
- Academic Editors
†These authors contributed equally.
Acute lung injury (ALI) triggered by sepsis continues to pose a significant difficulty in clinical practice. Due to its anti-inflammatory and antioxidant activities, calcitonin is considered a potential therapeutic option in sepsis.
Bioinformatics analysis was performed using the GSE89376 and GSE67652 datasets. Serum levels of CD3D and NLR family pyrin domain containing 3 (NLRP3) inflammasome, as well as high-mobility group box 1 (HMGB1)/myeloid differentiation primary response gene 88 (MyD88)/nuclear factor-κB (NF-κB) pathway components, were evaluated in sepsis/ALI patients. The effects of calcitonin and CD3D knockdown on human pulmonary microvascular endothelial cells (hPMECs) activated by lipopolysaccharide (LPS) were investigated in vitro. Experimental assays, including quantitative real-time polymerase chain reaction (qRT-PCR), Cell Counting Kit-8 (CCK-8) assay, western blotting (WB), enzyme-linked immunosorbent assay (ELISA), and flow cytometry, were used to assess cell viability, apoptosis, cell cycle, and oxidative stress markers.
CD3D was identified as a key sepsis/ALI-associated hub gene and correlated with NF-κB pathway activation in patients. CD3D silencing in hPMECs effectively suppressed LPS-induced inflammation, oxidative stress, apoptosis, and G1 phase arrest by downregulating the expression of NLRP3, phosphorylation (p)-NF-κB, MyD88, and HMGB1. Calcitonin alone mitigated LPS-induced injury in a dose-dependent way and further enhanced the protective impacts of CD3D knockdown. Co-treatment resulted in synergistic inhibition of interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF)-α, a reduction in oxidative markers, restoration of antioxidant capacity (superoxide dismutase (SOD) and glutathione (GSH)), improved endothelial cell viability, and attenuation of apoptosis. Notably, combined treatment more robustly suppressed the HMGB1/MyD88/NF-κB pathway than either intervention alone.
CD3D exacerbates sepsis-induced ALI by potentiating the HMGB1/MyD88/NF-κB pathway and NLRP3 inflammasome, driving inflammation and oxidative stress. Combined CD3D knockdown and calcitonin treatment offers a novel synergistic therapeutic strategy for mitigating pulmonary endothelial injury in sepsis.
Sepsis is a reaction of systemic inflammation triggered by invading pathogenic microorganisms such as bacteria. Its risk factors are diverse and include advanced age, immunosuppression, chronic comorbidities, and invasive medical procedures [1]. The acute systemic inflammation associated with sepsis frequently involves the respiratory system, with the lungs being particularly vulnerable [2]. Excessive inflammation and oxidative stress during sepsis contribute to the development of acute lung injury (ALI) and acute respiratory distress syndrome (ARDS), conditions characterized by severe outcomes and elevated mortality [3]. Additionally, acute inflammation in sepsis induces oxidative stress, which disrupts endothelial cell function and causes microvascular dysfunction [4]. Such alterations in endothelial responses promote pathological processes, including inflammation, coagulation, and cell adhesion, ultimately contributing to organ dysfunction [5]. Recent experimental evidence has demonstrated that oleuropein exerts protective effects against lipopolysaccharide (LPS)-induced ALI in rats through anti-inflammatory and antioxidant mechanisms [6], while a meta-analysis has highlighted the efficacy of terpenoids in reducing pulmonary edema in ALI animal models, supporting the therapeutic potential of natural compounds [7]. Furthermore, accumulating evidence suggests that antioxidant-based interventions may exert protective effects in sepsis, highlighting their promising therapeutic potential [8, 9]. Therefore, the development of innovative therapeutic strategies and reliable prognostic biomarkers is crucial for improving clinical outcomes in patients with sepsis.
Procalcitonin (PCT) is widely recognized as one of the most specific and sensitive biomarkers for diagnosing sepsis [10]. PCT, the prohormone of calcitonin, is generally processed by enzymatic cleavage to yield mature calcitonin and several smaller peptide fragments [11]. Calcitonin, a peptide hormone secreted primarily by the thyroid gland, is crucial in modulating calcium and phosphate metabolism [12]. Evidence suggests that severe infections and sepsis can induce skeletal destruction, tissue injury, and other physiological abnormalities, which may in turn elevate circulating calcium concentrations [13]. In such conditions, calcitonin functions to decrease circulating calcium concentrations through the inhibition of calcium release. Notably, studies in animal models and ex vivo human blood have demonstrated that calcitonin exerts a modest but deleterious effect in experimental septic shock, potentially mediated via calcitonin gene-related peptide (CGRP) receptors [14]. Therefore, investigating the specific mechanisms of action between calcitonin and sepsis is crucial for developing improved therapeutic strategies.
CD3D, also known as CD3 delta chain, is a protein encoded by the CD3D gene and constitutes an essential component of the CD3 complex [15]. As a key player in T cell growth and T cell receptor (TCR) signaling, this complex is essential for signal transmission in T cells. Deficiency or dysfunction of CD3D impairs T cell function and disrupts immune homeostasis, thereby contributing to various autoimmune disorders and immunodeficiencies [16]. In the context of sepsis, CD3D has been recognized as a potential marker of immune dysfunction and disease severity [17]. Yang et al. [18] conducted a bioinformatics analysis investigating the association between CD3D, CD247, and septic shock, demonstrating that reduced expression of both CD3D and CD247 correlated with poor prognosis. Nevertheless, the relationship between CD3D, sepsis, and calcitonin therapy is not yet well defined, underscoring the necessity of additional research to determine its value as a prognostic biomarker or therapeutic target in sepsis management.
Although CD3D has been recognized by recent transcriptome analysis as a possible immune-related biomarker in sepsis, its precise function in pulmonary endothelium damage remains unclear. This work combined bioinformatics analyses with experimental approaches to explore the role of CD3D in sepsis-induced ALI, focusing on its participation in oxidative stress, inflammatory, and apoptotic pathways in human pulmonary microvascular endothelial cells (hPMECs). In several inflammatory illness models, calcitonin has shown cytoprotective and anti-inflammatory qualities. Nevertheless, whether calcitonin contributes to the preservation of pulmonary microvascular endothelial integrity during sepsis, particularly through modulation of immune signaling pathways, remains unclear. Therefore, this research aims to comprehensively examine the function of CD3D and the possibility of calcitonin as a treatment for sepsis-induced ALI.
The Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/gds/) provided
the sepsis-related dataset GSE89376. A total of 12 sepsis and 12 control samples
were analyzed. Differential expression was assessed using the “limma” package
in R (version 4.0; R Foundation for Statistical Computing, Vienna, Austria), with
thresholds set at FC
DEGs were subjected to functional enrichment employing the Database for Annotation, Visualization, and Integrated Discovery (DAVID; https://davidbioinformatics.nih.gov/summary.jsp). Gene ontology, along with Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, was identified and visualized.
To explore interactions among DEGs, a PPI network was established using the STRING platform (https://string-db.org/). Key modules were identified in Cytoscape (version 3.8.1; Cytoscape Consortium San Diego, CA, USA) via CytoHubba with Degree, Maximum Neighborhood Component (MNC), and Maximal Clique Centrality (MCC) algorithms. Overlapping genes across modules were identified through the Venn analysis tool (https://bioinformatics.psb.ugent.be/webtools/Venn/). Their expression patterns were subsequently validated in the GSE89376 and GSE67652 datasets by comparing septic and control samples. Data analysis and visualization were performed with Sangerbox (http://vip.sangerbox.com/home.html).
A total of nine participants were enrolled in this investigation, including
three patients with sepsis-induced ALI, three patients with sepsis without
pulmonary involvement, and three non-septic control patients without pulmonary
inflammation. Sepsis patients were admitted to the Department of Intensive Care
Medicine. In contrast, control patients were recruited from the Department of
Hepatobiliary Surgery, the Third Affiliated Hospital of Naval Military Medical
University. All participants were age- and gender-matched. Sepsis was identified
using the Sepsis-3 criteria, which include life-threatening organ dysfunction and
a Sequential Organ Failure Assessment (SOFA) score rise of
hPMECs (Cat. no. CP-H001, Procell, Wuhan, China) were cultured in complete hPMEC medium (Cat. no. CM-H001, Procell, Wuhan, China) under standard conditions of 37 °C with 5% CO2. According to the supplier’s quality control statement, hPMECs were authenticated by CD31 immunofluorescence and tested to be over 90% pure. Additionally, the supplier confirmed that the cells were free from human immunodeficiency virus (HIV)-1, hepatitis B virus (HBV), hepatitis C virus (HCV), Mycoplasma, bacteria, fungi, and yeast contamination. To establish a sepsis-induced ALI model, cells were exposed to LPS (10 µg/mL; Cat. no. L8880, Solarbio, Beijing, China) for 24 h. For evaluating the protective role of calcitonin, LPS-treated hPMECs were further incubated with calcitonin (Cat. no. T3535, Sigma-Aldrich, St. Louis, MO, USA) at concentrations of 1, 5, and 10 nM for 24 h.
