
- Academic Editor
Faust Akl et al. revealed in Nature a paradigm-shifting mechanism distinct from myeloid-driven immunosuppression, whereby glioblastoma induces T-cell apoptosis via tumor-derived IL-11, prompting astrocytes to reprogram into immunosuppressive tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)+ effectors, thereby establishing astrocytes as active immunomodulators. Therapeutically, herpes simplex virus type 1 (HSV-1) (anti-TRAIL) achieves a dual therapeutic effect, offering novel strategies to overcome glioblastoma (GBM)’s evasion tactics.
Glioblastoma (GBM), the most aggressive primary brain tumor [1], characterized by genetic and epigenetic variations among tumor cell [2, 3], has long evaded immunotherapy due to its profoundly immunosuppressive microenvironment [4, 5]. While prior research focused on the key immune regulators, tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs), a transformative study by Faust et al. [6] in Nature recently redefines the cellular dogma of GBM immunosuppression by uncovering astrocyte—the brain’s most abundant glial cell that likely to establish direct contact with GBM cells [7]—as active architects of T cell dysfunction [6]. Not only does this work challenge the classical view of astrocyte as a passive bystander but also reveal a novel IL-11/signal transduction and activation of transcription factor 3 (STAT3)/tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) axis bridging tumor signaling to adaptive immune collapse, offering unprecedented therapeutic opportunities.
The study’s most striking innovation lies in the discovery of a TRAIL-expressing astrocyte-specialized subpopulation emerging under the direction of IL-11 secreted by GBM. By single-cell RNA sequencing of human GBM samples, the study reveals these TRAIL+ astrocytes cluster preferentially in patients with early recurrence and correlate with survival outcome—the finding validated in orthotopic mouse models. Different with earlier studies implicating astrocyte joins in GBM growth through metabolic support or structural remodeling, this work demonstrates their direct role in immune evasion.
Molecularly, GBM-derived IL-11 activates STAT3 signaling in astrocytes, promoting TRAIL expression and induced apoptosis in tumor-infiltrating T cells via death receptor death receptor 5 (DR5). Spatial transcriptomics and multiplex imaging revealed significant co-localization of TRAIL+ astrocytes with apoptotic CD8+ T cells in the tumor periphery, defining an immune-rejection zone formed by tumor domesticated astrocytes.
What sets the study apart from prior research is its resolution of a long-standing paradox: though abundant T cell infiltration in some GBM patients, functional anti-tumor responses remain rare. While myeloid cells have been blamed for creating an immunosuppressive milieu, the study proves astrocytes execute parallels, non-redundant mechanism of T cell elimination, which is exemplified by genetic experiments where astrocyte-specific TRAIL deletion—unlike manipulations targeting TAMs—restored T cell vitality and tumor control without changing myeloid populations. Thus, the IL-11/STAT3/TRAIL axis represents a previously unrecognized immune checkpoint distinct from the canonical programmed death receptor 1 (PD-1)/cytotoxic T lymphocyte-associated protein 4 (CTLA-4) pathway, potentially carrying profound clinical implications. Notably, bioinformatics analysis of TCGA data linked high IL-11/TRAIL expression to T-cell exhaustion markers (TOX, LAG3), suggesting this pathway may underlie resistance to existing immunotherapies.
Yet unanswered questions remain. What role does IL-11 play in controlling the immunosuppressive nature of the tumor microenvironment (TME) in GBM? GBM subtypes, based on tumor gene expression profiling studies, exhibit significant heterogeneity with at least three distinct subtypes, namely proneural (PN), classical (CL), and mesenchymal (MES), which can undergo mutual transformation, reflecting substantial immunological microenvironment heterogeneity [8]. Analysis of human GBM scRNA-seq and cell type deconvolution TCGA datasets in this study revealed that IL-11 was most highly expressed in malignant cells across all GBMs, while IL-11RA was more widely expressed in both malignant and non-malignant compartments. Spatially resolved transcriptomics analysis further demonstrated that pSTAT3 expression was significantly correlated with TRAIL expression in GBM samples. Though the original text noted that IL-11 expression was most closely associated with the MES GBM subprogram, it did not directly compare differences in IL-11/STAT3 pathway activity across different GBM subtypes. The universality of the IL-11/STAT3 axis across all subtypes and regulatory differences in this pathway across distinct molecular subtypes remain to be explored. Is the IL-11/STAT3/TRAIL axis universally present across GBM subtypes, or restricted to interstitial tumors with high IL-11 secretion? Should future immunotherapy incorporates GBM subtype-specific precision design, and if so, how? Whether small-molecule STAT3 inhibitors currently tested for bone marrow targeting could be repurposed to block astrocyte reprogramming [9]? Intriguingly, preliminary data suggest synergistic effects between TRAIL blockade and PD-1 inhibition, hinting at broader combination therapy paradigms. Furthermore, the blood-brain barrier (BBB) still remains an obstacle for systemic anti-TRAIL antibodies, however, which may be overcome through emerging nanoparticle platforms [10]. Perhaps we could encapsulate STAT3 inhibitors or IL-11 antibodies within liposomes or polymer nanoparticles, leveraging receptor-mediated endocytosis to penetrate the BBB, while simultaneously employing focused ultrasound (FUS) and microbubbles (MB) to temporarily open the BBB, thereby enhancing delivery efficiency. However, this remains purely speculative at present.
