 may indirectly influence SLC7A5/mTOR through mitochondrial function or oxidative stress pathways. It is important to emphasize that our current data, while demonstrating a strong correlative link between SENP5 deficiency and suppressed SLC7A5/mTOR signaling, cannot distinguish between these direct and indirect mechanisms. Future studies employing co-immunoprecipitation, SUMOylation assays, or rescue experiments will be necessary to elucidate the exact molecular link.
In summary, our research shed light on the role of SENP5, SLC7A5/mTOR signaling, and apoptosis in LPS-induced lung injury. We provided evidence that SLC7A5/mTOR signaling is a key downstream pathway through which SENP5 exerts its anti-apoptotic effects in lung epithelial cells. This study identified SENP5 as a potential therapeutic target and provides valuable molecular insights for acute lung injury.
This study has several limitations. For example, we will supplement relevant basic research in the future to establish a direct causal relationship between SLC7A5 and mTOR. Further functional validation is needed to establish causality within this pathway. Besides, the specific mechanism of SENP5 regulating SLC7A5 remains unclear. More studies are needed to evaluate whether SENP5 directly regulates SLC7A5 via deSUMOylation.
Our study demonstrates that SENP5 was significantly upregulated in both LPS-induced pulmonary epithelial cells and lung tissues. Furthermore, knockdown of SENP5 was found to exacerbate the LPS-induced inflammatory response and promote apoptosis both in vitro and in vivo. Mechanistic investigations revealed that the pro-apoptotic effect triggered by SENP5 deficiency was mediated through the inhibition of the SLC7A5/mTOR signaling pathway.
The data that support the findings of this study are available from the corresponding author upon reasonable request.
YH and HZ conceived and performed the experiment; JG provided experimental facilities and performed the data analysis; MJ and CZ contributed to designing the research study, project administration and manuscript writing—review and editing. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
All animal experiments were conducted in accordance with the UK Animals (Scientific Procedures) Act 1986. All animal experiments were approved by the Animal Review Committee of Zhongshan Hospital, Fudan University (2022/02/01).
Not applicable.
This research was supported by the Innovation Fund of Zhongshan Hospital, Fudan University (Grant No.2023-2ZSCX12), and the Shanghai Public Health Talent Development Program for Outstanding Discipline Leaders (Grant No. GWVI-11.2-XD36).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL45811.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.