After being seeded at a density of 2
RNA purity and concentration were determined with a NanoDrop spectrophotometer (Thermo Fisher Scientific, Shanghai, China). First-strand cDNA was synthesized from 1 µg of RNA using the PrimeScript RT Kit (Takara, Shiga, Japan). qRT-PCR was performed on a StepOnePlus Real-Time PCR System (Applied Biosystems, Shanghai, China) using SYBR Green PCR Master Mix (Cat. no. 4309155; Thermo Fisher Scientific, Waltham, MA, USA). All reactions were performed in triplicate. Relative gene expression levels were calculated using the 2-ΔΔCt method, with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) serving as the internal control. Primer sequences are provided in Table 1.
| Target | Direction | Sequence (5′-3′) |
| CD3D | Forward | AGACTGGACCTGGGAAAACG |
| CD3D | Reverse | AGACTCCCAAAGCAAGGAGC |
| p21 | Forward | AGTCAGTTCCTTGTGGAGCC |
| p21 | Reverse | CATTAGCGCATCACAGTCGC |
| CCND1 | Forward | GATGCCAACCTCCTCAACGA |
| CCND1 | Reverse | ACTTCTGTTCCTCGCAGACC |
| CDK2 | Forward | GACACGCTGCTGGATGTCA |
| CDK2 | Reverse | CTGGAGCAGCTGGAACAGAT |
| iNOS | Forward | AATGTGGAGAAAGCCCCCTG |
| iNOS | Reverse | TGCATCCAGCTTGACCAGAG |
| COX-2 | Forward | GGCCATGGGGTGGACTTAAA |
| COX-2 | Reverse | ACCGTAGATGCTCAGGGACT |
| HMGB1 | Forward | TCTCAGGGCCAAACCGATAG |
| HMGB1 | Reverse | TCGTGCACCGAAAGTTTCAA |
| MyD88 | Forward | ACTTGGAGATCCGGCAACTG |
| MyD88 | Reverse | ATCCGGCGGCACCAATG |
| NLRP3 | Forward | GCTGGCATCTGGGGAAACCT |
| NLRP3 | Reverse | CAAGTCCACATCCTCCAGGTC |
| Bax | Forward | TGATGGACGGGTCCGGG |
| Bax | Reverse | TGAGACACTCGCTCAGCTTC |
| Bcl-2 | Forward | AACCTTTCAGCATCACAGAGGAAGT |
| Bcl-2 | Reverse | AGGGGGTGTCTTCAATCACG |
| CASP3 | Forward | TGTGAGGCGGTTGTAGAAGAGT |
| CASP3 | Reverse | CTTTATTAACGAAAACCAGAGCGCC |
| GAPDH | Forward | AATGGGCAGCCGTTAGGAAA |
| GAPDH | Reverse | GCGCCCAATACGACCAAATC |
qRT-PCR, quantitative real-time polymerase chain reaction; CD3D, CD3 delta subunit of T-cell receptor complex; p21, cyclin-dependent kinase inhibitor 1A (CDKN1A); CCND1, cyclin D1; CDK2, cyclin dependent kinase 2; iNOS, inducible Nitric Oxide Synthase; COX-2, cyclooxygenase-2; HMGB1, high-mobility group box 1; MyD88, myeloid differentiation primary response gene 88; NLRP3, NLR family pyrin domain containing 3; Bax, Bcl-2 associated X; Bcl-2, B-cell lymphoma-2; CASP3, caspase-3; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
hPMEC proteins were isolated using radioimmunoprecipitation assay (RIPA) buffer
(Cat. no. 89900; Thermo Fisher Scientific, Waltham, MA, USA) supplemented with
protease and phosphatase inhibitors. Protein levels were measured using a
bicinchoninic acid (BCA) assay (Cat. no. P0011; Beyotime, Shanghai, China). Equal
quantities of protein were subjected to sodium dodecyl sulfate polyacrylamide gel
electrophoresis (SDS-PAGE) and subsequently blotted onto poly-vinylidene
difluoride (PVDF) membranes (Cat. no. FFP39; Beyotime, Shanghai, China). The
membranes were blocked and subsequently incubated with primary antibodies against
CD3D (1:1000, ab109531), p21 (1:2000, ab109520), Cyclin D1 (1:1000, ab109531),
cyclin dependent kinase 2 (CDK2) (1:1000, ab32147), cyclooxygenase-2 (COX-2)
(1:2000, ab179800), inducible Nitric Oxide Synthase (iNOS) (1:2000, ab178945),
HMGB1 (1:1000, ab92310), MyD88 (1:1000, ab133739), p-NF-
Cell proliferation and viability were determined by CCK-8 assay (Cat. no. C0042;
Beyotime, China). Briefly, hPMECs were seeded in 96-well plates at a density of 5
Apoptosis and cell-cycle profiles of hPMECs were determined by flow cytometry.
After 24 h culture in 24-well plates (1
Tumor necrosis factor (TNF)-
To determine cellular metabolic and oxidative stress markers, hPMECs were
cultured in 24-well plates (1
At least three independent experiments were carried out for each dataset.
Statistical evaluation was conducted in R. Differences between the two groups
were determined using Student’s t-test. In contrast, multiple-group
comparisons were analyzed by one-way analysis of variance (ANOVA) with Tukey’s
post-hoc test. Data are shown as mean
From the GSE89376 dataset, 182 genes were found to be upregulated and 178 downregulated (Fig. 1A). DEGs were significantly enriched in biological processes (BP)-related GO terms, mainly involving “Positive regulation of apoptotic process”, “Regulation of T cell proliferation”, and “Positive regulation of vascular endothelial growth factor production” (Fig. 1B). For cellular components (CC), enrichment was observed in “Coated vesicle membrane”, “ER-to-Golgi transport vesicle membrane”, and “Endocytic vesicle membrane” (Fig. 1C). Regarding molecular function (MF), DEGs were enriched in “Protein kinase activator activity”, “Exopeptidase activity”, and “Calcium channel regulator activity” (Fig. 1D). KEGG pathway analysis further indicated enrichment in “Human T-cell leukemia virus 1 infection”, “Cell adhesion molecules”, as well as “Hematopoietic cell lineage” (Fig. 1E).
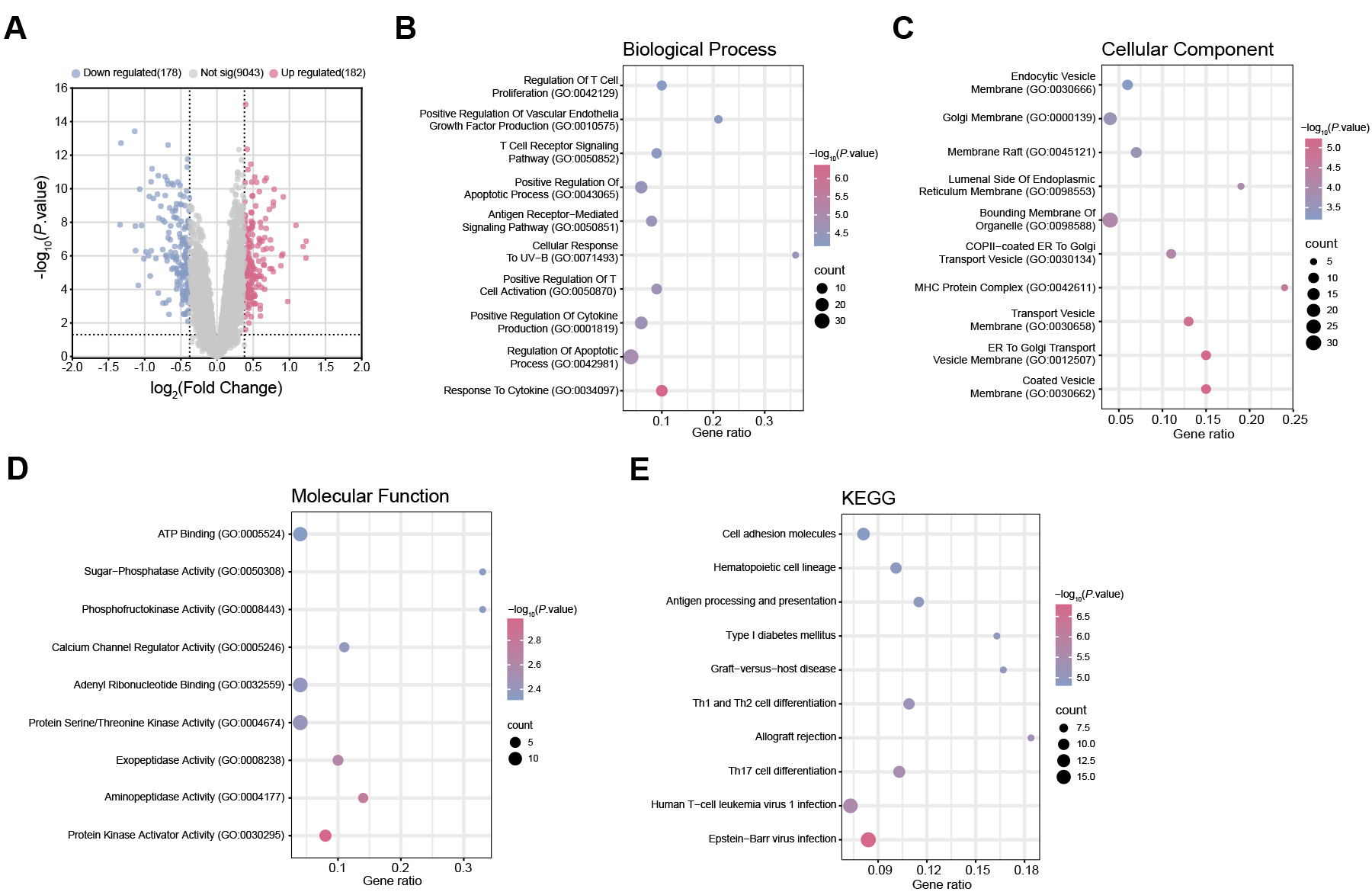 Fig. 1.
Fig. 1.
Differential gene expression and functional enrichment analysis. (A) Volcano plot displaying identified DEGs from the GSE89376 dataset. Each point represents an individual gene. Up-regulated DEGs are depicted in pink, and down-regulated DEGs are depicted in blue. (B) GO enrichment analysis of BP terms associated with DEGs. The x-axis represents the GeneRatio, and the y-axis indicates the enriched terms. The size of the dots corresponds to the number of genes, and the color reflects the adjusted p value. (C) GO enrichment analysis of CC terms of DEGs. (D) GO enrichment analysis of MF terms of DEGs. (E) KEGG enrichment analysis predicted the pathways of DEGs involvement. DEGs, Differential Expressed Genes; GO, Gene Ontology; BP, Biological process; CC, Cell component; MF, Molecular function; KEGG, Kyoto Encyclopedia of Genes and Genomes.