The primary treatment for GBM is surgical intervention supplemented by radiotherapy and chemotherapy, while cutting-edge therapeutic technology continues to emerge, including targeted therapy, molecular immunotherapy, tumor treating fields (TTF), and laser-induced thermal therapy (LITT). Distinct with the conventional immunotherapy focusing solely on tumor cells or T cell activation [11], herpes simplex virus type 1 (HSV-1) (anti-TRAIL) engineered virus simultaneously attacks both tumors and their glial protective shields (Fig. 1). By expressing a single-chain antibody neutralizing astrocyte-derived TRAIL while maintaining viral cytotoxicity, the construct achieves dual therapeutic effects, namely disrupting the immunosuppressive niche while enhancing tumor-specific T cell responses together. In mouse GBM models, HSV-1 (anti-TRAIL) achieved a 68% reduction in tumor volume, significantly exceeding the empty vector controls, as it triples the influx of functional CD8+ T cells. Strikingly, survival benefits persisted even in PD-1-refractory tumors, underscoring the axis’s independence from traditional checkpoint pathways, where the specificity aligns with the study’s broader thesis that astrocytes and myeloid cells occupy non-overlapping roles in GBM immunosuppression, necessitating combinatorial strategies.
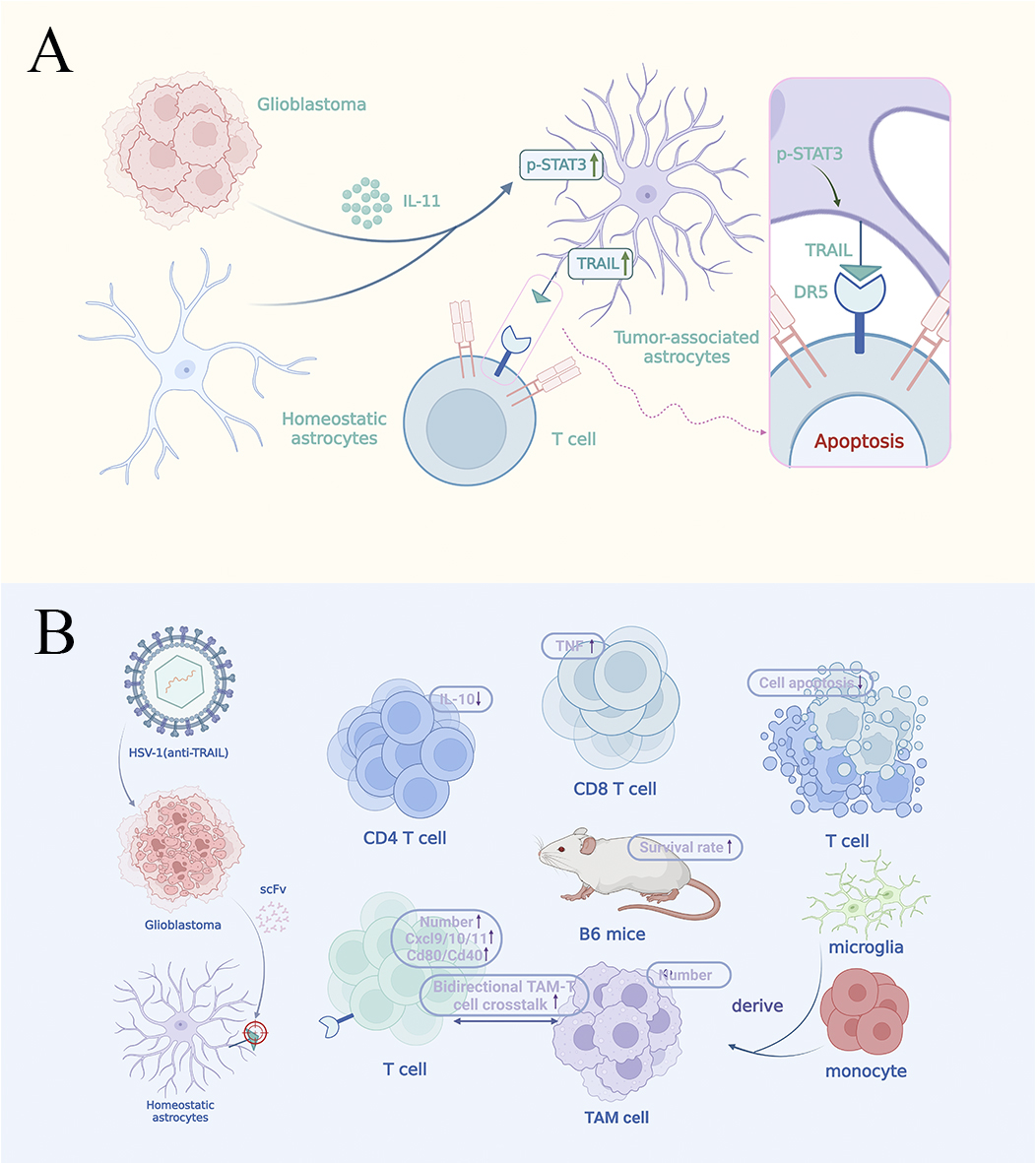 Fig. 1.