Candidate genes associated with sepsis were identified using PPI network
analysis. Three topological algorithms (MNC, MCC, and Degree) were applied to
screen for hub genes, and the top 10 genes ranked by each algorithm were selected
for comparison (Fig. 2A–C). Venn diagram analysis revealed four overlapping
genes across all three algorithms: CR7, CD247, CD3D,
and CD69 (Fig. 2D). Expression profiling using the GSE89376 dataset
showed significant upregulation of all four genes in sepsis patients compared
with controls (Fig. 2E). Validation in an independent dataset (GSE67652)
confirmed their consistently elevated expression (Fig. 2F). In the GSE89376
dataset, the Cohen’s d for CD3D was 3.36, with a 95% confidence
interval (CI) of 0.43–0.71 (p
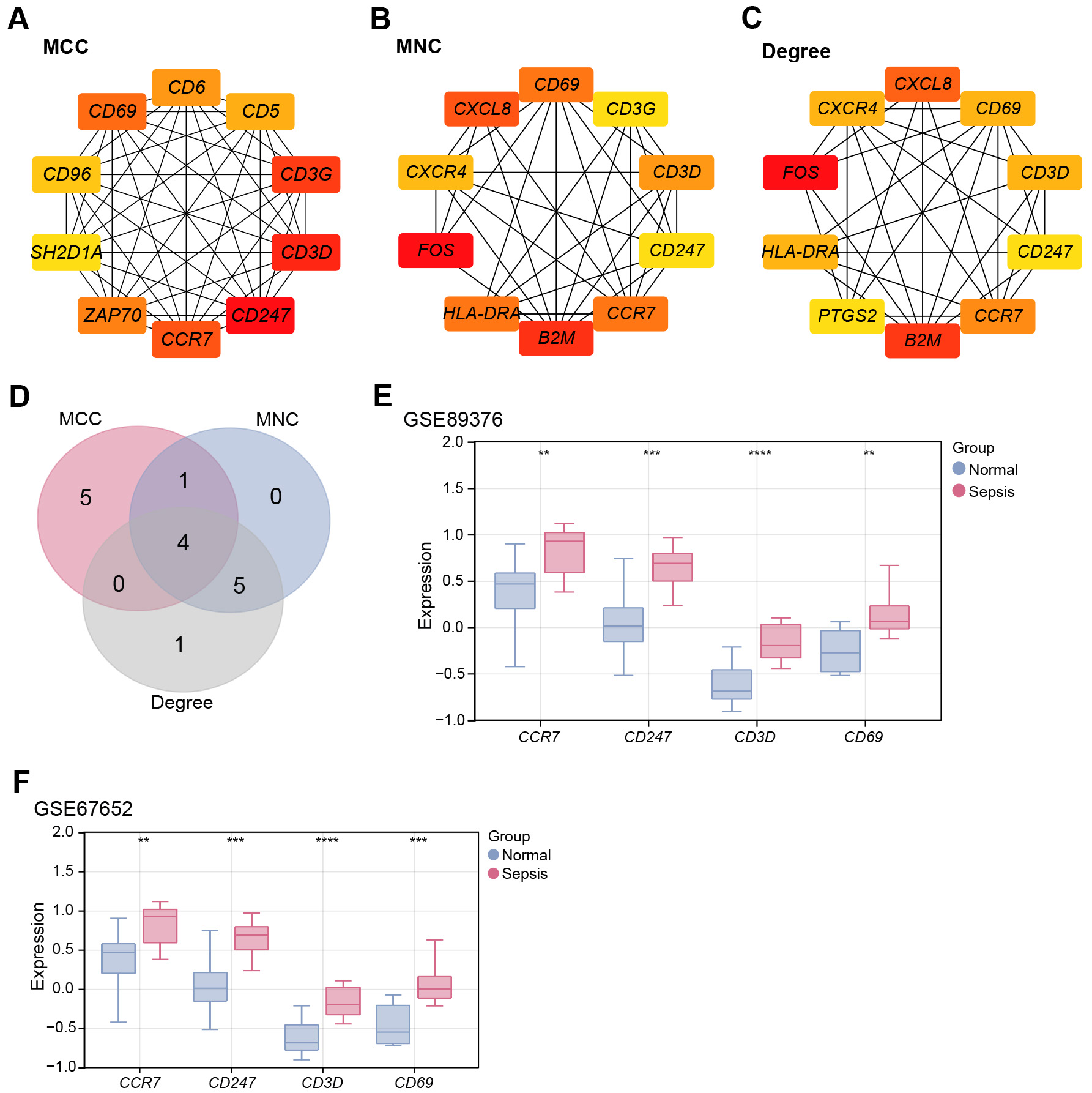 Fig. 2.
Fig. 2.
Identification of CD3D as a key hub gene in sepsis
pathogenesis. (A) PPI network constructed using the MCC algorithm, showing the
top ten highly interconnected genes (10 nodes and 45 edges). (B) PPI network
constructed using the MNC algorithm, displaying the top ten hub genes (10 nodes
and 34 edges). (C) PPI network constructed using the Degree algorithm,
highlighting the top ten hub genes (10 nodes and 33 edges). (D) Venn diagram of
the top ten genes identified by the MCC, MNC, and Degree algorithms, with four
overlapping hub genes obtained. (E) Box plots of the expression levels of
overlapping hub genes (CCR7, CD247, CD3D, and
CD69) in sepsis and normal samples from the GSE89376 dataset. (F) Validation of
the expression of overlapping hub genes (CCR7, CD247,
CD3D, and CD69) in the GSE67652 dataset. **p
HMGB1 has been reported to activate NF-
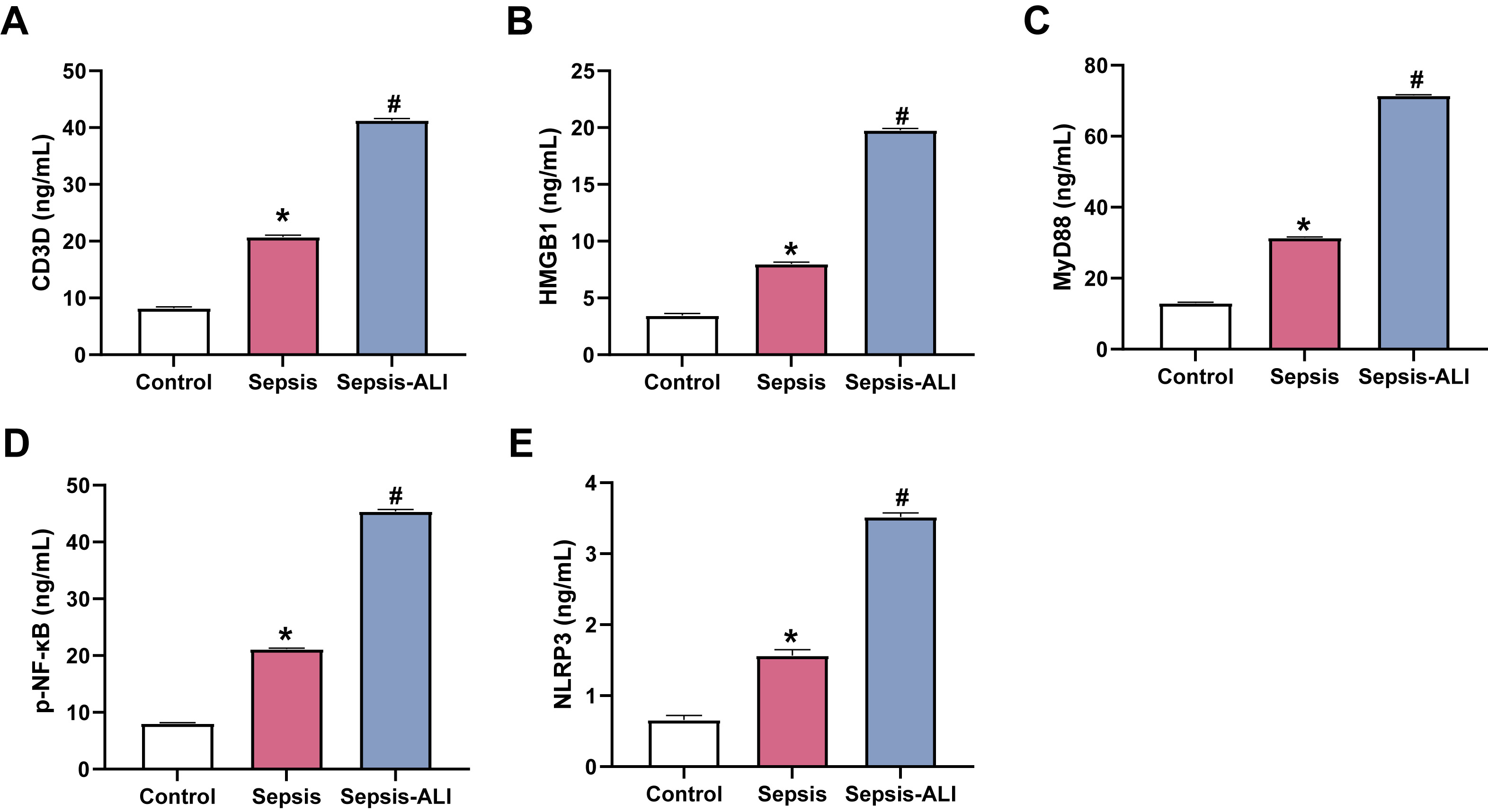 Fig. 3.
Fig. 3.
Sepsis and sepsis-induced ALI are associated with elevated serum
levels of CD3D, HMGB1, MyD88, p-NF-
The knockdown efficiency of two CD3D-targeting siRNAs in hPMECs was
assessed by qRT-PCR and WB. Both siRNAs effectively reduced CD3D
expression, with si-CD3D-2 showing the most substantial effect and
therefore used for subsequent experiments (Fig. 4A–C). To mimic the septic
inflammatory microenvironment, hPMECs were stimulated with LPS. LPS significantly
increased CD3D expression compared with controls, whereas CD3D
knockdown suppressed this induction (Fig. 4D–F). ELISA analysis revealed that
LPS stimulation significantly promoted the secretion of TNF-
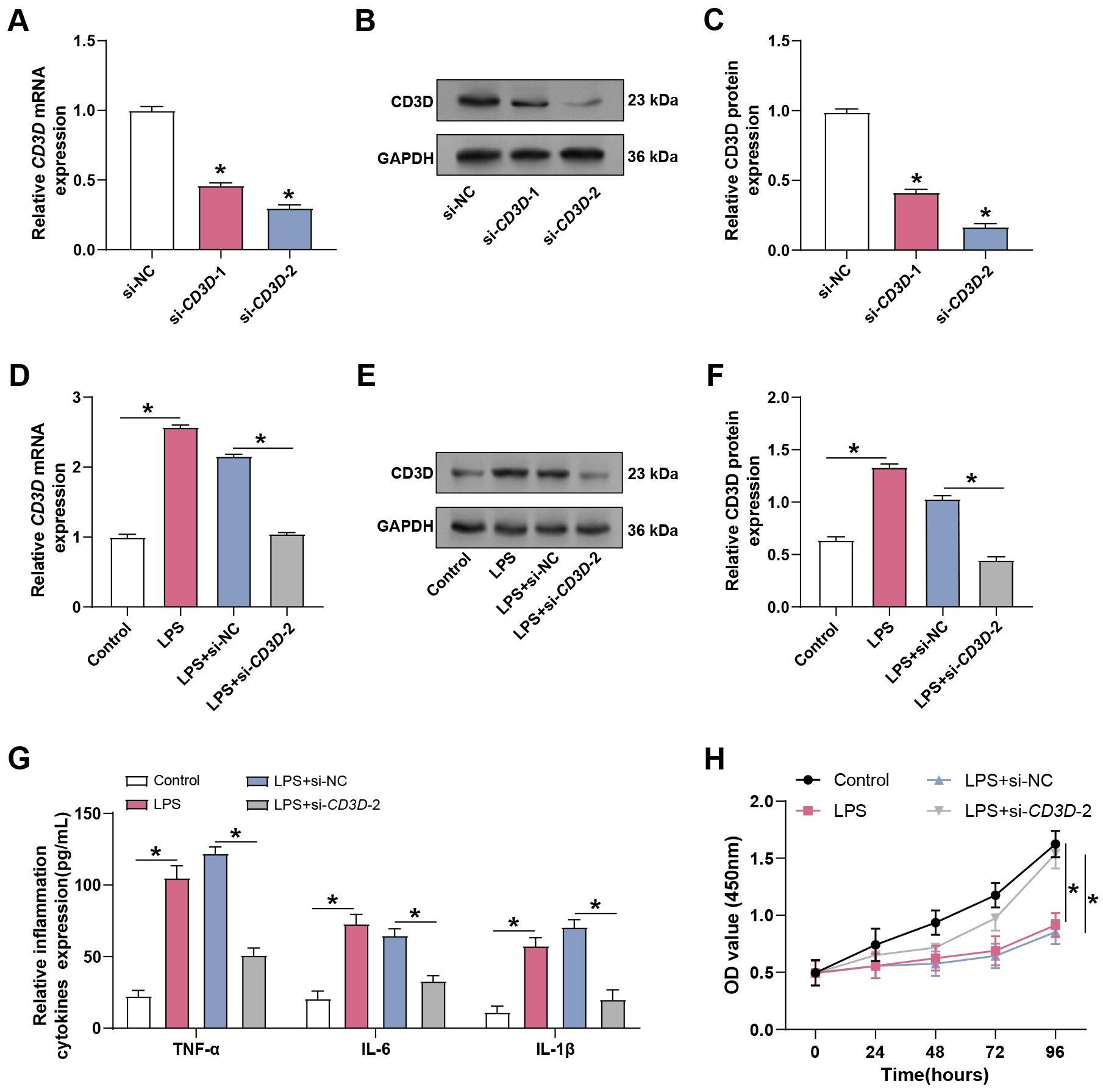 Fig. 4.
Fig. 4.
Knockdown of CD3D alleviates LPS-induced ALI in
hPMECs. (A) qRT-PCR analysis of CD3D mRNA expression in hPMECs
following transfection with CD3D-specific siRNA. *p
To investigate the effect of CD3D knockdown on cell cycle progression, flow cytometry was performed. LPS exposure reduced the S-phase population and increased the proportion of cells in the G1 phase, indicating G1 arrest. CD3D knockdown partially reversed this effect, reducing G1 accumulation and increasing the S-phase fraction (Fig. 5A). Consistently, qRT-PCR showed that LPS treatment upregulated p21 expression while downregulating Cyclin D1 and CDK2. These transcriptional changes were reversed by CD3D knockdown and further confirmed at the protein level by WB (Fig. 5B–D). These findings suggest that CD3D silencing alleviates LPS-induced G1 arrest by modulating cell cycle regulators.
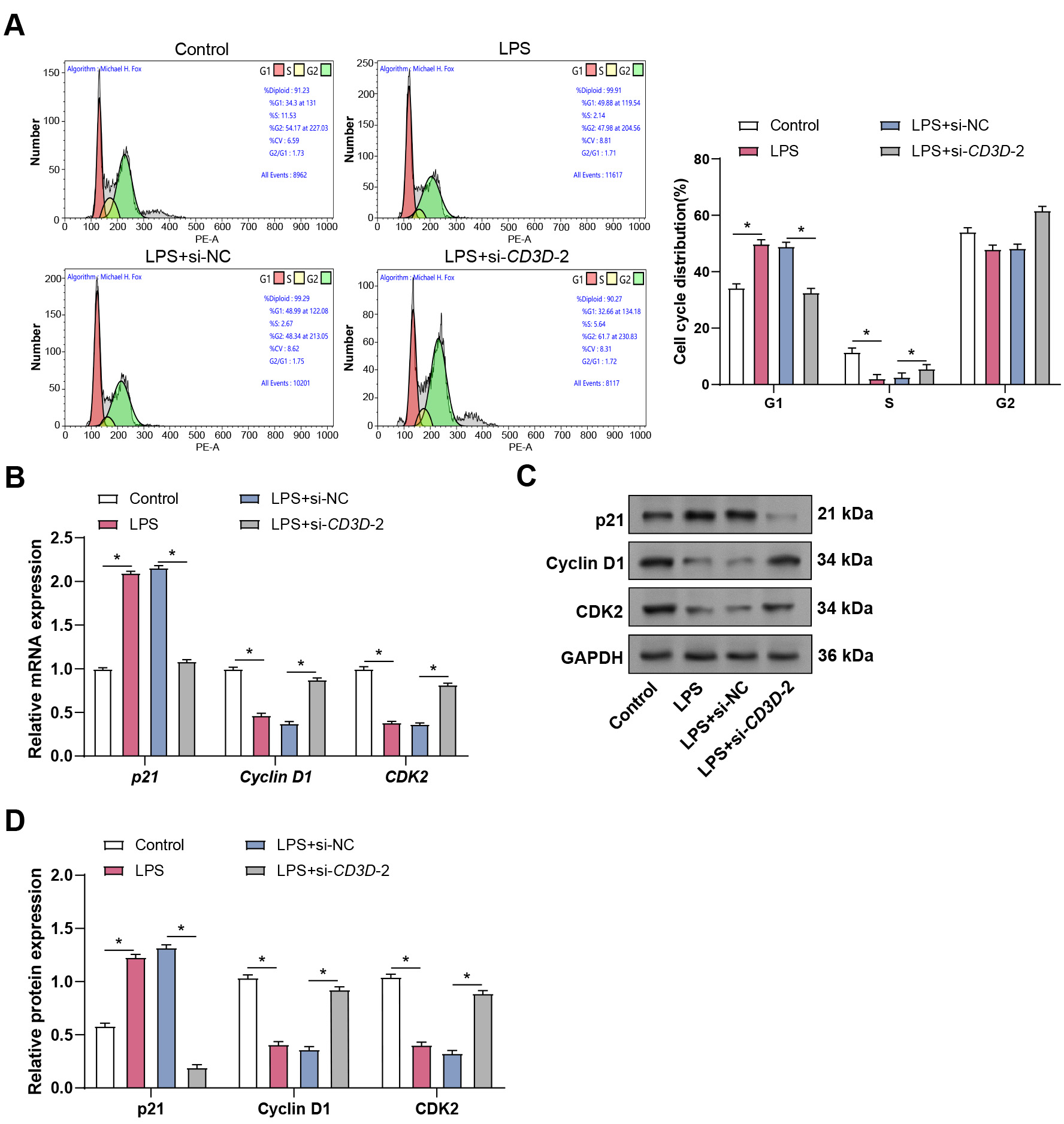 Fig. 5.
Fig. 5.
Effect of CD3D knockdown on sepsis-related LPS-induced
hPMECs cell cycle. (A) Flow cytometry analysis depicts the cell cycle
distribution in hPMECs stimulated with LPS alone or in combination with
CD3D knockdown. (B) qRT-PCR analysis of p21, Cyclin
D1, and CDK2 mRNA expression in hPMECs treated with 10 µg/mL LPS
alone or in combination with CD3D knockdown. (C) WB analysis of p21,
Cyclin D1, and CDK2 protein levels in hPMECs following 24-hour LPS stimulation
with or without CD3D knockdown. (D) Quantification of p21, Cyclin D1,
and CDK2 protein expression levels based on WB results. Each assay was performed
in triplicate to ensure accuracy and reproducibility. Statistical significance
was determined by comparing the mean values of each group. *p
To further evaluate the role of CD3D in sepsis-induced ALI, particularly in relation to oxidative stress, ELISA was used to measure LDH, MDA, SOD, and GSH levels. LPS stimulation significantly increased LDH and MDA, while decreasing SOD and GSH, indicating enhanced oxidative stress and damage. CD3D knockdown reversed these effects by lowering LDH and MDA and restoring GSH and SOD (Fig. 6A–D). In addition, LPS strongly upregulated COX-2 and iNOS, two key inflammatory mediators, at both mRNA and protein levels, whereas CD3D knockdown suppressed this induction (Fig. 6E–I). These results suggest that CD3D plays a role in LPS-induced oxidative stress and inflammatory responses in hPMECs.