Fig. 1.
Glioblastoma (GBM) reprograms astrocytes for immunosuppression
via the IL-11/STAT3/TRAIL axis. This figure illustrates the functional transformation of astrocytes in the Glioblastoma (GBM) microenvironment. Section A of the figure reveals GBM-induced functional reprogramming. This process was validated through spatial transcriptomics and multicolor fluorescence
imaging: tumor-derived IL-11 prompts astrocytes to express TRAIL through
activation of the STAT3 signaling pathway, which binds to the DR5 receptor on the
surface of CD8+ T cells to induce apoptosis and the formation of tumor peripheral
immune rejection zone. Section B of the figure presents the targeted
therapeutic strategy. The engineered oncolytic HSV-1 (anti-TRAIL) treatment
reduces CD4+ and CD8+ T cell apoptosis in the tumor microenvironment (TME) while
increasing TNF-
The finding demonstrates how GBM cells rewrite astrocyte identity through instructive signaling to serve tumor interests and reveals the precise molecular syntax of such reprogramming, which as the same name of cellular function emerging from intrinsic programs modified by microenvironmental cues [12]. Tumor-derived IL-11 acts as the extrinsic instructor, STAT3 phosphorylation acts as the intracellular decoder, and TRAIL expression acts as the emergent immunosuppressive phenotype. This mechanism extends cellular dogma to neuroimmunooncology, demonstrating that astrocyte, too, exhibits significant functional plasticity when hijacked by malignant tumors, just as neuron and immune cell adapts their functions in response to environmental signals [12]. Crucially, the work establishes causal hierarchy in cellular instruction, proving astrocytic TRAIL expression is neither intrinsic nor stochastic, but rather a deterministic outcome of tumor-derived signals, revealing how pathological niches disrupt normal cellular properties at molecular resolution to sustain the disease ecosystem, with implications extending beyond GBM. Consider this: if a single cytokine, such as IL-11 as described in this paper, can transform supportive astrocytes into immune effectors, might other neuronal cell types in cancer also be similarly reprogrammed? The study thus provides both a specific mechanism and a generalizable paradigm for understanding how tumor manipulates TME through cellular re-education, assisting with decoding similar pathological rewiring across diseases.
The discovery of GBM-induced immunosuppressive astrocytes has transformed our understanding of tumor immune evasion, representing a paradigm shift in neuro-oncology. The previous predominantly focused on myeloid cell-mediated immunosuppression [13], whereas this study reveals that astrocyte actively forms an immunosuppressive TME through a TRAIL-dependent T cell clearance mechanism. And its HSV-1 (anti-TRAIL) therapy, targeting the novel IL-11/STAT3/ TRAIL immune checkpoint axis, overcomes the limitation of classical pathways like PD-1/CTLA-4, whose innovation lies in its dual mechanism, whereby it utilizes oncolytic viruses to selectively lyse tumor cells as it creates a pro-inflammatory microenvironment, inducing protective T cell responses against both the virus and tumor antigens—not only directly killing tumor cells but also breaking through the immune barrier of astrocytes.
Recent studies have demonstrated that astrocytes exhibit remarkable plasticity within the GBM microenvironment, transitioning from the traditional supportive role to an active immunometabolic hub regulating tumor progression. For instance, on one hand, they recruit and polarize TAMs by secreting factors such as CCL2 and CSF1, while supporting tumor cell survival through cholesterol efflux mediated by ABCA1 [14]. On the other hand, GBM-derived IL-11 induces astrocyte expression of TRAIL via STAT3 signaling, directly inducing T cell apoptosis. These findings reveal astrocytes’ dual immunometabolic regulatory functions within the TME, which not only reshape the understanding of tumor-neural microenvironment interactions but also provide a theoretical foundation for developing novel combination immunotherapies targeting astrocytic plasticity mechanisms and underscore the importance of effective immunotherapy targeting both tumor cells and their co-conspirators within the neural microenvironment simultaneously. Plus, the study’s value extends beyond GBM treatment, suggesting similar neuro-immune interaction mechanisms may underlie immune therapy resistance across multiple central nervous system tumors, shifting our perception of the TME as a dynamic ecosystem where non-malignant cells are systematically hijacked to sustain tumor progression.
YYW was responsible for Conceptualization, Visualization, and Writing – original draft. WLJ was responsible for Conceptualization, Funding acquisition, Supervision, and Writing – review & editing. Both authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We apologize to colleagues whose important work could not be cited due to space constraints. We thank the members of Jin Laboratory for their comments and discussions.
This work was financially supported by the High Level Talent Introduction Funds from the First Hospital of Lanzhou University.
The authors declare no conflict of interest. Given his role as the Editorial Board member, Wei-Lin Jin had no involvement in the peer-review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Giuseppe Murdaca.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.