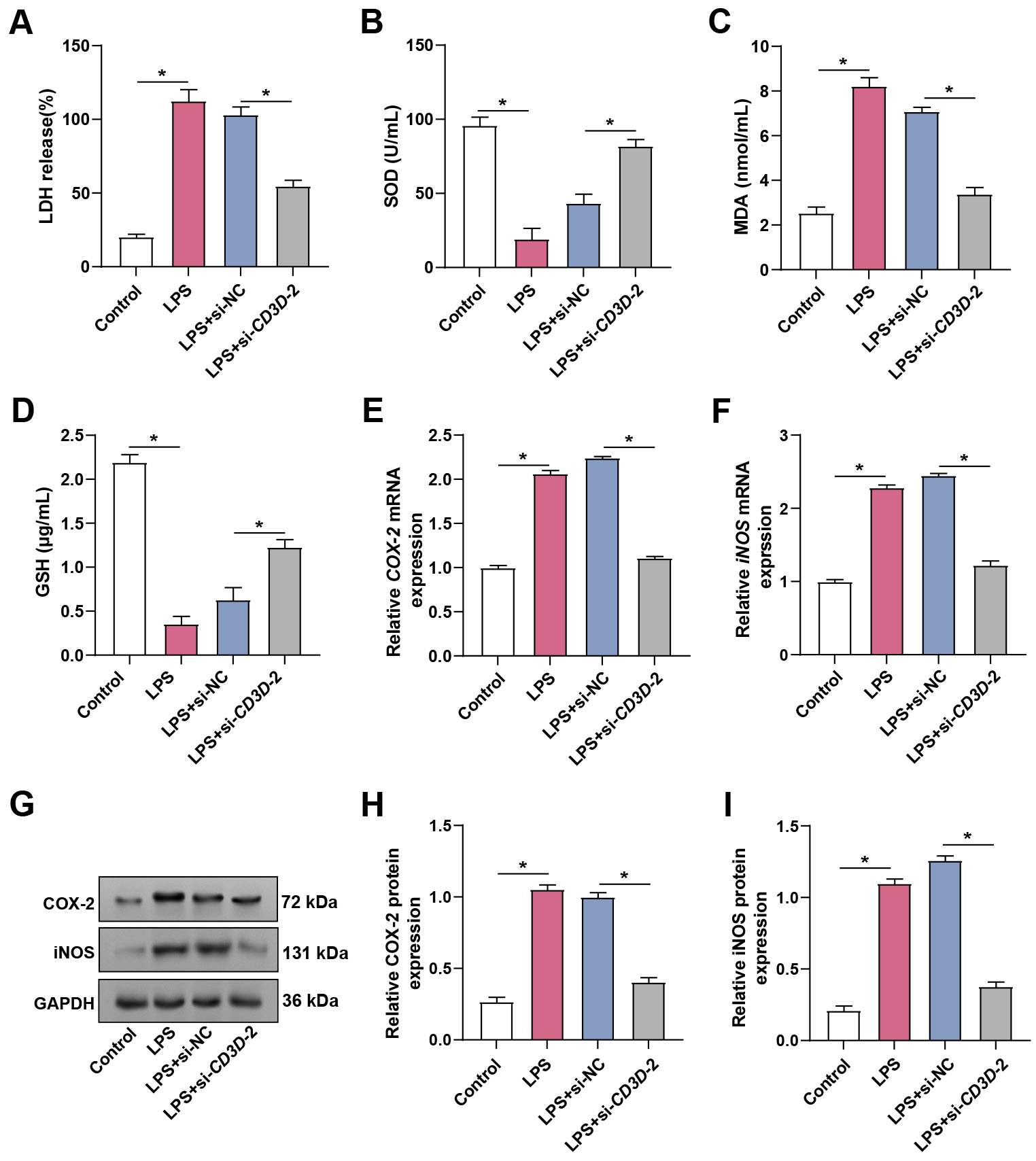 Fig. 6.
Fig. 6.
Effects of CD3D knockdown on oxidative stress and
inflammatory markers in LPS-treated hPMECs. (A) LDH release in hPMECs after
treatment with 10 µg/mL LPS alone or in combination with CD3D
knockdown for 24 hours. (B) SOD activity in hPMECs under the indicated
treatments. (C) MDA levels measured in hPMECs under the indicated treatments. (D)
GSH levels in hPMECs under the indicated treatments. (E) qRT-PCR analysis
of COX-2 mRNA expression in hPMECs treated with LPS alone or combined
with CD3D knockdown. (F) qRT-PCR analysis of iNOS mRNA
expression in hPMECs under the same conditions. (G) Representative WB images
showing COX-2 and iNOS protein levels in hPMECs stimulated with LPS with or
without CD3D knockdown. GAPDH served as the loading control. (H)
Quantification of COX-2 protein expression from WB analysis. (I) Quantification
of iNOS protein expression from WB analysis. LDH, Lactate Dehydrogenase; SOD,
Superoxide Dismutase; MDA, Malondialdehyde; GSH, Glutathione. Each assay was
performed in triplicate to ensure accuracy and reproducibility. Statistical
significance was determined by comparing the mean values of each group.
*p
Calcitonin, secreted by thyroid parafollicular C cells, is known to reduce serum
calcium levels [22]. To explore its protective role, hPMECs were treated with
calcitonin at concentrations of 1, 5, and 10 nM under LPS stimulation. Calcitonin
alone did not significantly alter cell viability. However, LPS markedly reduced
viability, which was restored by calcitonin in a dose-dependent manner (Fig. 7A).
WB further revealed that calcitonin had no significant effect on CD3D protein
expression in control cells; however, in LPS-stimulated hPMECs, calcitonin
suppressed CD3D expression in a concentration-dependent manner (Fig. 7B,C). Flow
cytometry analysis further showed that calcitonin attenuated LPS-induced
apoptosis in a concentration-dependent manner (Fig. 7D,E). Consistently, ELISA
results indicated that calcitonin suppressed LPS-induced secretion of
TNF-
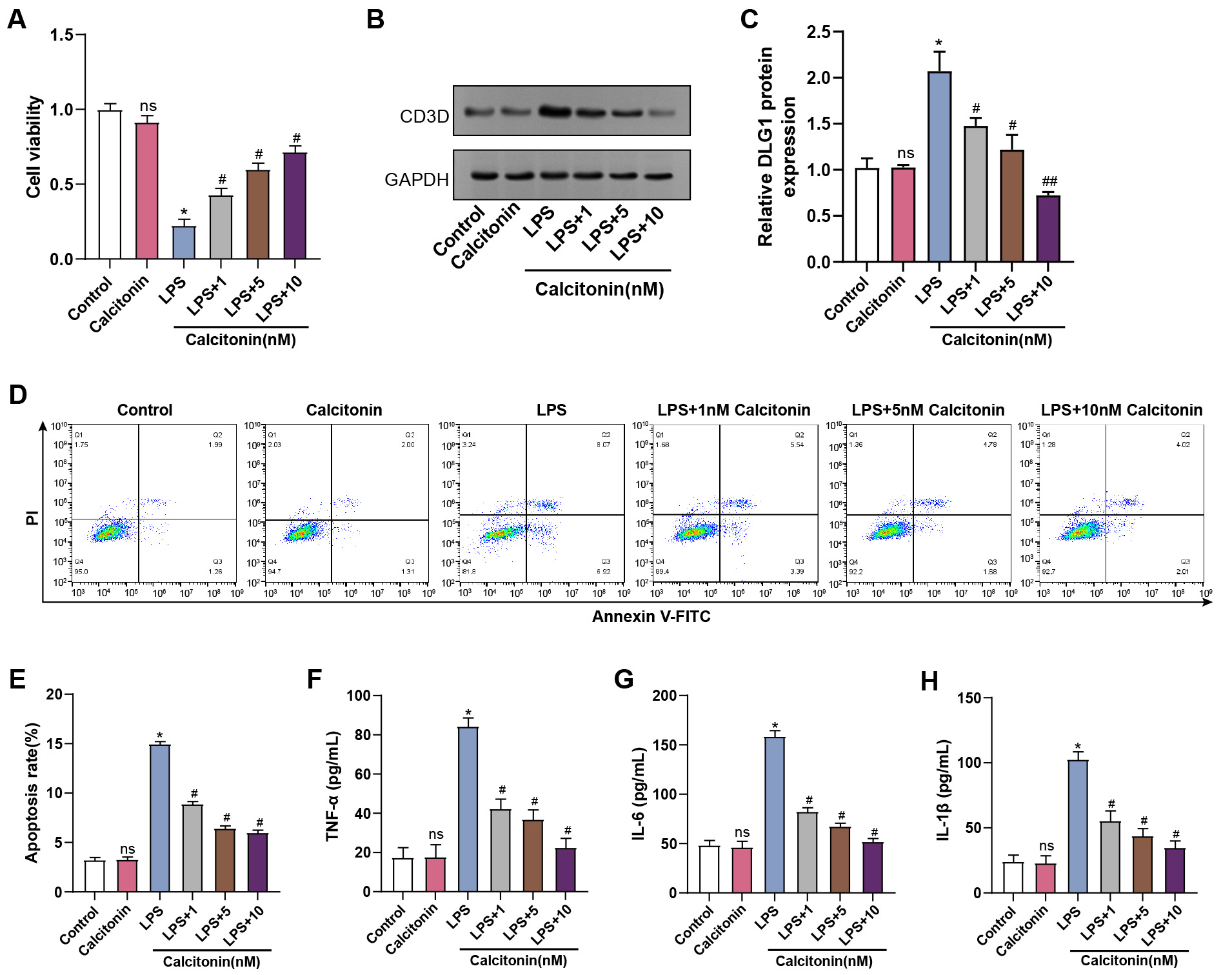 Fig. 7.
Fig. 7.
Effect of calcitonin on cell viability, apoptosis, and
inflammatory cytokine production in LPS-treated hPMECs. (A) Cell viability was
assessed using CCK-8 assay after 24 hours of stimulation with LPS and treatment
with different concentrations of calcitonin (1, 5, 10 nM). The y-axis represents
the cell viability, and the x-axis represents different treatment conditions. (B)
WB analysis of CD3D protein levels in hPMECs after 24 h of LPS stimulation and
treatment with different concentrations of calcitonin (1, 5, 10 nM). (C)
Quantification of CD3D protein expression levels from (B), normalized to GAPDH.
(D) Flow cytometry plots showing apoptosis in hPMECs after 24 hours of
stimulation with LPS and treatment with different concentrations of calcitonin
(1, 5, 10 nM). (E) Quantification of the apoptosis rate in hPMECs under the
indicated treatment conditions. (F) ELISA analysis of TNF-
To evaluate the effects of calcitonin on oxidative stress, ELISA was used to measure SOD, MDA, and GSH levels. LPS stimulation significantly increased MDA, while decreasing SOD and GSH, indicating oxidative stress and injury. Co-treatment with calcitonin reversed these changes in a dose-dependent manner, reducing MDA levels and restoring SOD and GSH activity (Fig. 8A–C).
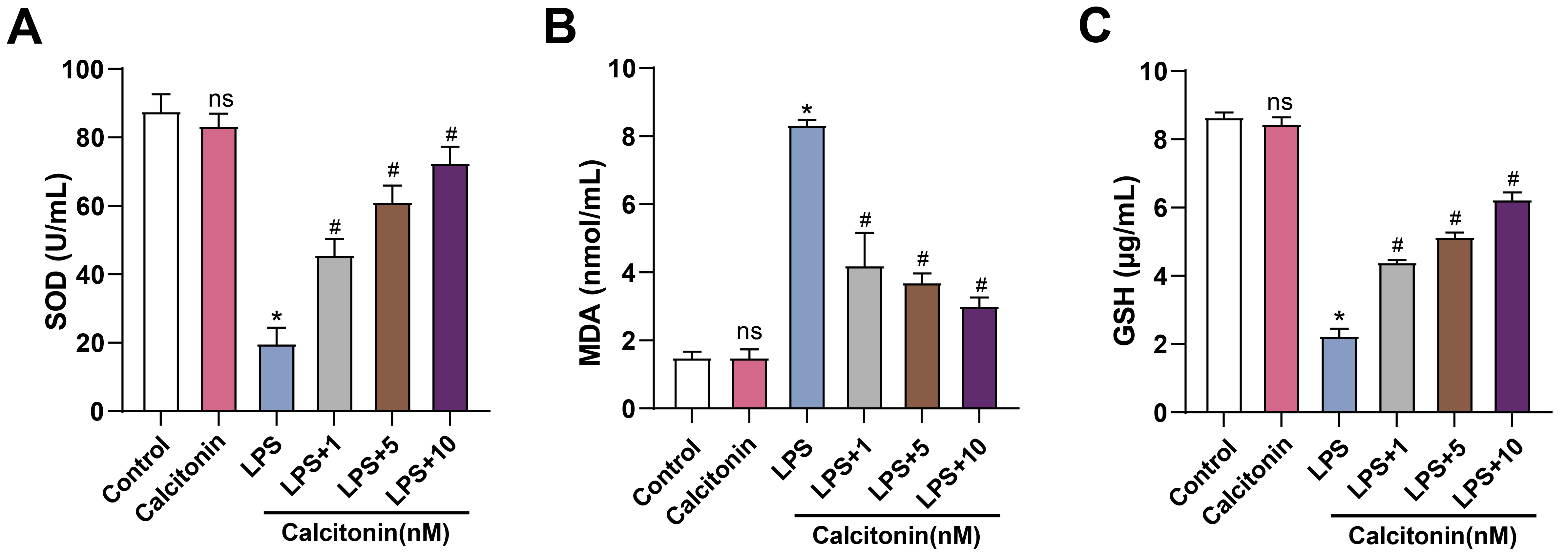 Fig. 8.
Fig. 8.
Calcitonin modulates oxidative stress markers in LPS-stimulated
hPMECs. (A) SOD activity in hPMECs was measured using a commercial SOD assay kit
after 24 h of stimulation with 10 µg/mL LPS, with or without calcitonin
treatment at concentrations of 1, 5, or 10 nM. The y-axis represents SOD activity
(U/mL). (B) MDA levels in hPMECs were determined using a lipid peroxidation assay
kit under the same treatment conditions. The y-axis represents MDA content
(nmol/mL). (C) GSH levels in hPMECs were quantified using a GSH detection kit
under the same treatment conditions. The y-axis represents GSH concentration
(µg/mL). Each assay was performed in triplicate to ensure accuracy and
reproducibility. Statistical significance was determined by comparing the mean
values of each group. *p
To further explore the relationship between calcitonin and inflammatory
signaling, the mRNA levels of key pathway components were assessed in hPMECs
stimulated with LPS, with or without increasing concentrations of calcitonin.
qRT-PCR analysis revealed that calcitonin alone did not alter HMGB1,
MyD88, NF-
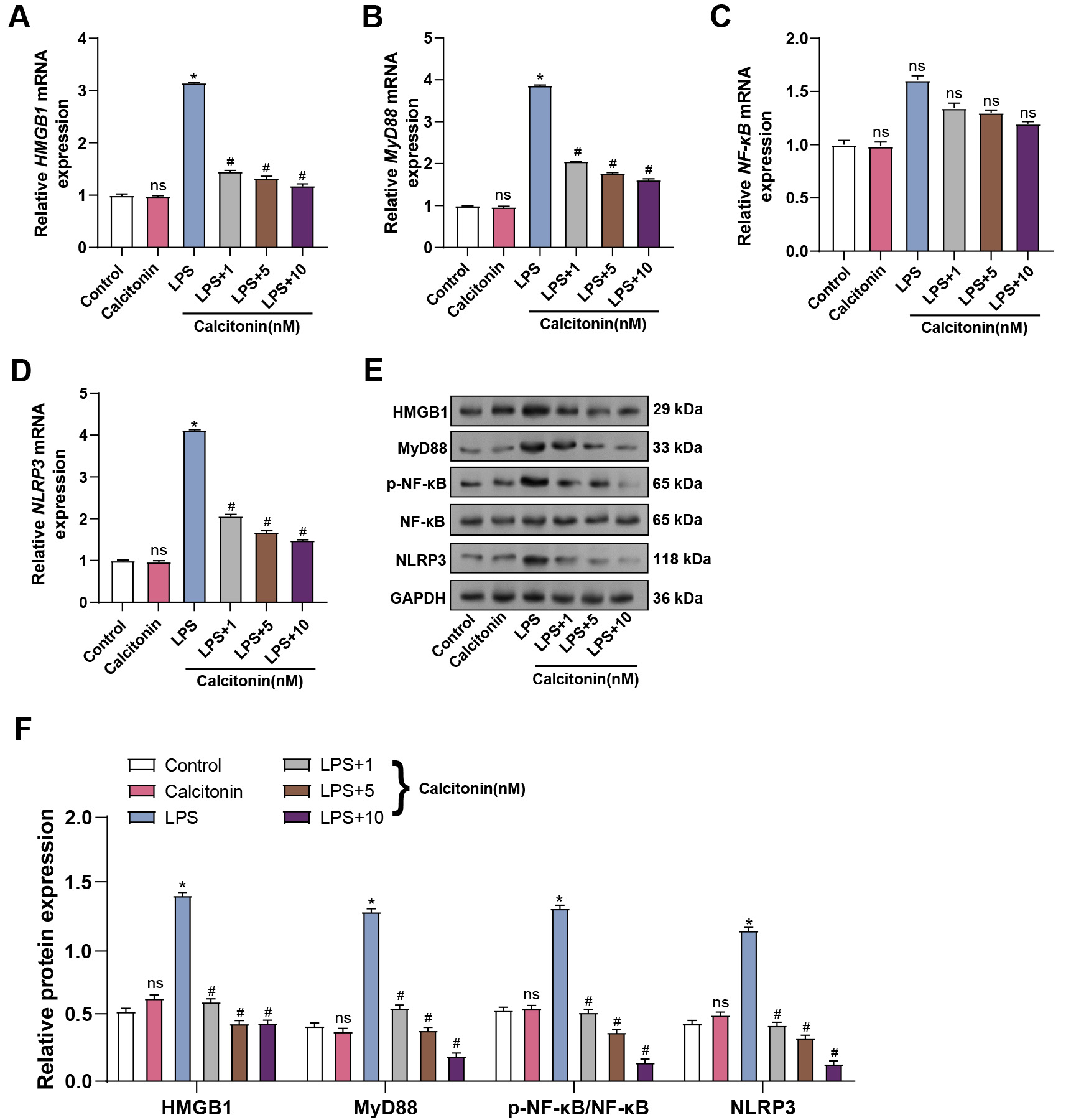 Fig. 9.
Fig. 9.
Calcitonin attenuates LPS-induced inflammatory response via the
NF-
Among the investigated doses, 10 nM calcitonin had the most antioxidative impact and was thus chosen for further investigations. To determine the combined effect of CD3D knockdown and calcitonin, hPMECs were exposed to LPS in the presence or absence of si-CD3D-2 and/or 10 nM calcitonin. CCK-8 assays showed that both CD3D knockdown and calcitonin treatment significantly restored LPS-impaired cell viability, while their combined application further enhanced viability, suggesting a synergistic effect (Fig. 10A). Flow cytometry confirmed that apoptosis was most effectively reduced in the co-treatment group (Fig. 10B). Both qRT-PCR and WB confirmed that combined therapy enhanced Bcl-2 levels while more effectively downregulating Bax and Caspase-3 compared to single-agent treatments (Fig. 10C–G). These data suggest that CD3D depletion and calcitonin act in concert to suppress LPS-triggered apoptosis and enhance endothelial resilience.
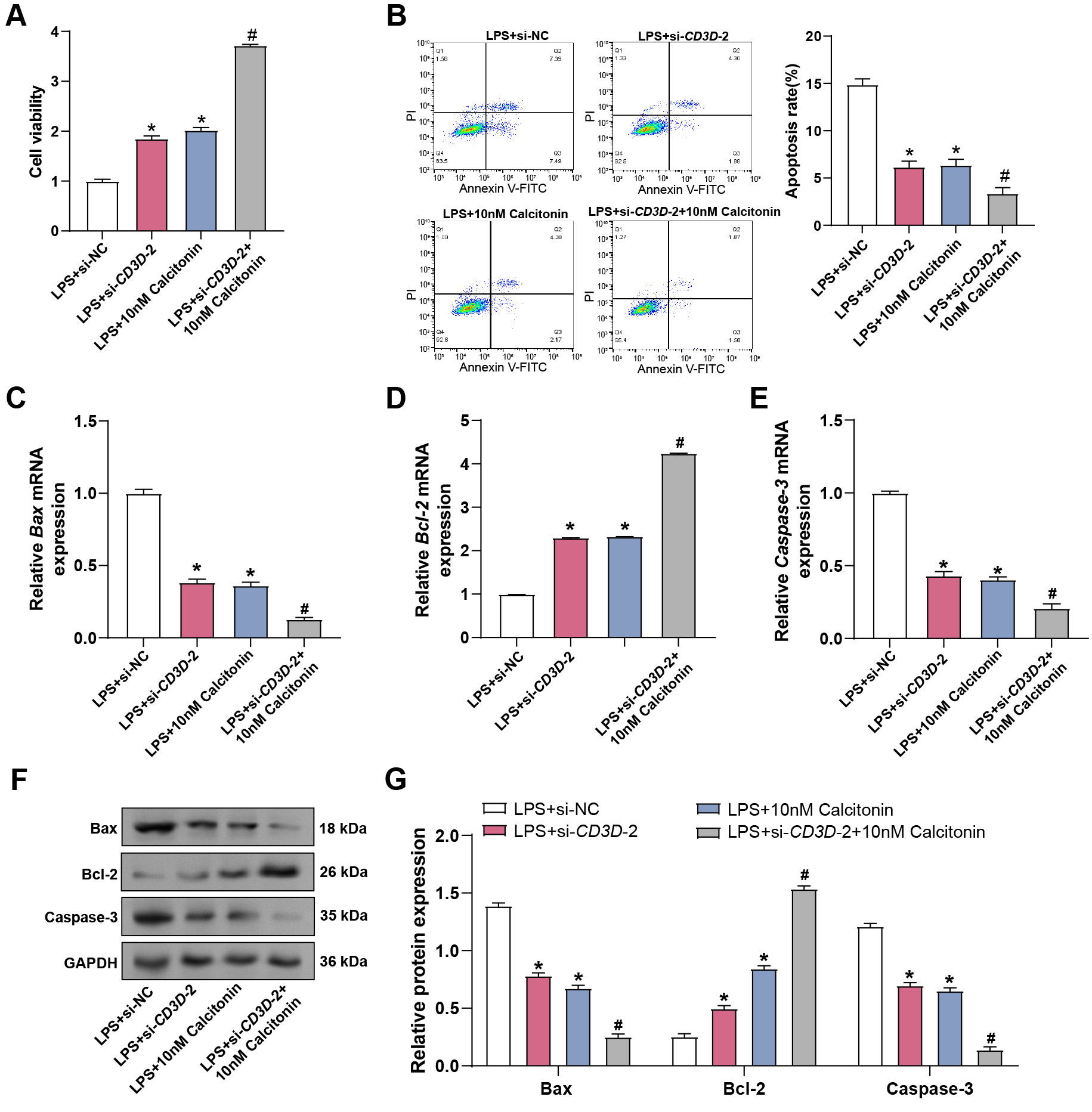 Fig. 10.
Fig. 10.
Combined effect of calcitonin and CD3D silencing on
LPS-induced viability and apoptosis in hPMECs. (A) Cell viability assessed by
CCK-8 assay in hPMECs stimulated with LPS for 24 hours, co-treated with 10 nM
calcitonin and subjected to CD3D knockdown. The x-axis represents
different treatment conditions, and the y-axis represents cell viability. (B)
Apoptosis was detected by flow cytometry in hPMECs stimulated with LPS for 24
hours, co-treated with 10 nM calcitonin, and subjected to CD3D
knockdown. (C) qRT-PCR analysis of Bax mRNA levels in hPMECs stimulated
with 10 µg/mL LPS for 24 hours, with or without CD3D knockdown
and/or 10 nM calcitonin treatment. (D) qRT-PCR analysis of Bcl-2 mRNA
expression in hPMECs under the same conditions. (E) qRT-PCR analysis of
Caspase-3 mRNA expression in hPMECs under the same conditions. (F)
Representative WB images showing protein expression levels of Bax, Bcl-2, and
Caspase-3 in hPMECs under the same treatment conditions. GAPDH was used as an
internal loading control. (G) Quantitative analysis of protein expression levels
of Bax, Bcl-2, and Caspase-3 from WB results in (F), normalized to GAPDH.
*p
To further assess the regulatory effects of CD3D knockdown and
calcitonin on inflammatory and oxidative responses in hPMECs under septic
conditions, ELISA analysis was performed. CD3D knockdown substantially
decreased LPS-induced TNF-
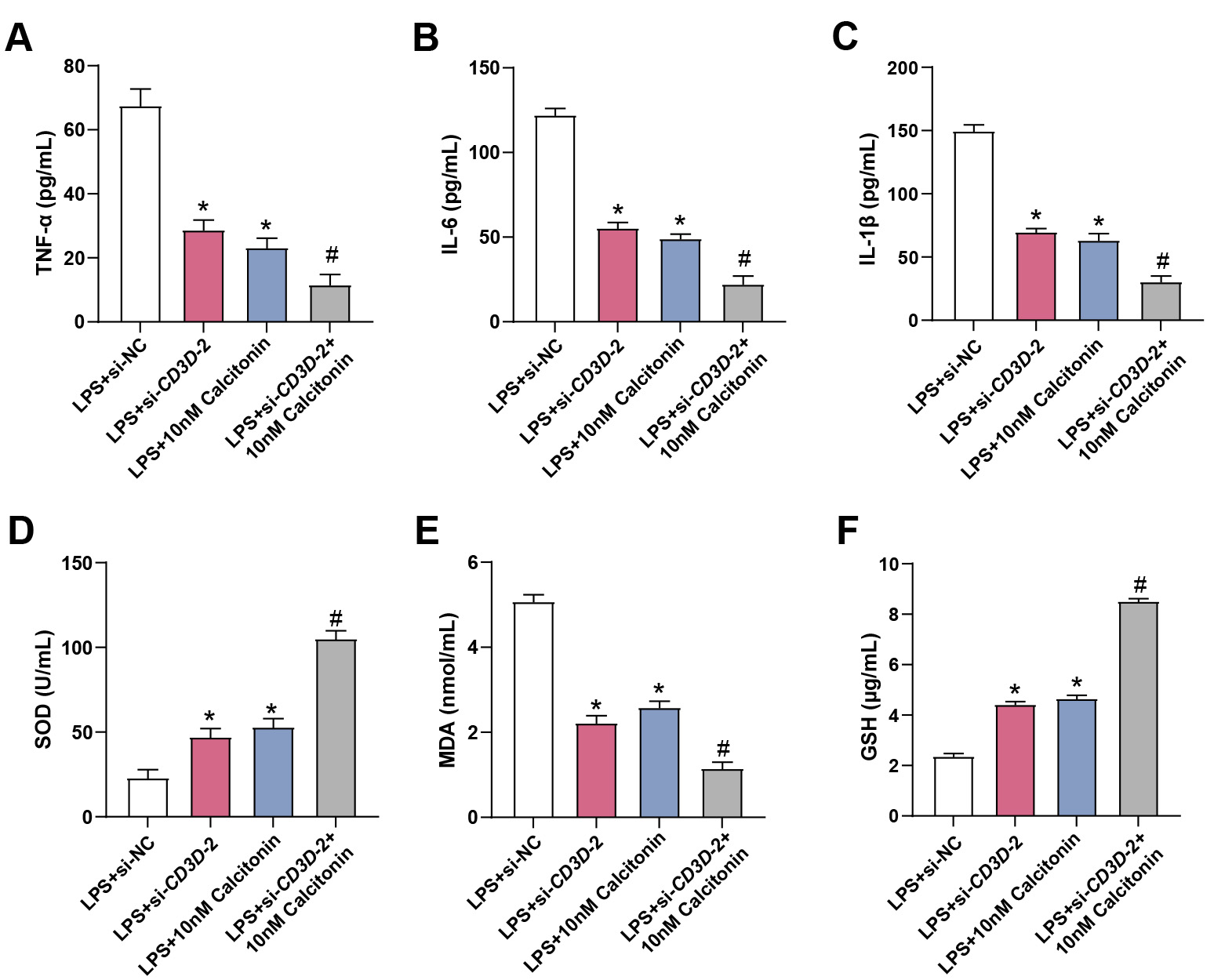 Fig. 11.
Fig. 11.
Effects of calcitonin and CD3D knockdown on
inflammatory cytokine production and oxidative stress in LPS-treated hPMECs. (A)
ELISA analysis of TNF-
To elucidate the underlying mechanisms, qRT-PCR analysis was performed.
CD3D knockdown reduced LPS-induced expression of HMGB1,
MyD88, and NLRP3, and co-treatment with calcitonin further
enhanced this suppression, while NF-
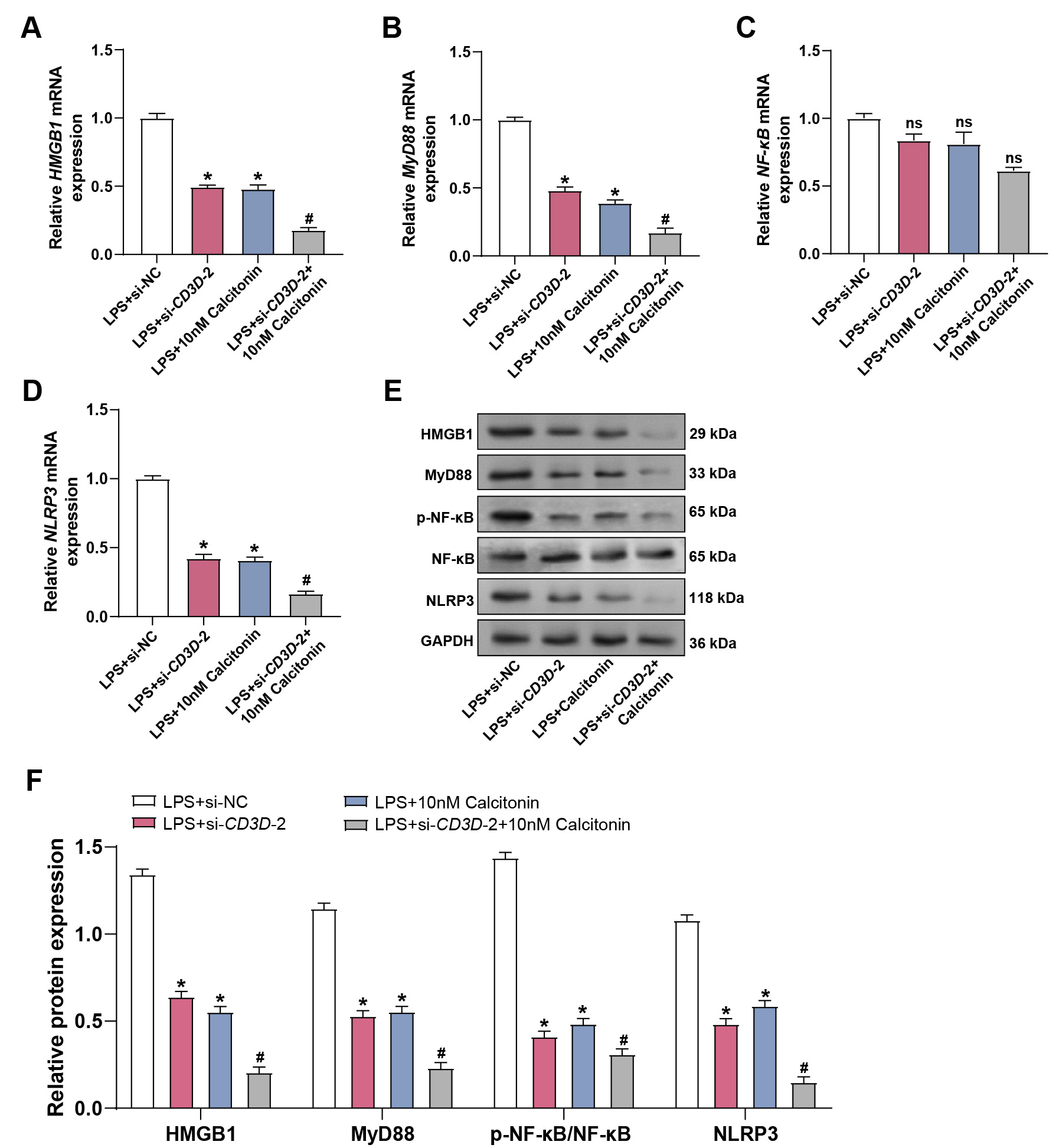 Fig. 12.
Fig. 12.
CD3D knockdown combined with calcitonin suppresses
LPS-induced activation of the HMGB1/MyD88/NF-
Our research comprehensively explored the function of CD3D in sepsis
and sepsis-induced ALI using bioinformatic analyses, clinical samples,
and in vitro experiments. Analysis of sepsis-related transcriptomic
datasets identified CD3D as a hub gene, characterized by elevated
expression in sepsis and further upregulation in patients with ALI. Functional
assays in hPMECs demonstrated that CD3D knockdown mitigated LPS-induced
inflammatory responses, oxidative stress, cell cycle arrest, and apoptosis.
Furthermore, calcitonin was shown to exert protective effects against LPS-induced
endothelial injury in a dose-dependent manner. Importantly, combined treatment
with CD3D knockdown and calcitonin produced synergistic effects in
suppressing inflammation, oxidative stress, and HMGB1/MyD88/NF-
Sepsis can progress to severe infection and complications. Vigilant monitoring and management of organ dysfunction, circulatory failure, and metabolic disturbances are crucial therapeutic measures [23]. In this research, differential expression and functional enrichment analyses of sepsis-related gene datasets were performed, identifying key pathways such as TCR Signaling Pathway, Calcium Channel Regulator Activity, and Endocytic Vesicle Membrane. Our study identified four significantly expressed genes (CCR7, CD247, CD3D, and CD69) through PPI network analysis. Notably, Liang et al. [24] reported that CCR7 and CD3D were closely linked to sepsis. Furthermore, Jiang et al. [25] found reduced expression of FYN and CD247 in septic shock, connecting them to impaired immune responses and decreased T-cell activity. At the same time, Goswami et al. [26] identified CD69 and CD64 as upregulated markers for rapid sepsis detection. In this study, CD3D was identified as the most significantly differentially expressed gene across two independent sepsis-related datasets. In vitro experiments revealed that LPS stimulation markedly upregulated CD3D expression in hPMECs. LPS exposure induced G1 phase cell cycle arrest, whereas CD3D knockdown partially reversed this effect by downregulating p21 and restoring the expression of Cyclin D1 and CDK2, thereby facilitating cell cycle progression.
Immune cells emit important pro-inflammatory cytokines, such as TNF-
In sepsis, excessive release of bacterial endotoxins and cytokines leads to
increased oxidative stress in the host organism. Oxidative stress plays a vital
role in the development of sepsis, causing cellular damage and organ failure
[32]. By preventing endoplasmic reticulum stress and enhancing mitochondrial
activity, Sang et al. [33] showed that quercetin reduced oxidative
stress damage and shielded mice against sepsis-induced ALI. Key indicators of
cellular metabolism and oxidative stress, such as LDH, SOD, MDA, and GSH, are
essential in disease research, providing insights into intracellular mechanisms.
Liu et al. [34] further revealed that elevated LDH levels at admission
were strongly related to increased 30-day mortality in septic patients. This
finding underscores the potential of LDH as an important prognostic indicator for
sepsis outcomes. Therapeutic strategies that focus on oxidative stress and
inflammation have demonstrated promising effects in sepsis-induced ALI. Le
et al. [35] demonstrated that N-acetylcysteine (NAC) mitigates ALI
severity in septic rats by suppressing TNF-
Calcitonin has demonstrated strong anti-inflammatory properties in a range of inflammatory conditions, including sepsis [37]. Its ability to modulate the inflammatory response is critical for reducing tissue damage and improving survival rates in septic models. Baranowsky et al. [14] demonstrated that PCT exerts pro-inflammatory effects in sepsis-induced shock by acting through the CGPR receptor. This mechanism aggravates septic conditions by promoting immune cell production of the inflammatory cytokine IL-17A. In the present study, LPS stimulation of hPMECs exacerbated intracellular oxidative stress, as evidenced by increased MDA and LDH levels, along with reduced activities of the antioxidant enzymes SOD and GSH. Knockdown of the CD3D gene effectively reversed these alterations, thereby alleviating oxidative damage. Moreover, calcitonin exerted a dose-dependent protective effect against LPS-induced cellular injury, progressively restoring cell viability, reducing apoptosis, and significantly inhibiting the release of pro-inflammatory cytokines. Additionally, calcitonin reduced intracellular MDA levels while enhancing SOD and GSH content, collectively mitigating oxidative stress. Moreover, knockdown of CD3D also attenuated LPS-induced cytokine production and oxidative stress, and its combination with calcitonin further potentiated these protective effects, synergistically preserving pulmonary microvascular endothelial cells from injury.
In patients with sepsis and sepsis-induced ALI, serum analysis showed marked
increases in CD3D, HMGB1, p-NF-
Our study demonstrates that both calcitonin treatment and CD3D knockdown reduce inflammatory and oxidative stress responses in vitro. However, the precise mechanistic relationship between calcitonin and CD3D remains unclear. It is not yet determined whether calcitonin directly modulates CD3D expression or whether these interventions act through distinct, convergent pathways to achieve similar protective effects. Further molecular investigations are warranted to clarify this relationship and to elucidate the signaling cascades involved. From a clinical perspective, the in vitro concentrations of calcitonin used in this study (1–10 nM) exhibited pronounced protective effects. Nevertheless, it remains uncertain whether such concentrations can be safely achieved in patients. Careful evaluation of dosing strategies, pharmacokinetics, and potential side effects is therefore essential before clinical translation. Moreover, the present study was primarily conducted in vitro, which may not fully recapitulate the complex immune and metabolic environment of sepsis in vivo. Additionally, the patient serum analysis included an extremely small sample size (n = 3 per group), which is insufficient for drawing robust statistical conclusions and substantially limits the generalizability of our findings. Although Cohen’s d effect sizes were reported to better illustrate the magnitude of group differences, the confidence intervals for several comparisons were wide, reflecting the limited precision caused by the small sample size. This further underscores the need for cautious interpretation and highlights that these preliminary findings should be validated in larger and independent patient cohorts to yield more stable effect size estimates and narrower confidence intervals. The reported dual role of calcitonin in other contexts, including potential pro-inflammatory effects, further underscores the need for cautious interpretation. Future studies with larger patient cohorts and in vivo models are necessary to validate our findings and to optimize therapeutic strategies for targeting inflammation and oxidative stress in sepsis.
In conclusion, this study identifies CD3D as a key pro-inflammatory
mediator contributing to endothelial dysfunction in sepsis and sepsis-induced
ALI. Functional experiments demonstrated that CD3D knockdown alleviated
LPS-induced inflammation, cell cycle arrest, and oxidative stress in hPMECs.
Concurrently, calcitonin exhibited dose-dependent protective effects against
LPS-induced injury, including the suppression of inflammatory cytokine
production, oxidative damage, and apoptosis. Notably, combined CD3D
silencing and calcitonin treatment exerted synergistic benefits by further
restoring endothelial viability and suppressing activation of the
HMGB1/MyD88/NF-
The datasets used and analyzed in this article are accessible from the corresponding author upon reasonable request.
Conception and design: HZ, RZ, JH; Data acquisition: RZ, HW; Data analysis: HZ, RZ, JJ, HW; Manuscript drafting: HZ, RZ; Critical revision: All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Our study is approved by Academic Committee of the Third Affiliated Hospital of Naval Medical University (approval number: KY2024081). All participants and their legal guardians provided written informed consent. The study was carried out in accordance with the guidelines of the Declaration of Helsinki.
Not applicable.
This research was supported by the research on the mechanism of alveolar epithelial cell-derived exosomes loaded with Isthmin-1 in regulating pulmonary capillary leakage in sepsis (2023 QN098) and the research on the mechanism of alveolar epithelial cell-derived exosomes loaded with Isthmin-1 in regulating pulmonary capillary leakage in sepsis (2023 QN008).
The authors declare no conflict of interest.
In this dissertation, the authors used AI tools to assist in literature retrieval and organization/diagram and audio/video production/code debugging and analysis/grammar proofreading and format checking, etc. The core ideas, research design, conclusions and innovations of the dissertation were all completed independently by us. We conducted a legal review and reasonable editing of the content completed with the assistance of this tool, and I assume all legal and academic ethical responsibilities for the content of this dissertation.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.